



 CC-BY 4.0
CC-BY 4.0
1. Introduction
Magnetic resonance imaging (MRI) is currently one of the most widely used and powerful diagnostic imaging modalities in clinical practice, and it has also become an important tool in biomedical research. MRI offers high spatial and temporal resolution and is capable of visualizing soft tissue alterations in order to detect, for instance, chronic inflammation, tumors, tissue perfusion, and other abnormalities, making it an invaluable tool for diagnosing various diseases [1, 2, 3]. This non-invasive technique is based on applying an external magnetic field to align the spin moments of protons in the water molecules within the tissues or organs of interest. After low-energy radio frequency irradiation, the longitudinal (T1) and transverse (T2) relaxation properties of these protons are exploited to produce detailed images of internal structures [2].
Thus, the images generated by MRI result from differences in the density (i.e., concentration) and the nuclear relaxation properties of protons, mainly of water, which produce variations in signal intensities. However, the inherent contrast is often insufficient for accurate diagnosis, and therefore contrast enhancing agents are needed. These paramagnetic or superparamagnetic drugs can reduce the longitudinal and transverse relaxation times of the surrounding water protons, thereby considerably improving the diagnostic quality of the images [4, 5, 6].
All commercial MRI contrast agents as well as the overwhelming majority of potential probes reported in the literature are gadolinium(III) (Gd) chelates, which involve the use of polyamino-polycarboxylate ligands. Gd(III) complexes formed with macrocyclic (gadoteric acid, gadobutrol, gadopiclenol) or linear (gadopentetic acid) structures have been thus far the golden standard in clinical practice. They have been efficiently used for instance to highlight morphological and functional abnormalities, enhance blood vessels in MR angiography, and to assess blood–brain barrier degradation [7, 8].
In recent years, with the overall aims of improving sensitivity, specificity, and safety of MRI contrast agents to promote earlier and more reliable detection of diseases such as cancer and neurodegenerative disorders, research has been pursued in several domains: (a) increasing the relaxation efficiency of the complexes [6]; (b) addressing contrast agents to specific biological targets [3, 9]; (c) detecting variations in tissue physical–chemical properties like pH, temperature, cation concentration, and redox state with smart probes [10, 11, 12, 13]; and (d) substituting Gd(III) with more biocompatible paramagnetic metal ions such as Mn(II), Mn(III), and Fe(III) [14, 15, 16, 17, 18]. Coordination chemistry is fundamental to addressing these challenges. Within this general context, this review aims to focus on the collaborative achievements between research groups in the Centre of Molecular Biophysics, CNRS, Orléans and in the Coimbra Chemistry Centre - Institute of Molecular Sciences (CQC-IMS), Chemistry Department of the University of Coimbra, Portugal over the past two decades. This highly fruitful partnership began with the investigation of basic coordination properties of macrocyclic lanthanide chelates, and has been later extended to the design of MRI agents with increased efficiency or various targeting capabilities. In particular, a lot of effort has been dedicated to the visualization of amyloid peptides, mostly in relation to Alzheimer’s disease. More recently, the monitoring of the tissue redox environment, using Mn-complexes of tetrapyrrolic macrocycles, has become a major focus. Finally, detection has been also extended to 19F MRI, which offers complementary advantages over classical 1H MRI. The excellence of this collaborative research, leading to 36 joint publications altogether, was recognized by the Portuguese and French Academies of Sciences with the attribution of the Mariano Gago Prize in 2022. This minireview illustrates, without the intention to be exhaustive, some of the main achievements of the Coimbra–Orléans collaboration via a selected number of representative examples.
2. Strategies to increase the efficiency of MRI agents
The MRI efficiency of a paramagnetic metal chelate is expressed by its proton relaxivity, r1, defined as the paramagnetic enhancement of the longitudinal water proton relaxation rate referred to 1 mM concentration. Proton relaxivity is the sum of inner- and outer-sphere contributions originating from dipolar interactions between the electron spin of the paramagnetic metal ion and the nuclear spins of the water protons in the inner coordination sphere or of those diffusing in the proximity of the complex (outer sphere), respectively. The inner-sphere term can be modulated by chemical design, as it is determined by structural and dynamic properties of the metal complex. Among these, the most important are the number of inner-sphere coordinated water molecules (q), their exchange rate with bulk water (kex), the rotational correlation time of the chelate (𝜏R), and the electron spin relaxation times of the metal ion (T1,2e) [20]. For small molecular weight Gd(III) complexes, such as clinical agents, relaxivity is limited by fast rotation. Therefore, many different approaches have been explored in the past in order to slow down the motional dynamics of the chelates and increase relaxivity. Slow rotation promoted by macromolecular systems can indeed lead to a remarkable (5–10 fold) relaxivity improvement between 20 and 60 MHz. However, this effect vanishes at higher frequencies. Given the great increase in detection sensitivity (and consequently of image resolution) with magnetic field, there is a clear interest in performing MRI at high fields (4.7–9.4 T, corresponding to 200–400 MHz proton Larmor frequency) especially for preclinical studies, where the small animal size calls for high sensitivity. The relaxivity optimization at these magnetic fields requires a different strategy, in particular, appropriate fine-tuning of the rotational motion by using medium-sized complexes instead of macromolecules. In addition, the rigidity of the molecules is another key element in order to exclude internal flexibility, which could be detrimental to relaxivity gain. We have achieved this with various molecular design approaches, such as the creation of a metallostar [21, 22] or a benzene-core trimer [19, 23] (Figure 1), both bearing bis(hydrated) Gd(III) chelates to maximize the relaxivity. Indeed, inner-sphere relaxivity is linearly proportional to the hydration number; thus increasing q has a direct effect, which is independent of the magnetic field. Thanks to the combined effects of the optimal molecular size, the rigidity, and the presence of two inner-sphere water molecules, these complexes are endowed with remarkably high proton relaxivities at high magnetic fields as compared to clinical agents (Figure 1). This excellent relaxation efficiency is retained in vivo as evidenced by MRI experiments on healthy mice [19, 21]. For both compounds, the MRI data have been further complemented by biodistribution studies performed in γ-scintigraphy imaging in rats using the 153Sm analogue. Both MRI and γ-scintigraphy confirmed fast renal elimination of the two compounds, leakage to the extracellular space in the muscle tissue, and no permeation to the brain with intact blood–brain barrier, all important for the development of general-purpose MRI agents.
