



 CC-BY 4.0
CC-BY 4.0
1. Introduction
1.1. Definitions
This review refers to several types of fluorescent probes and sensors. To avoid any confusion, we define a few terms that can sometimes be found with different meanings in articles. A fluorescent chemosensor refers to a synthetic small molecule for sensing ions or other chemical species with a change in their fluorescent properties (e.g., intensity, wavelength, lifetime) [1]. Fluorogenic probes or fluorogens are small molecular fluorophores that bind biomolecules (herein proteins) with an increase in their fluorescence [2]. Biosensors refer to fluorescent protein (FP) based sensors of biological activity. Semisynthetic biosensors are fluorescent reporters based on the combination of a small molecular fluorescent indicator and a genetically encoded nonfluorescent protein; they are also called chemogenetic [3] or chemigenetic sensors [4].
1.2. Fluorescent sensors for bioimaging
Genetically encoded FPs have revolutionized bioimaging in living systems by providing unparalleled selectivity with cell- or organelle-specific expression [5]. Significant efforts have also been put into engineering FPs to develop biosensors with diverse photophysical properties and target analytes using evolved recognition motifs [6, 7]. However, the development and optimization of FP-based biosensors is complex especially for long wavelength emitting ones, and numerous biological substances remain challenging or even impossible to detect using protein-based recognition motifs. Alternatively, fluorescent chemosensors based on organic fluorophores benefit from a long research history and offer virtually endless versatility thanks to the power of modern synthetic organic chemistry and well-identified sensing mechanisms and recognition groups (Figure 1A) [8, 9]. However, the precise control of the intracellular localization of chemosensors in cells to visualize biological processes with high spatial selectivity remains an issue and explains the success of FPs and biosensors. Small organic molecules usually accumulate in a given organelle or cellular compartment depending on their charge and lipophilicity (e.g., cationic rhodamines in mitochondria), but it is hard to predict and modulate. Chemical targeting strategies have been put in place with functional groups that can direct chemosensors towards specific cellular locations, but again these strategies are not completely general and can fail due to competition with spontaneous localization of the parent molecule [10, 11]. Recently, hybrid chemical and genetic systems have emerged as an alternative that combines the best of both approaches. They are based on the targeting of small chemical compounds towards genetically encoded proteins called self-labeling protein (SLP) tags and benefit from both the targeting selectivity of genetically encoded proteins and from the diversity and versatility