(a) Structure of the metallostar and of the Gd3L1 trimer having optimized relaxivity for high-field MRI applications. (b) Relaxivities at different frequencies and temperatures compared to the clinical agents GdDOTA and GdDTPA. (c) Dynamic contrast enhanced magnetic resonance images acquired at 9.4 T (400 MHz proton Larmor frequency) after GdDOTA or Gd3L1 injections at 8 mmol⋅Gd⋅kg−1 dose. For both complexes, marked signal intensity increase is observed in the vascular system, renal cortex, and medulla; the signal enhancement is considerably higher with Gd3L1 due to its threefold higher relaxivity (adapted with permission from Contrast Media Mol. Imag. [19]).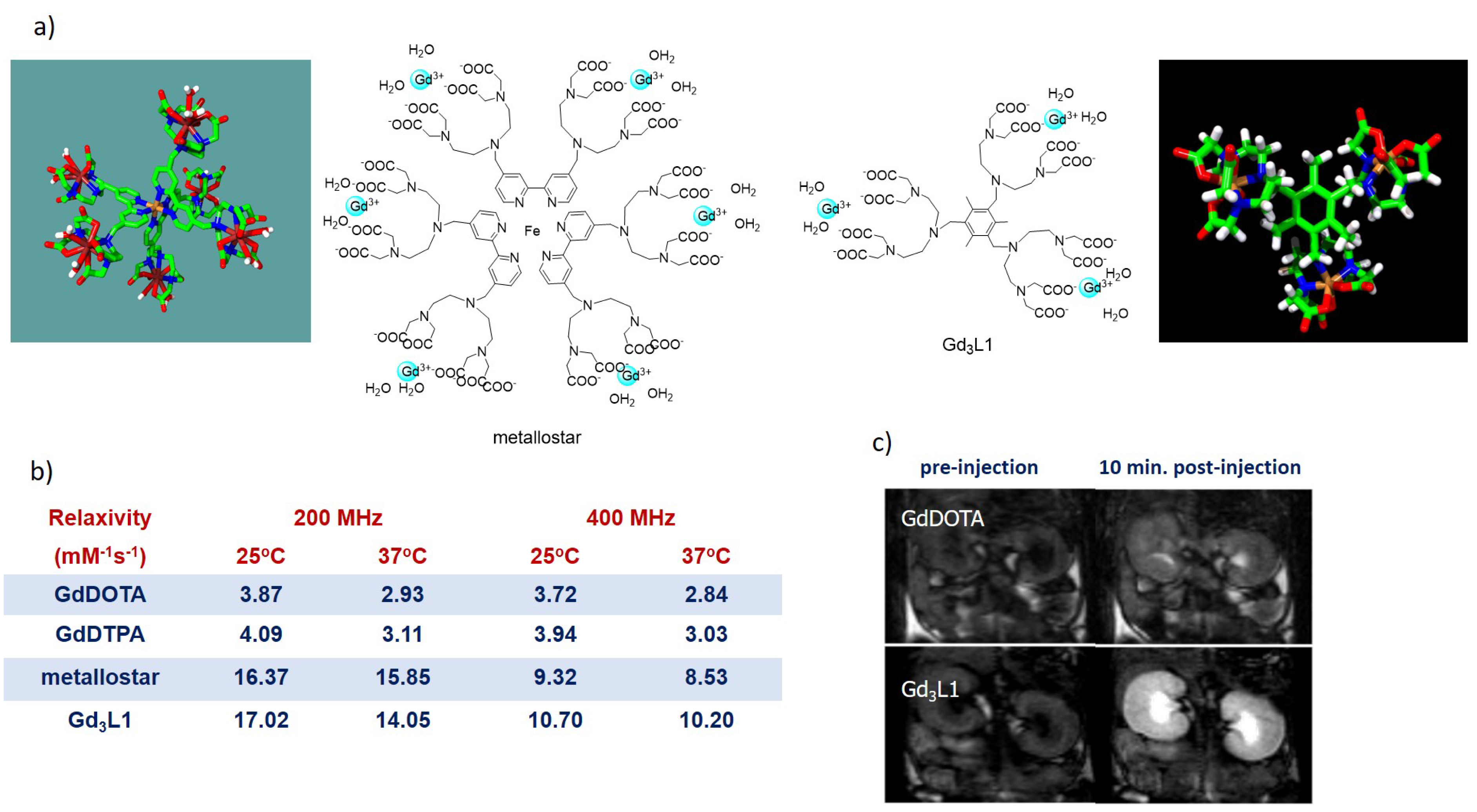
In the search for higher MRI efficiency, many efforts have been also dedicated to the optimization of the water exchange rate. While for typical DOTA- or DTPA-type small molecular weight Gd(III) complexes, the water exchange process is fast enough not to be limiting for proton relaxivity, for slowly rotating macromolecular systems with optimized rotational dynamics, water exchange can become the limiting parameter [24]. Consequently, it is important to design chelates with accelerated water exchange. It has been shown that nine-coordinate, monohydrated Gd(III) complexes undergo dissociative water exchange [20], implying that the rate-determining step is the dissociation of the leaving water molecule before the entry of the incoming water. For such water exchange processes, steric crowding around the water binding site was found to be the key element determining the exchange rate. The steric crowding could be increased by replacing an ethylene by a propylene bridge in the DTPA backbone (EPTPA) [25, 26] or an acetate with a propionate pendant arm in DOTA (DO3A-prop) [27], leading to two and one orders of magnitude increase in the water exchange rate, respectively (Scheme 1). Both of these approaches have been further combined with simultaneous optimization of the rotational motion of the chelates. For instance, a long hydrophobic chain has been attached to the ligand (EPTPAC16; Scheme 1). This promotes the formation of micelles in aqueous solution of the Gd(III) chelate, endowed with slow rotation and consequently improved proton relaxivities [26, 28]. Indeed, the relaxivity of Gd(EPTPAC16) amounts to 9.1 mM−1⋅s−1 in the monomeric and to 20.6 mM−1⋅s−1 in the micellar form (measured at 0.2 mM and 2.0 mM, below and above the critical micellar concentration [cmc], respectively) as compared to 3.3 mM−1⋅s−1 for GdDTPA (20 MHz, 25 °C). Some of the complexes have been also successfully evaluated in vivo in MRI experiments performed on rats [29].
Accelerating water exchange for DOTA- and DTPA-type Gd(III) complexes by increased steric crowding around the water binding site. The water exchange rates, kex, indicated refer to 298 K.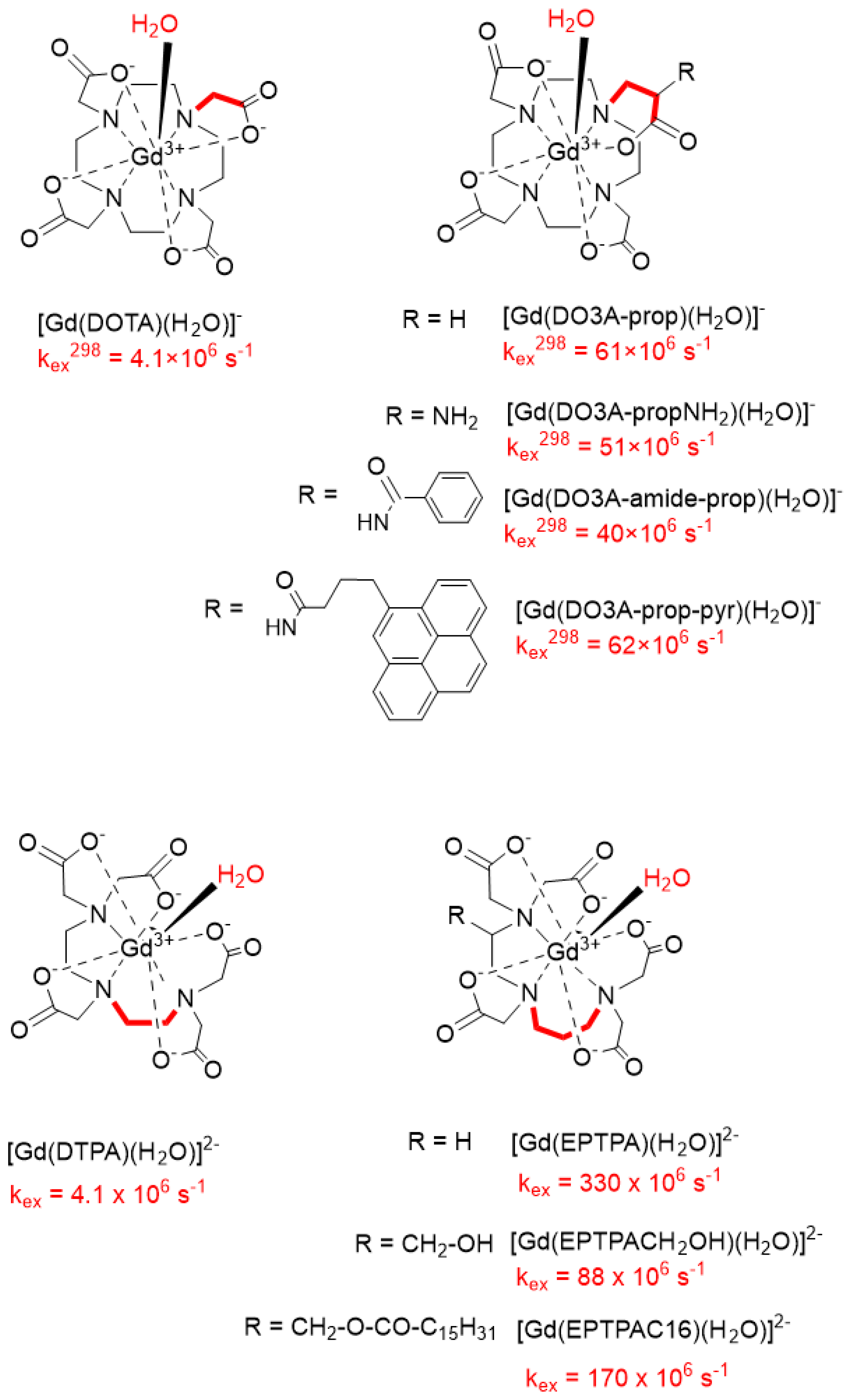
3. Design of lanthanide-based bimodal MRI and optical imaging probes
In biomedical imaging, there has been a continuous interest in combining two (or more) detection modalities in bimodal (or multimodal) approaches in order to ascertain the results [30]. Ideally, a bimodal imaging probe should incorporate, within the same molecular entity, detection capabilities in both modalities. Since lanthanide ions have very similar coordination chemistry but highly versatile magnetic and optical properties, lanthanide complexes provide an ideal platform for the development of bimodal optical and MRI agents [31, 32, 33, 34, 35, 36, 37, 38]. Indeed, several lanthanide ions can have luminescence emission in the visible region or in the near-infrared region (NIR), provided they are sensitized via an appropriate chromophore that is able to absorb the exciting light and transfer the energy to the lanthanide ion to generate characteristic lanthanide luminescence [39]. The very sharp emission bands that are independent of experimental conditions, the long emission lifetimes, or the high resistance to photo-bleaching are all advantageous features of luminescent lanthanide probes with respect to organic fluorophores [40]. The same ligand can be applied to complex Gd(III), adapted to MRI detection, and another luminescent lanthanide ion for optical detection, and the two complexes are expected to have an identical biodistribution, thereby allowing for straightforward overlapping of the results obtained with the two different imaging modalities. However, while the coordination of one inner-sphere water molecule in the Gd(III) chelate is indispensable to MRI, it has negative consequences for the optical properties of the luminescent analogue given the non-radiative energy dissipation due to O–H resonances. This has to be therefore compensated by efficient sensitization of the luminescence using well-adapted chromophore antennas. The situation is even more challenging with bishydrated complexes, which provide increased relaxation efficacy for the Gd(III) analogues or for lanthanide ions with near-infrared emission.
We have investigated different ligand systems in the context of developing lanthanide-based bimodal, optical, and MRI agents. For instance, a pyrene derivative of the DO3A-prop ligand (DO3A-prop-pyr, Scheme 1) has been complexed to Gd(III) as well as to the near-infrared emitting Nd(III) and Yb(III) ions [28]. Here the pyrene itself can be used as a fluorophore, but it was also demonstrated that it can also sensitize the near-infrared luminescence of Nd(III) and Yb(III). The aggregated micellar form of the Gd(III) complex displays an impressive relaxivity (32 mM−1⋅s−1, 20 MHz, 25 °C) resulting from the simultaneous optimization of the rotational dynamics and of the water exchange.
In another approach, the pyridine N-oxide coordinating function was used to replace two acetate arms in trans position of the DOTA to obtain the DO2A-trans-(pyNO)2 ligand (Scheme 2), and its Ln(III) complexes were investigated as potential bimodal probes. Interestingly, the water exchange rate on the Gd(III) analogue is only 50% higher than that on GdDOTA despite the increased steric hindrance induced by the two six-membered chelate rings. This limited increase was attributed to the positive charge of the chelate, which is known to slow down the dissociation of the leaving water molecule at the rate-determining step. The luminescence properties have been assessed for several lanthanide complexes (Eu(III) and Tb(III) emitting in the visible, and Yb(III), Nd(III), Ho(III), and Pr(III) emitting in the NIR region). Remarkably, the pyridine N-oxide was found to be a very efficient chromophore antenna for all these metal ions in aqueous solution, with a particularly interesting quantum yield for Yb(III) (0.73% in H2O) [41].