of chemical probes and chemosensors (Figure 1B). Efforts in the development of SLP-tags have led to two categories of covalent (SNAP-tag, HaloTag, CLIP-tag, PYP-tag, BL-tag, and TMP-tag) and noncovalent (FAP and FAST) binding systems [12]. Semisynthetic biosensors can be obtained by conjugating chemosensors to an SLP-tag [13]. Two different possibilities exist depending on whether the chemosensor is fluorogenic (i.e., activated by reaction with the protein, Figure 1C) or not (Figure 1B). Numerous examples of the latter class have been reported for the genetically targeted sensing of Zn2+ [14, 15, 16, 17], Ca2+ [18, 19, 20], K+ [21], pH [22, 23], nitric oxide [24, 25], viscosity [26], membrane tension [27], and voltage [28]. In all these examples however, the chemosensor is sensitive to its analyte or to the biological process whether or not it is linked to its target protein, which requires extensive washing steps to remove the excess of unbound molecules and ensure the selectivity of the signal (Figure 1B).
General principle of the different classes of sensors for bioimaging: (A) chemosensors, (B) semisynthetic biosensors, and (C) locally activated semisynthetic biosensors.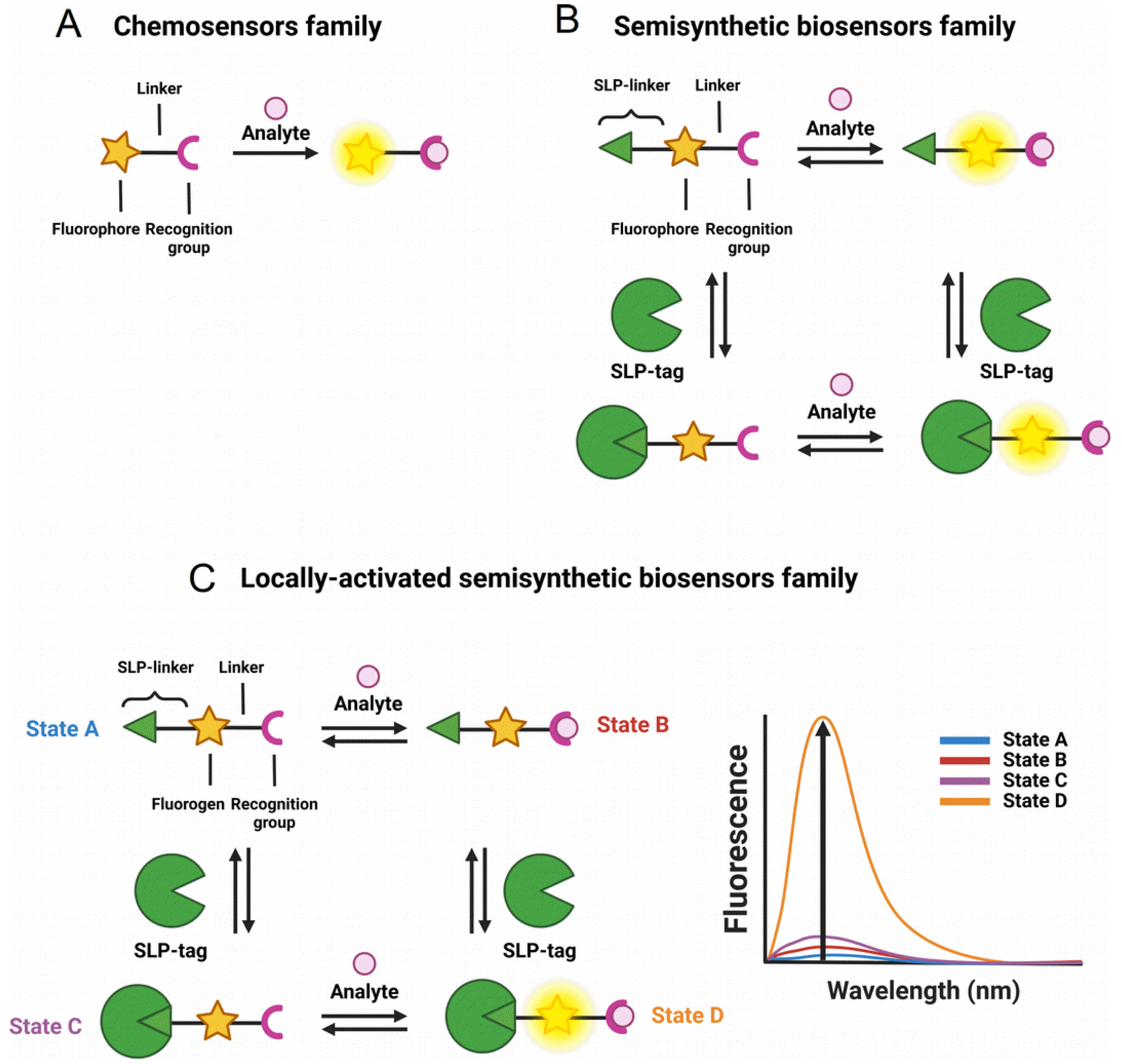
To provide an alternative to FP-based biosensors with comparable selectivity, semisynthetic biosensors should be built on fluorogenic probes that only become fluorescent when they are bound to the target protein (Figure 1C). To achieve such fluorogenic semisynthetic sensors, two strategies have been used. The first one consists in using fluorogenic probes targeted at modified SLP-tags that incorporate a sensing moiety [4, 29, 30, 31]. In that case, the detection is performed by the protein moiety, and it does not avoid some drawbacks associated with FP-based biosensors such as complex protein evolution to ensure efficient detection and slower detection kinetics for fast biological processes such as calcium oscillations compared to synthetic chemosensors [32]. This approach has been recently reviewed by Broch et al. [3]. This minireview focuses on the few reported examples of the second strategy, which is all-molecular and consists in incorporating two fluorescence control mechanisms within a single molecule. The first ensures the pre-activation of the fluorescence upon binding to the cognate SLP-tag so that it is able to become fluorescent upon detection of the target analyte (Figure 1C). The most commonly encountered SLP-tags are SNAP-tag and HaloTag, which enable highly selective and rapid protein labeling (Figure 2). HaloTag in particular has established itself as the most promising technology with a chemically simple linker that facilitates the synthesis of conjugates as well as their cell penetration and a very fast reaction rate. For a detailed review of HaloTag applications in bioimaging, the readers are referred to a recent review by Cook et al. [33].