Chemical structure of the DO2A-trans-(pyNO)2 ligand.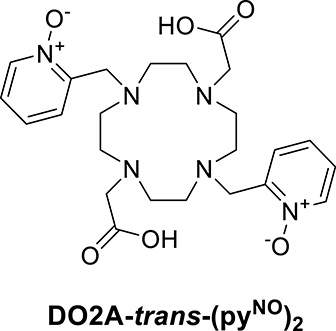
4. Metal complexes targeted at amyloid peptides
Even though the ability to form amyloid fibrils is nowadays considered a natural property of polypeptide chains, it has been recognized early on that amyloid forms of certain proteins are characteristic of various pathologies and can be considered relevant biomarkers for their imaging detection. The most well-known examples are Aβ in the context of Alzheimer’s disease, amylin in type 2 diabetes mellitus, and α synuclein in Parkinson’s disease, just to name a few. Amyloidogenesis involves all different processes corresponding to the unfolding and misfolding of proteins that are not corrected by protein quality control and degradation mechanisms, and thus lead to misassembled, soluble or insoluble, toxic, and oligomeric or polymeric fibrillary aggregates. The presence of these protein forms affects cell or organ function, potentially leading to cell death. Amyloidogenesis often occurs well before the clinical onset of diseases. Therefore, the detection of misassembled protein forms represents an important goal for molecular imaging diagnosis of amyloid-related pathologies.
Metal-based imaging agents capable of visualizing amyloid peptides have been the focus of very intensive research for many years now, with the most attention being directed at Alzheimer’s disease by using nuclear imaging techniques [42, 43, 44]. We have been long engaged in this research via the development of potential imaging agents for Aβ and amylin. Our probes are constructed in a modular way, consisting of a macrocyclic, DOTA-type metal chelate, an amyloid recognition moiety based on the benzothiazole derivative Pittsburg compound B (PiB), and a linker between the two. The modularity allows for a better understanding and control of the role of each element in amyloid binding properties. The DOTA-type chelator makes the probes easily adaptable to various imaging modalities, as it can efficiently complex various metal ions suitable for MRI (Gd(III)), for optical detection (luminescent lanthanides), and for nuclear imaging (e.g., 111In for single-photon emission computed tomography and 68Ga for positron emission tomography [PET]).
We have investigated the influence of different structural variations on peptide binding properties: the length and the chemical nature of the linker, the orientation of the benzothiazole targeting unit, and the charge of the metal complex. The first generation of compounds (L1–L4, Scheme 3) [42, 45, 46, 47, 48, 49, 50] contained a short spacer between the metal chelate and the PiB, and their Gd(III) complexes displayed dissociation constants (Kd) in the range of 67–194 μM determined by surface plasmon resonance (SPR) for the binding with Aβ1–40 aggregates. Neutral complexes were found to have higher affinity than negatively charged complexes. Overall, the decrease in affinity was around five orders of magnitude for these complexes as compared to the PiB itself. Proton relaxivity measurements also evidenced binding to amyloid Aβ1–40 as well as to human serum albumin (HSA), yielding, respectively, two- and fourfold relaxivity increase upon binding of GdL1 for instance. Ex vivo immunohistochemistry revealed selective targeting of Aβ plaques in human brain tissue. Octanol/water partition coefficients varied between −0.15 (for GdL1) and +0.32 (for GdL4), and such relatively hydrophilic molecules do not readily cross the blood–brain barrier, which would be important in the context of Alzheimer’s disease. In vivo brain uptake was assessed for the L1 ligand labeled with 111In in wild-type mice and showed 0.36 and 0.5%ID/g in the cortex and in the cerebellum (at 2 min), respectively, and rapid clearance, which might be sufficient for nuclear imaging if the probe is retained in the Alzheimer brain. In vivo PET imaging using the 68Ga-labeled analogues showed somewhat higher brain uptake in transgenic Alzheimer mice than in control animals with 68GaL2 but not with 68GaL1. However, autoradiography on human Alzheimer brain tissues failed to detect amyloid deposits [47].
Amyloid-targeted PiB-derivative ligands.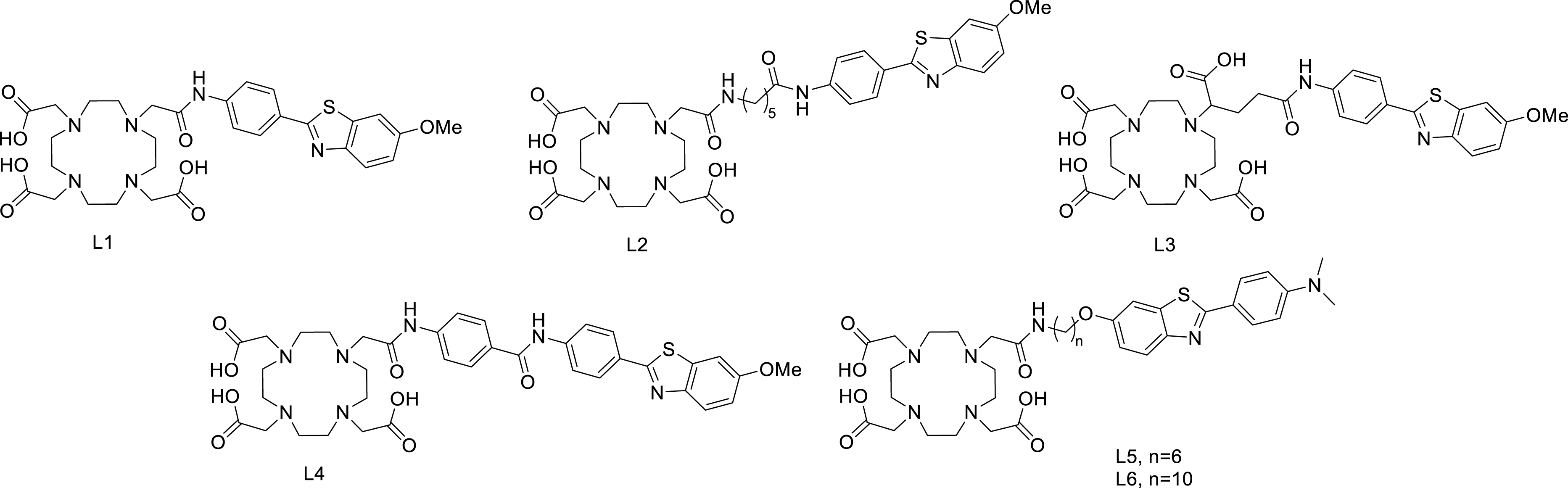
With the objective of increasing lipophilicity, a second family was later designed by changing the orientation of the PiB and using longer spacers (L5 and L6, Scheme 3). Despite its higher lipophilicity (log Poct/water = 0.63), 111InL5 did not have much enhanced brain uptake (0.12%ID/g at 30 min post-injection vs 0.07%ID/g for 111InL2), which was explained by its higher molecular weight [51].