Comparison of HaloTag and SNAP-tag strategies for subcellular bioimaging.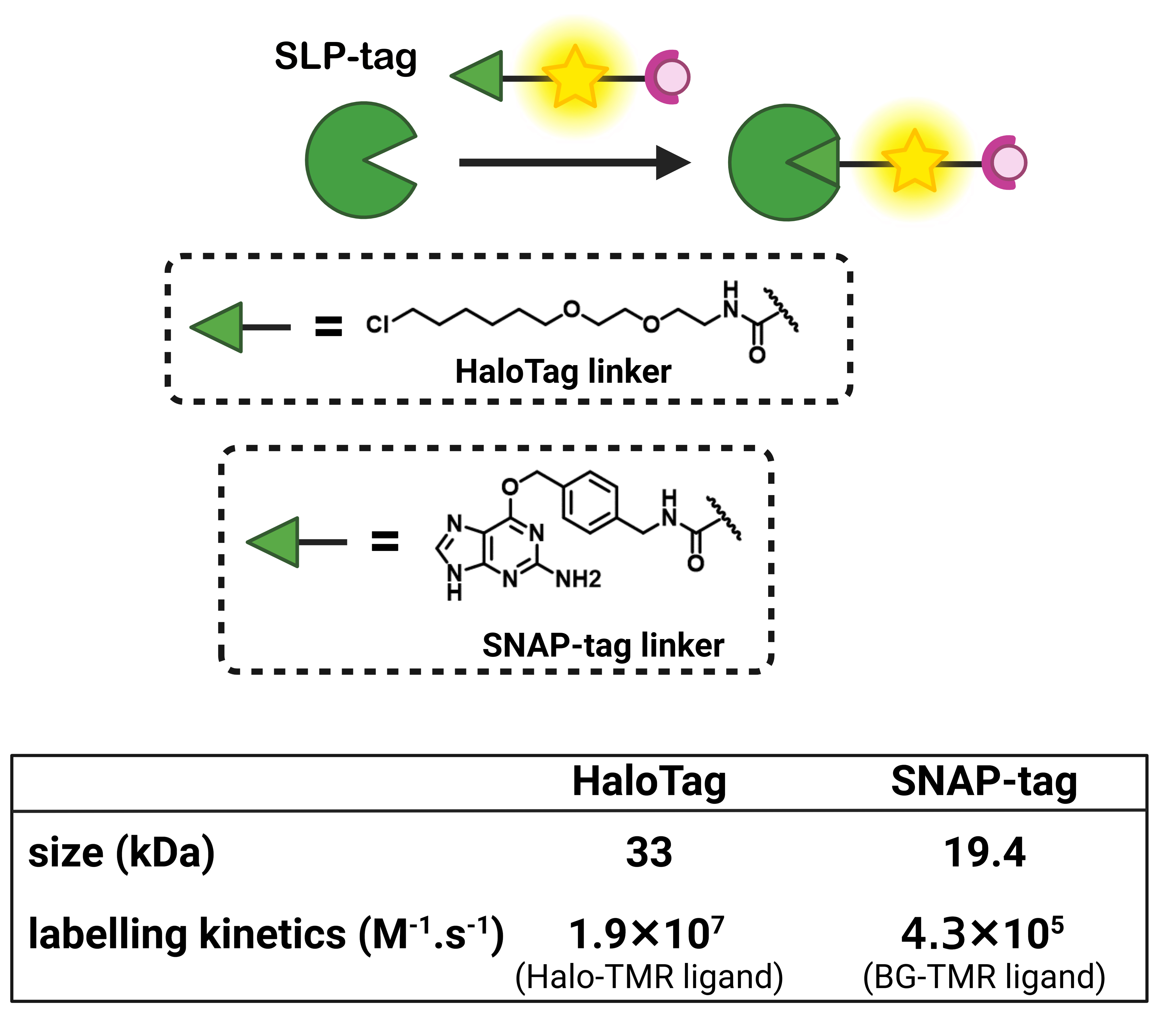
In practice, these locally activatable chemosensors that we also refer to as dual-input probes due to the dual control of the fluorescence are based on the incorporation of an analyte-sensing group on a fluorogenic protein probe sometimes called fluorogen. This approach has the advantage of using well-established SLP-tags and is based on small-molecule engineering. The different fluorogenic chemosensors are classified depending on the type of fluorogens on which they are built rather than on the target analytes, considering that the sensing strategies are relatively well known and that the originality of these research works lies in the efficient combination of the two fluorescence control mechanisms within the same molecule. The common theme of the works covered by this minireview is thus the local activation of fluorescent chemosensors by genetically encoded proteins. It encompasses diverse structures with different target analytes and includes some articles where the local activation was not necessarily underlined, especially for early works. We thus aim to show the common character of all these molecules and identify some guidelines for the design of future fluorogenic semisynthetic biosensors. In doing so, we also recall some generalities on the design of fluorescent chemosensors and fluorogenic probes.
1.3. Analyte detection mechanisms
To develop dual-input probes for genetically targeted analyte sensing, the protein fluorogen targeting a specific SLP-tag must be combined with an analyte-sensing mechanism. As previously stated, the field of chemosensors benefits from a very large body of work, with numerous well-identified analyte receptors (e.g., ion chelators, pH- or ROS-sensitive groups) that can be coupled to a fluorophore to control its fluorescence with different photophysical mechanisms [9, 34, 35, 36, 37]. The mechanisms that have been implemented in the design of locally activated semisynthetic sensors are the three most commonly encountered: photo-induced electron transfer (PeT), intramolecular charge transfer (ICT), and Förster resonance energy transfer (FRET) (Figure 3). Chemosensors based on PeT or ICT are composed of three distinct parts: a receptor for analyte detection, a fluorophore, and a linker to couple them. Depending on the nature of this linker, the chemosensors operate via PeT or ICT [38].