Amylin fibrils, accumulated in pancreatic islets in the case of type 2 diabetes, represent a biomarker that is more accessible for the imaging agent than Aβ in the brain. We have therefore assessed the binding properties of GdL1, GdL2, GdL5, and GdL6 to amyloid amylin as well, in comparison to Aβ1–40. These studies have been performed by SPR at different peptide immobilization levels on the SPR chips, standard ∼4000 RU and very high ∼9000 RU, and the data were analyzed by fitting a 1:1 Langmuir isotherm. We could show that the binding affinity increases when the metal complex is placed further away from the peptide recognition unit (Table 1). In particular, in the case of GdL6 containing the longest C10 spacer and when using high peptide immobilization levels on the SPR chips, the data revealed nanomolar affinity for both peptides (Kd = 4.4 nM and 4.5 nM for Aβ1–40 and amylin, respectively), similar to that of the PiB alone. This was the first time that such high affinity was reported for a metal chelate. On the other hand, none of these systems have shown any notable selectivity for one peptide over the other.
Dissociation constant (Kd) determined by fitting the SPR data to 1:1 Langmuir isotherms [51]
| Kd | Aβ1–40 | Amylin | HSA |
|---|---|---|---|
| GdL1 | 170 μMc | 154 μM | 1100 μMd |
| GdL2 | 71 μM | 8.3 μM | 1700 μM |
| GdL5 | 16 μM | 17.2 μM | 300 μM |
| GdL6a | 5 μM | 3 μM | 36 μM |
| GdL6b | 4.4 nM (from data between 0 and 125 nM) | 4.5 nM (from data between 0 and 62.5 nM) | - |
aImmobilization: ∼4000 RU; bImmobilization: ∼9000 RU; cMeasured for LaL1; dMeasured by relaxometric titration.
Due to their amphiphilic nature, all these LnL complexes themselves undergo important aggregation in aqueous solution. These “micellization” processes have been investigated over a large concentration range, from micro- to millimolar, of amphiphilic LnL chelates. For these studies, different techniques have been applied, each of them adapted to a different concentration range: fluorescence, UV–vis, and relaxometry. The results when combined revealed more than one cmc value, implying that different aggregation regimes exist over the entire concentration range. The existence of different aggregation states of the LnL complexes proved to be very important for their peptide binding affinity. Indeed, by carrying out a Scatchard linearization analysis of the SPR data, we could evidence different affinity regimes for each of the complexes as indicated by clear breakpoints on the linearized Scatchard plots (Figure 2). These regimes are characteristic of the complex while they remain independent of the peptide. Most importantly, the breakpoints correspond to the cmc values previously determined. These findings evidence for the first time that the aggregation of the probe itself is important for its binding interaction with the peptide. Our data indicate that the monomer form has the highest affinity. The interaction of the probes with HSA has been also investigated by SPR. While all complexes bind HSA, the affinities are one to two orders of magnitude lower towards Aβ1–40 or amylin (Table 1).
Top: SPR binding plots fitted as 1:1 Langmuir isotherm functions for the interaction of GdL6 with Aβ1–40 (A, C) and amylin (B, D). Peptide immobilization was 4000 RU (A, B) and 9000 RU (C, D). For C and D, points recorded only at low concentrations are represented. E and F are respectively the Scatchard linearization plots of A and B. Reproduced with permission from Sens. Diagn. [43]. Bottom: Sketch of the aggregation of amyloid-targeted, PiB-derivative probes and its influence on peptide binding affinities. Reproduced with permission from Chem. Eur. J. [52].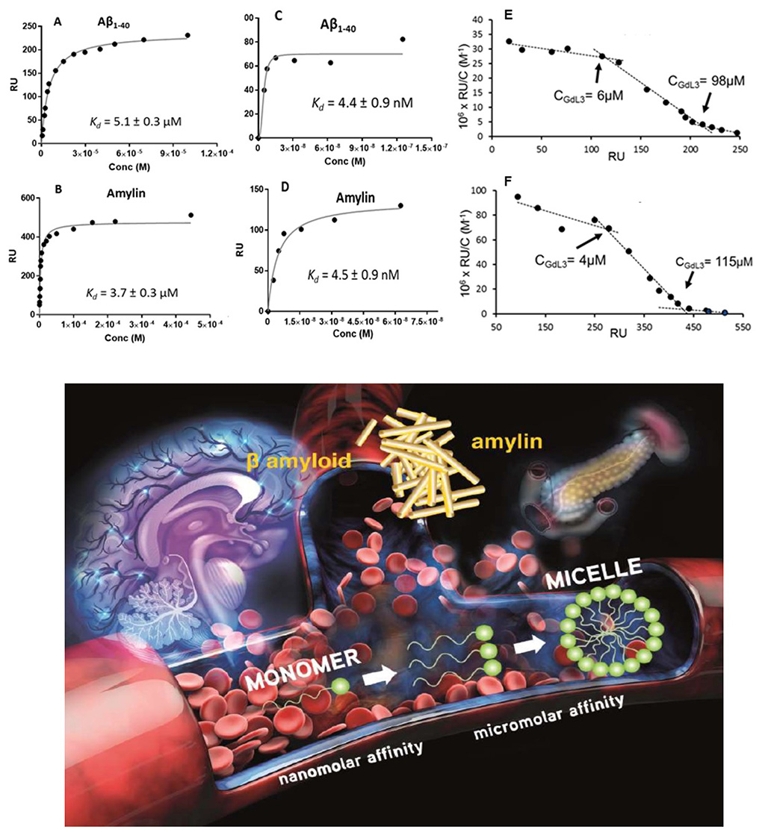
The presence of complexes also influences the aggregation process of the peptide as has been shown for Aβ1–40. In the case of LnL1–L3, combined UV–vis spectroscopy, circular dichroism, dynamic light scattering, and transmission electron spectroscopy data indicated that the small differences between these structurally similar molecules can promote different behavior as LnL1 and LnL3 accelerated while LnL2 inhibited the aggregation of Aβ1–40 [50]. Fluorescent assays using thioflavin T (ThT) were later applied to compare the effects of GdL2, GdL5, and GdL6. The intercalation of this fluorescent dye into the β-sheets of amyloid fibrils yields a fluorescence change that can be used to follow the aggregation process. Aggregation was slower in the presence of GdL5 or GdL6, and these studies also showed competition of these complexes with ThT for the same binding sites on the peptide fibrils [51].
Finally, the biodistribution of radiolabeled 111InL5 and 111InL6 has been assessed in wild-type mice. As for the previous complexes, brain uptake was low. There was no specific organ retention in these healthy mice. The probes distributed to the pancreas at an interesting level (2.9%ID/g and 3.7%ID/g for 111InL5 and 111InL6, respectively), which is very promising for potential amylin imaging in diabetic pancreas [51].
5. Manganese complexes of tetrapyrrolic macrocycles as potential contrast agents
Although the use of Gd(III)-based MRI agents to improve image quality has been a key element in the clinical success of MRI, lately there have been concerns about the safety of these Gd(III) complexes. First, a new and potentially lethal disease, nephrogenic systemic fibrosis (NSF) was discovered mostly in kidney impaired patients and linked to Gd injections [53]. It has been rapidly understood that in the case of slow kidney elimination, open-chain Gd(III) complexes endowed with lower kinetic inertness might undergo partial dissociation in vivo, and the free Gd(III) ions released are the cause of NSF disease. This has led to the withdrawal of several linear Gd(III) chelates from clinical practice and warnings issued by drug safety authorities concerning MRI examinations in kidney impaired patients. In parallel, Gd deposits have been also detected in human bones and brains following Gd-enhanced MRI, though without toxic effects [54, 55]. Furthermore, recent years have witnessed increasing awareness about the presence of gadolinium not only in wastewater but also in natural water [56]. All these concerns promote important research towards the replacement of Gd by more biocompatible paramagnetic metal ions. Most of the research in this field has been directed towards Mn(II) complexes. Although Mn is more paramagnetic in the +2 redox state (S = 5/2), the high spin state of the +3 redox form is also a good relaxation agent (S = 2). Manganese is an essential metal ion that clearly alleviates toxicity problems. Even if in a non-complexed form, Mn(II) or Mn(III) would be also toxic at the doses required for MRI, implying that it has to be chelated in thermodynamically stable and kinetically inert complexes. Mn(III) is very well stabilized by tetrapyrrolic macrocycles, such as porphyrins and phthalocyanines. Indeed, Mn(III) porphyrin complexes have been explored early on as potential MRI agents, however, without further clinical development [57]. It was demonstrated that they can have interestingly high proton relaxivities, presenting a different magnetic field dependence from that of small molecular weight Gd(III) or Mn(II) chelates. Nevertheless, low water solubility and aggregation in aqueous solution can often be a problem.
As the first approach, we have reported the synthesis of biocompatible manganese(III) phthalocyanines and their evaluation as potential contrast agents [58]. The synthetic strategy is depicted in Scheme 4. It starts with the functionalization of a phthalonitrile with polyethylene glycol (PEG) or dimethylaminoethoxy moieties by performing a nucleophilic substitution reaction between these nucleophiles and 4-nitrophthalonitrile using ultrasound as an alternative heating source, yielding 1a and b [59, 60]. The condensation of 1a or 1b phthalonitriles was performed using a mixture of DMAE/n-butanol as the solvent and Zn(OAc)2 as the template at room temperature and 100 °C, respectively, yielding the desired biocompatible phthalocyanines 2a and 2b in ≈60% yield. Aiming at the preparation of manganese(III) phthalocyanines, the demetalation of 3a and 3b was pursued in the reflux of pyridine and pyridinium HCl salt. The final step was the complexation of free-base phthalocyanines 3a and 3b, with Mn(OAc)2 in DMF as the solvent, under air atmosphere. Mn(III)-phthalocyanines 4a and 4b were obtained following spontaneous oxidation of Mn(II) to Mn(III) by air oxygen in 36% and 61% overall yields. To turn the phthalocyanine 4b water soluble, alkylation with iodomethane was additionally performed. Both phthalocyanines presented relatively low r1 relaxivities (4.0–5.7 mM−1⋅s−1 at 20 MHz) due to aggregation in aqueous solution. No significant cytotoxicity to HeLa cell cultures was observed after 2 h incubation and up to 2 mM concentration. The reduction of Mn(III) to Mn(II) was also investigated in both phthalocyanines, but the Mn(III) oxidation state proved to be quite stable.
Synthesis of Mn(III) phthalocyanines.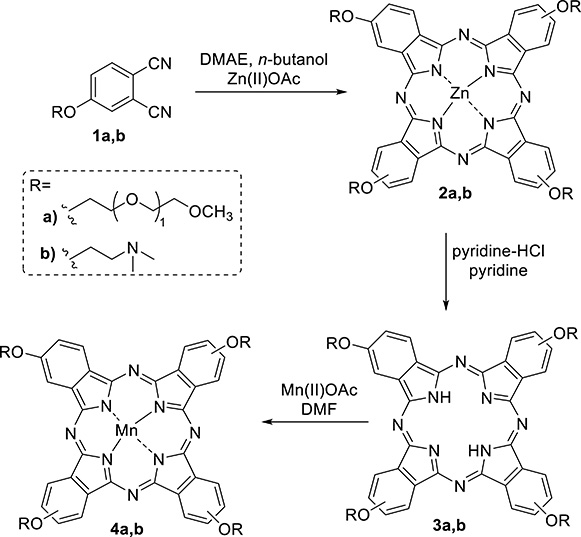
Building on the extensive experience on porphyrins in Coimbra [61, 62, 63] and on responsive MRI probes in Orléans [13, 64, 65], we have designed a family of porphyrins with the objective of creating Mn-based redox MRI probes [66].
In living systems, the redox environments, both extra- and intracellular are tightly regulated via the presence of different biological redox couples, among which glutathione is a major actor representing the highest concentration (1–11 mM in the intracellular space). Many pathological states, including chronic inflammation, ischemia, cancer, and so on, are associated with redox imbalance, which makes the redox state an important biomarker for imaging detection [67].