Schematic illustration of the principle of (A) photo-induced electron transfer (PeT), (B) intramolecular charge transfer (ICT; EWG: electron-withdrawing group, EDG: electron-donating group separated by a π-conjugated linker), and (C) FRET (D: FRET donor, A: FRET acceptor) detection mechanisms used in the design of chemosensors.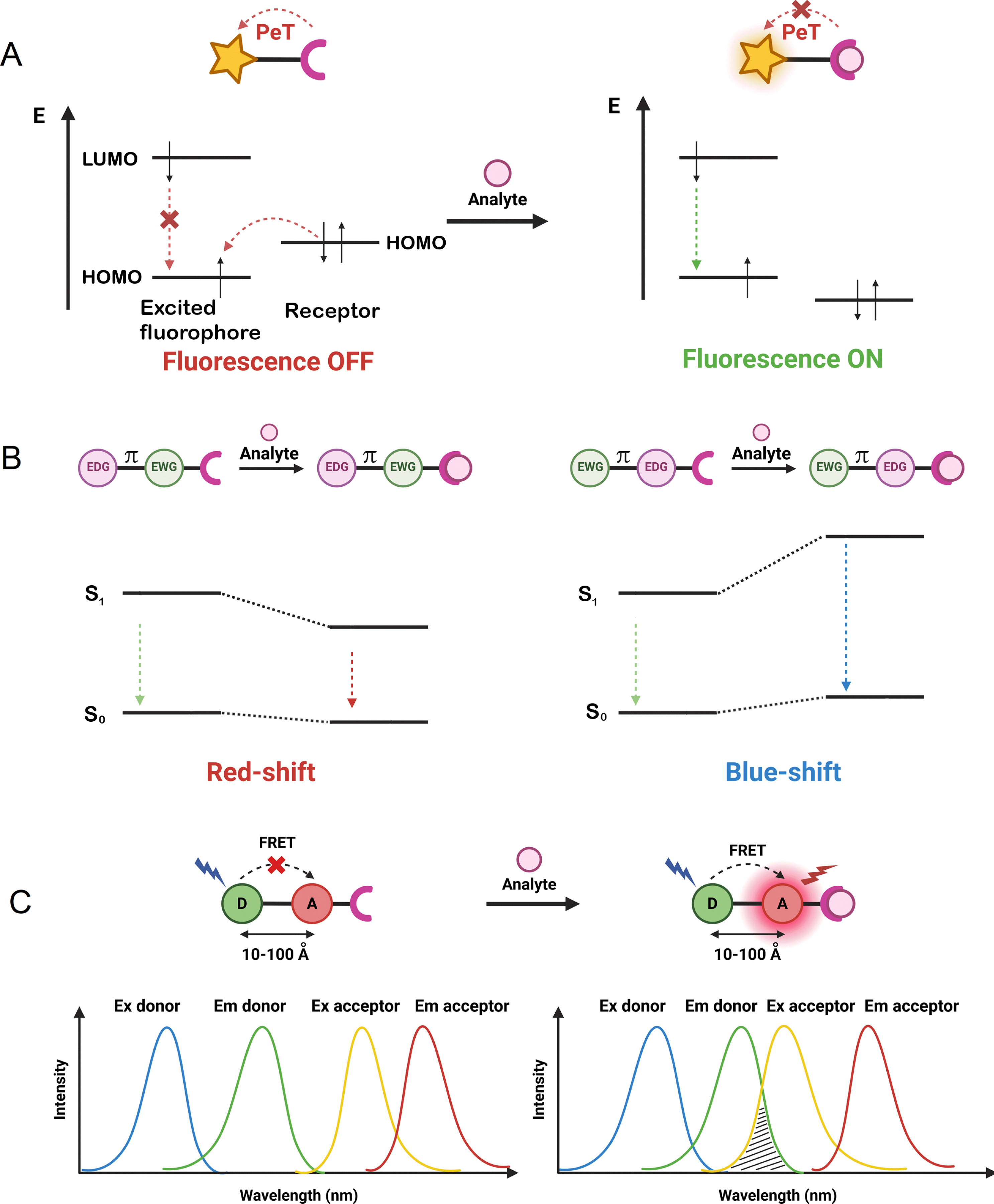
If the linker is not conjugated, the receptor and fluorophore can essentially be considered electronically independent and the receptor can transfer an electron to the excited state fluorophore, thus effectively quenching the fluorophore. Upon analyte binding, the energy of the electron donor (the analyte receptor) is lowered, thus inhibiting the PeT process and restoring the fluorescence (Figure 3A). Although the opposite is possible, reductive PeT is the most common mechanism found in the literature [39]. A variety of fluorescent chemosensors based on PeT have been developed for targeting a variety of biological analytes [40, 41]. The difficulty in the design of PeT sensors lies in the necessity of having properly positioned orbital energy levels to enable electron transfer. The probability of PeT between an electron donor and acceptor is governed by the Rehm–Weller equation [39].
If the receptor and fluorophore are electronically conjugated, they cannot be considered independent and PeT will be replaced by ICT. Upon analyte binding, the change in the electron density of the receptor will cause an ICT within the molecule that can affect the absorption properties and/or the fluorescence emission and result in a fluorescence turn-on or in a bathochromic or hypsochromic shift depending on whether the receptor is the electron donor or the electron acceptor (Figure 3B). ICT is another classical mechanism for the development of chemosensors, with a more complex response than the off/on PeT sensors that can be exploited to develop ratiometric sensors [42].
FRET-based fluorescent chemosensors rely on two distinct fluorophores, a donor and an acceptor in close proximity (10–100 Å) (Figure 3C) [37]. FRET requires a spectral overlap between the emission of the donor and the absorption of the acceptor and is highly sensitive to the distance between the two. In FP-based FRET sensors, the most usual sensing mechanism is based on a change in the distance between the donor and the acceptor [6]. However, for chemosensors, it is very common that the analyte binding will induce a change in the photophysical properties of the donor or the acceptor, usually via one of the two previous photophysical mechanisms, PeT or ICT (Figure 3C) [37].
2. Design of locally activated semisynthetic biosensors
2.1. Early examples based on the biarsenical FlAsH/tetracysteine tag system
In 2007, the group of Roger Tsien reported on the first example of a locally activated semisynthetic fluorogenic biosensor for Ca2+ CaGF (Scheme 1) [43]. It is based on the fluorescein derivative FlAsH, which reacts selectively in cells with a tetracysteine tag (TC-tag), coupled to a BAPTA-derived calcium chelator with a low affinity [44]. The chelator controls the fluorescence by PeT, and the fluorescein becomes fluorescent in the presence of calcium. Although the dual activation properties were not discussed in detail in this paper, the reaction of FlAsH with the TC-tag is intrinsically fluorogenic, and so CaGF can be considered a locally activated semisynthetic biosensor. It was targeted at the connexin 43 protein in HeLa cells and shown to report on depolarization-evoked calcium waves at gap junctions. More recently, Fujii et al. reported another example of a dual-input sensor for magnesium ions based on FlAsH (Scheme 1) [45]. KMG-104-AsH is based on a biarsenical difluorofluorescein, for lower pH sensitivity, combined with a magnesium chelator selective for Mg2+ over Ca2+. As for the previous example, this semisynthetic fluorogenic biosensor only becomes fluorescent when bound to both the TC-tag and Mg2+ with more than 10-fold fluorescence activation due to fluorogenic binding with the TC-tag and PeT quenching of the chelating moiety. It was used to visualize intracellular magnesium by targeting the mitochondria or actin. The TC-tag combined with FlAsH was a pioneering approach opening the era of hybrid chemogenetic labeling strategies. It has the advantage of being intrinsically fluorogenic and based on a small nonperturbing peptide tag, but it suffers from poor selectivity due to the competition with other endogenous dithiols. The excess of probes must thus be quenched with 1,2-ethanedithiol to avoid nonspecific signals. Moreover, the TC-tag cannot be easily adapted to other fluorophores in order to tune photophysical properties. Due to these limitations, the TC-tag was outperformed by the development of SLP-tags, such as SNAP-tag or HaloTag, which enable rapid and selective labeling of proteins with virtually any organic dye. However, the reaction of usual fluorophores such as rhodamines and fluoresceins with SLP-tags is not fluorogenic, and the development of locally activated semisynthetic biosensors first required the development of adequate fluorogens targeted at these SLP-tags. Such fluorogens have been developed based mostly on rhodamines and small viscosity-sensitive molecular rotors. In the following sections of this review, we describe these two classes of fluorogenic probes and how they can be combined with analyte-sensing groups.