The switch of the Mn(III)/Mn(II) couple might offer an efficient way to report on the tissue redox state [68, 69, 70, 71, 72]. The two forms have different relaxation mechanisms obeying different magnetic field dependencies [71, 73, 74], which can provide substantial relaxivity differences with detectable signal change on MR images. Such a redox MRI probe has to fulfill several requirements, such as rapid redox transformation with a biologically relevant redox potential accompanied by a significant relaxivity variation and stable complexation of both Mn(II) and Mn(III) states. With respect to the latter, porphyrins are quite interesting since they are among the very few ligand systems capable of forming highly stable complexes with both Mn(II) and Mn(III) ions.
The first porphyrin that we developed as a potential redox-responsive MRI contrast agent was compound 7 (Scheme 5) [66]. Its synthesis started by PEGylation of free-base porphyrin 5 through nucleophilic aromatic substitution using NaH as the base and DMF as the solvent, yielding 6 with 65% isolated yield. The complexation reaction was performed in sodium acetate/acetic acid buffer with Mn(OAc)2 as the metal salt at 120 °C. The final Mn(III) porphyrin 7 was purified using extraction processes and was isolated with 82% yield. This new biocompatible fluorinated manganese porphyrin is capable of stabilizing both Mn(III) and Mn(II) oxidation states, and in aqueous solution, Mn(III)/Mn(II)-7 is able to undergo reversible reduction/oxidation reactions using ascorbic acid for the reduction and O2 for the oxidation. With ascorbic acid as the biological reductant, a rate constant k2 of 46.1 M1⋅s−1 was obtained, which corresponds to a half-life of few minutes in blood plasma, where ascorbic acid concentration reaches 20–40 μM. In addition, it was also observed that with glutathione, it was not possible to achieve the reduction of Mn(III) since glutathione carboxylate groups coordinate to the metal in the axial position as was demonstrated by nuclear magnetic resonance (NMR) data. Upon reduction of Mn(III) to Mn(II), a large increase in proton relaxivity was observed at low and medium magnetic fields (<60 MHz). Although the difference vanishes at higher fields with a reversion of the order for very high fields, even at 80 MHz, the relaxivity of the reduced form remains 30% higher than that of the oxidized form, which would correspond in MR images to remarkable “turn-on” response to a reducing environment (Figure 3).
Synthesis of PEGylated Mn(III) porphyrin.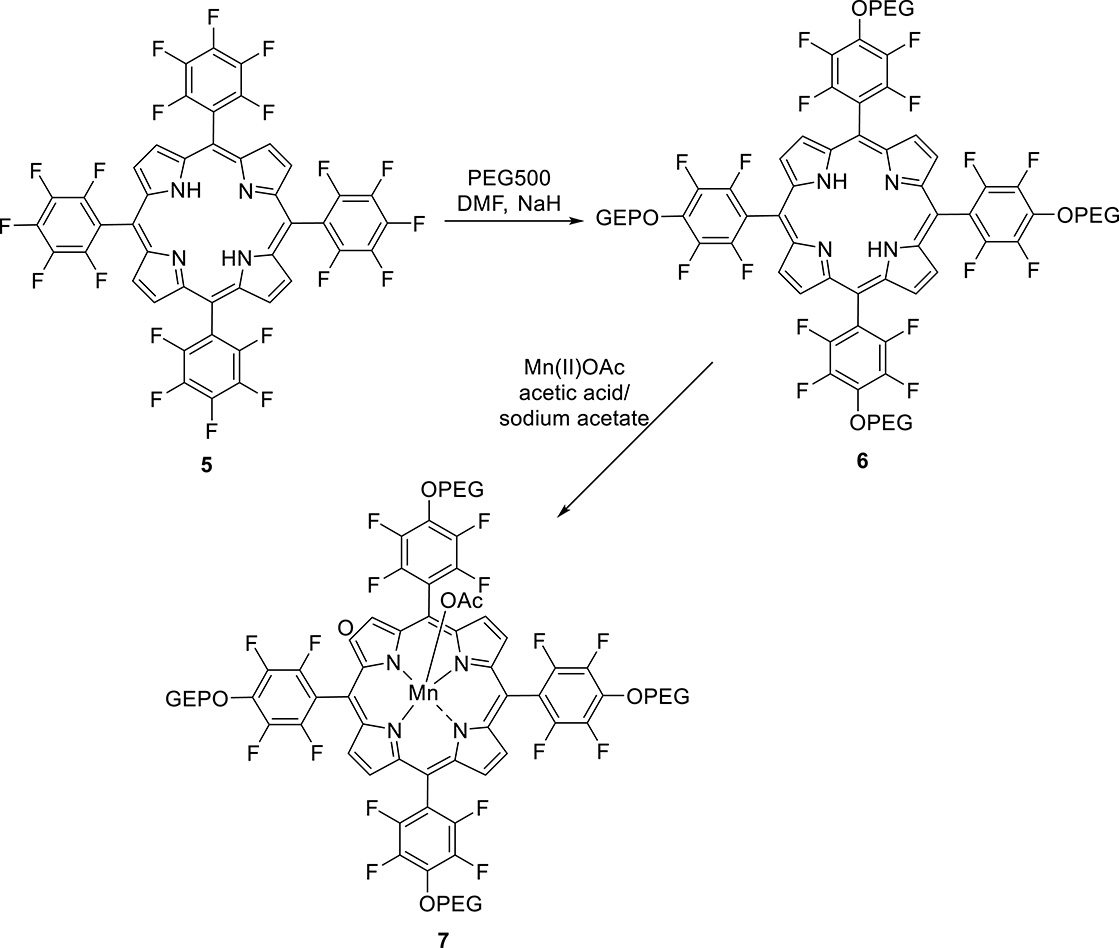
(a) UV–vis reduction titration of 0.041 mM Mn(III)-7 with ascorbic acid, recorded in PBS (25 °C, pH 7.4). Number of equivalents of ascorbic acid added: black, 0; red, 0.28; light blue, 0.57; light green, 0.85; pink, 6.4; dark green, 11.4; dark blue, 25.3. (b) UV–vis spectra of the reoxidation of Mn(II)-7 upon air exposure: black, Mn(II)-7 before exposure; red, after 10 min exposure; blue, after 4 h exposure; green, complete conversion to Mn(III)-7 (after 24 h exposure). (c) Proton NMRD profiles recorded at 298 K for Mn(III)-7 (red) and Mn(II)-7 (blue). Adapted with permission from Dalton Trans. (Ref. [66]).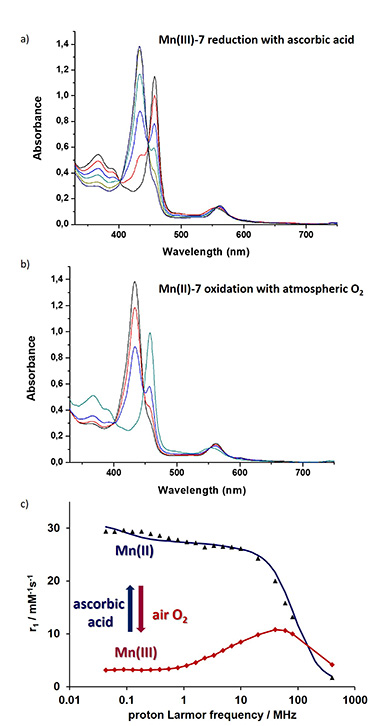
The presence of 19F atoms on Mn porphyrins, which is important to modulate their redox potential allowing reduction by ascorbate, opens further opportunities in MRI detection. 19F MRI has complementary advantages to classical 1H MRI, including the lack of any background signal in biological systems (hotspot imaging) or a wide range of accessible 19F chemical shifts allowing for multiplex detection. In the case of responsive probes, monitoring biomarker-dependent variations in two independent imaging modalities can further ascertain the findings.