Structure of dual-input calcium and magnesium sensors based on the FlAsH fluorogenic probe.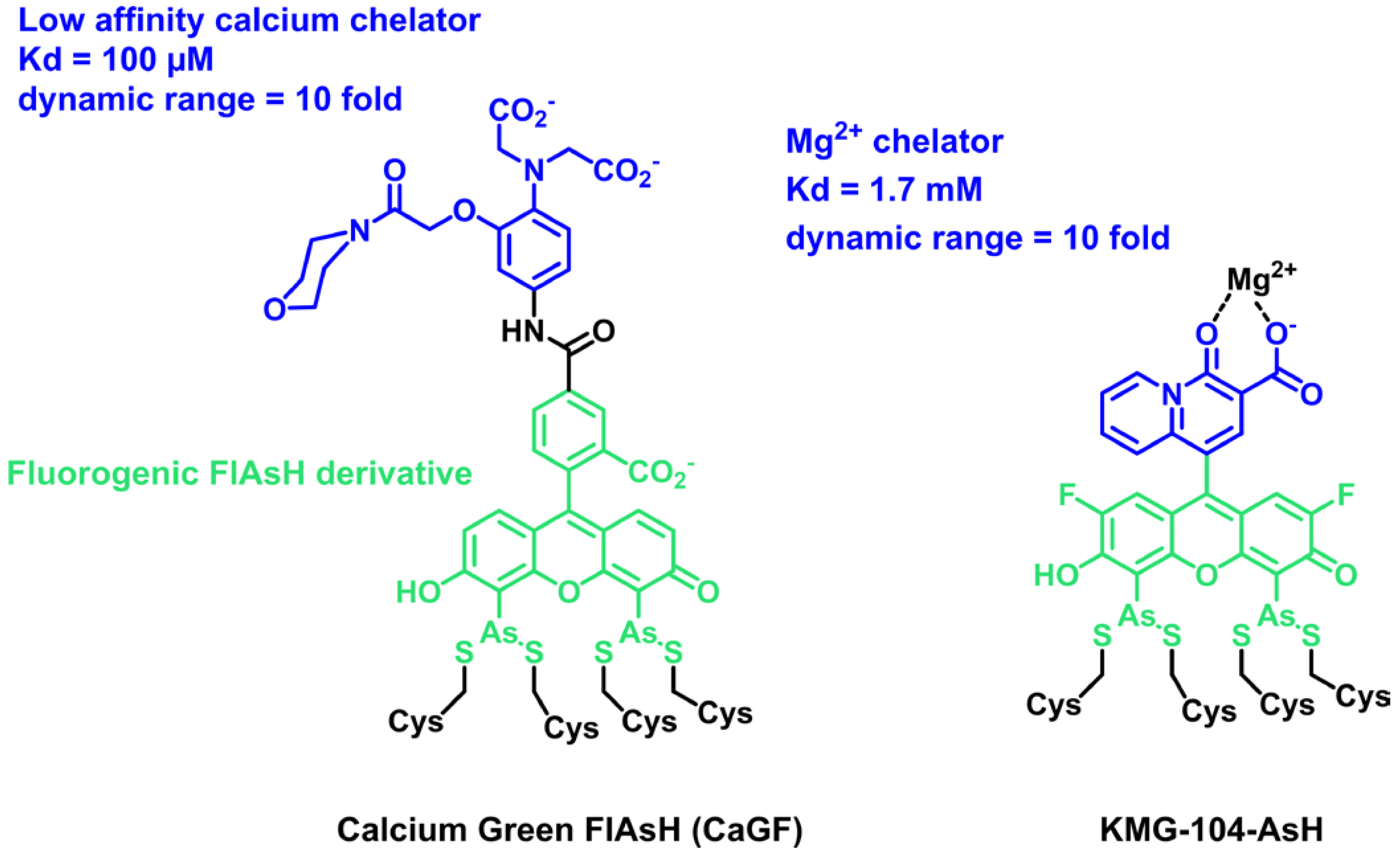
2.2. Fluorogenic semisynthetic biosensors based on rhodamines
2.2.1. Fluorogenic rhodamine probes
Rhodamines are a class of xanthene-derived dyes with excellent photophysical properties, such as high brightness and good photostability (Figure 4). Rhodamines can undergo polarity-sensitive equilibrium between a closed nonfluorescent and colorless spirocyclic form and an open fluorescent quinoid red-emitting form. This equilibrium has not only been exploited to develop chemosensors but also to fluorogenically target biomolecules [46, 47, 48, 49, 50, 51]. The Johnsson group pioneered this strategy by developing fluorogenic silarhodamines (SiRs), which transformed from a closed form in water to an open fluorescent form in the presence of a biomolecular target [52, 53]. It is assumed that the SiR fluorogens form hydrophobic aggregates in their closed form in water and that upon reaction with a biomolecule, they move from a hydrophobic environment to a polar aqueous medium in which they are open and fluorescent [54]. It is worth noting that this cyclization is also sensitive to pH. By using electron-poor amides such as cyanamide and sulfonamide, it has been possible to tune the spirocyclization reaction and adapt it to rhodamine or carborhodamine, leading to a palette of bright fluorogenic rhodamine dyes (Figure 4) [48, 55]. Given the excellent properties of the rhodamine scaffold, it has become a platform of choice to develop fluorogenic probes for bioimaging. The Lavis group has for instance played around with aniline auxochrome groups to improve brightness and photostability [46, 50]. Since the HaloTag protein was optimized with the tetramethylrhodamine dye, it has been proved to be excellent in reacting rapidly and activating the fluorescence of fluorogenic rhodamines (HaloTag7 version, k2 = 1.9 × 107 M−1⋅s−1 for TMR-Halo at 25 °C) [56, 57].
Structure of fluorogenic rhodamine derivatives.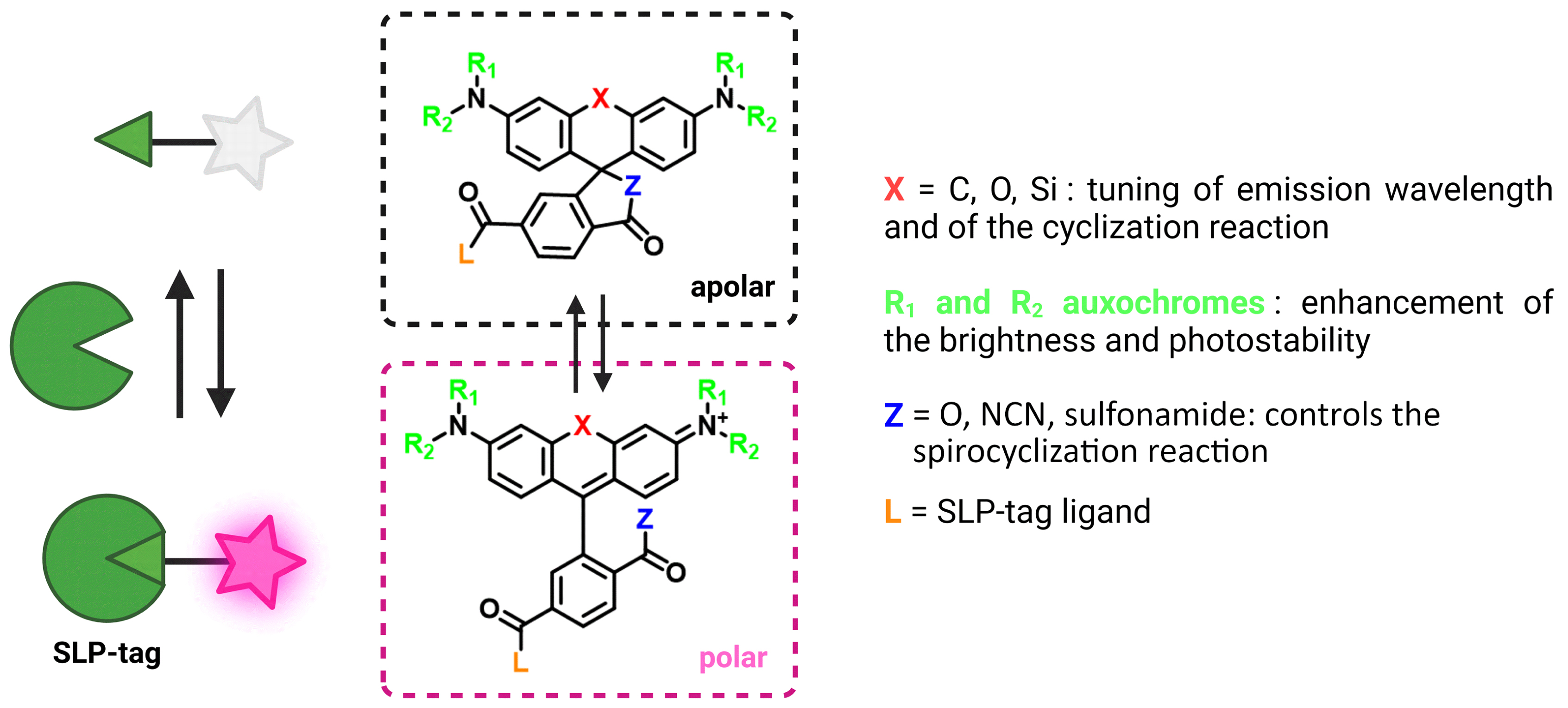
2.2.2. Combination of fluorogenic rhodamines with analyte-sensing modalities
Fluorogenic rhodamines in combination with HaloTag have been used to develop semisynthetic biosensors. The first attempt was made by the Lavis group that combined a fluorogenic rhodamine with the calcium chelator BAPTA, yielding molecule 13 (Figure 5A) [58]. BAPTA was discovered by Roger Tsien and has become the gold standard for building up fluorescent calcium chemosensors since it has suitable micromolar affinity and good selectivity for calcium over magnesium [60]. As discussed above, it can control the fluorescence of an associated fluorophore via PeT or ICT depending on the conjugation. In that case, it is a PeT process that creates an off/on trigger actuated by calcium. The resulting probe has good calcium sensitivity, but it is poorly fluorogenic upon reaction with HaloTag with only a 2.4-fold increase in fluorescence, which does not allow performing wash-free imaging. This first example testifies to the difficulty in combining efficiently several fluorescent control mechanisms within the same molecular scaffold.