With this objective, we have recently reported a novel fluorinated manganese porphyrin capable of providing dual 1H relaxivity and 19F MRI response to redox changes [75]. The synthesis of this porphyrin started by the condensation of 4-(trifluoromethyl)benzaldehyde 8 with 1H-pyrrole 9 in a mixture of acetic acid/nitrobenzene, using NaY zeolite as the Lewis acid (Scheme 6). Porphyrin 10 was obtained in 20% isolated yield. Complexation with manganese was performed using the strategy described above, giving 11 in 85% yield with an HPLC purity of 98%. By using experimental methods and density functional theory (DFT) calculations, porphyrin 11 was completely characterized and presented properties that made it a good starting point for the future development of 19F redox contrast agents. Despite its low solubility in aqueous medium, this fluorinated Mn porphyrin evidenced 1H relaxometric and 19F NMR properties in DMSO-d6/H2O, which are interesting for differential redox MRI detection. DFT calculations and experimental 19F relaxation data allowed us to conclude that an Mn–F distance of 9.7–10 Å, as in Mn(III)-11, enables adequate paramagnetic effect of Mn(III) on 19F T1 and T2 relaxation times, providing sufficiently fast longitudinal relaxation without excessive line broadening. Similarly to the previous porphyrin 7, porphyrin 11 was also readily reduced in the presence of ascorbic acid, evidenced by the broadening of the 19F NMR signal for the Mn(II) form. By using phantom 19F MR images, a clear decrease in MRI signal intensity was observed upon reduction from Mn(III) to Mn(II), which was subsequently regenerated upon reoxidation in air within 24 h (Figure 4).
Synthesis of fluorinated Mn(III) porphyrin.
(a) Most stable structure obtained through DFT optimization at the B3LYP/6-31G(d,p) level of Mn(III)-11. (b) 19F MRI phantom images acquired at 1.1 mM Mn(III)/(II)-11 concentration in DMSO. (c) Monitoring reoxidation of Mn(II)-11 (cMnL = 1.6 mM) by air oxygen; signal-to-noise ratio (SNR; with respect to the Mn(III)-11 peak) as a function of oxidized Mn(III)-11 content (%). B: 7 T; TR: 60 ms; TE: 1.3 ms; FA: 90°; NA: 256; acquisition time: 8 min 11 s. Reproduced with permission from Ref. [75].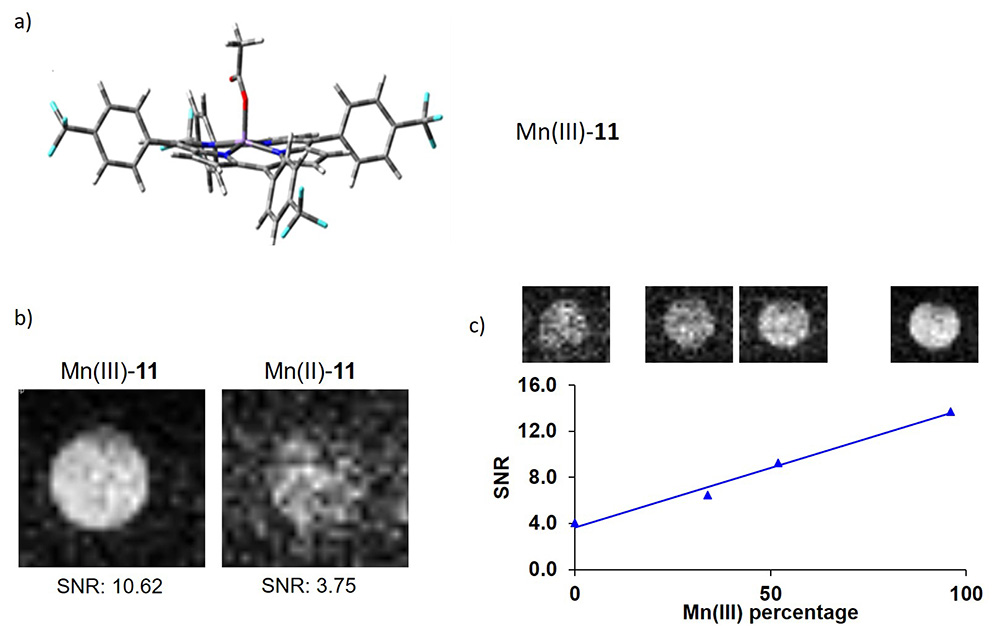
6. Conclusion
The collaborative work between the research groups at the Centre of Molecular Biophysics, CNRS, Orléans in France and the CQC-IMS, Department of Chemistry, University of Coimbra in Portugal has successfully contributed to addressing major challenges in the development of MRI agents over the past 20 years. Among the most significant recent achievements, a large family of imaging agents targeted at amyloid peptides have been designed and their in vitro characterization and in vivo preclinical imaging investigation have yielded novel insights into the understanding of amyloid binding properties of metal complexes. More recently, redox-responsive probes based on Mn(III)/Mn(II) porphyrin complexes have been developed with detection potential of the tissue redox state in both 1H and 19F MRI. The complementary expertise of the two groups in the physical chemistry of MRI agents and in the synthetic chemistry of porphyrins is a major asset for the success of future work.
Declaration of interests
The authors do not work for, advise, own shares in, or receive funds from any organization that could benefit from this article, and have declared no affiliations other than their research organizations.