Fluorogenic calcium probes targeted at HaloTag. (A) First design of rhodamine-based fluorogenic and chemogenetic Ca2+ reported in Ref. [58]. (B) Structures of MaPCa indicators. (C) Calcium titrations of MaPCahigh and MaPCalow. (D) Fluorescence microscopy images of HEK293 cells expressing and nonexpressing HaloTag–NLS incubated with 1 μM of MaPCa-558high (left), MaPCa-619high (middle), and MaPCa-656high (right) for 2 h imaged under wash-free conditions. Turn-on numbers indicate average of n = 200 cells. Scale bar = 20 μm. Images (B), (C), and (D) adapted from Ref. [59] under the CC BY 4.0 license.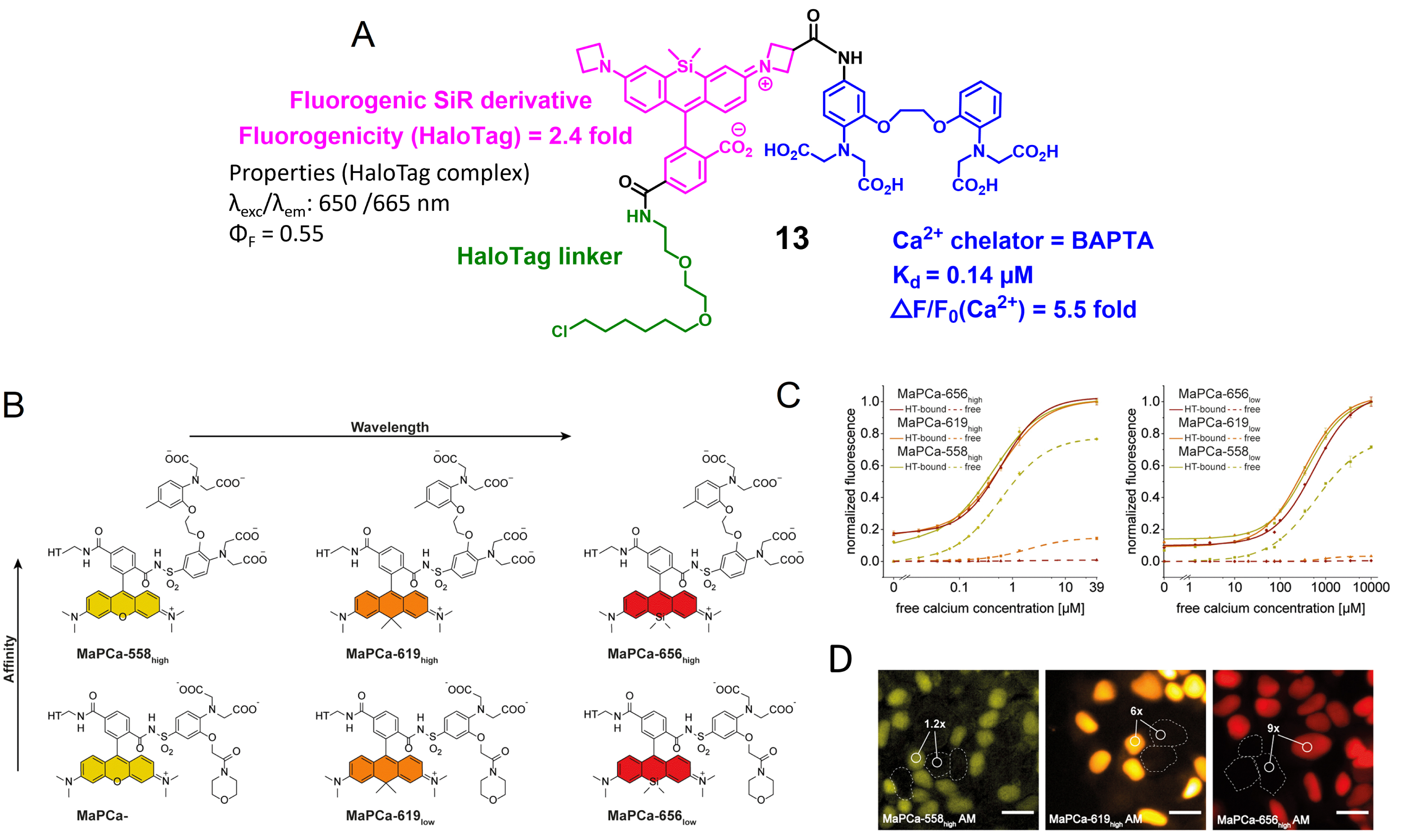
A few years later, the Johnsson group developed another series of locally activated fluorescent semisynthetic calcium indicators called “MaPCa indicators” with good fluorogenicity and tunable emission wavelength and affinity (KD from 0.41 to 457 μM) (Figure 5B) [59]. MaPCa-656high (𝜆exc = 656 nm) was identified as the best candidate with the highest fluorogenicity in the presence of calcium (6-fold increase) and HaloTag (120-fold increase) and the highest contrast in wash-free live-cell imaging (Figure 5B–D). It exhibits calcium affinity in a suitable range for cytosolic measurement (KD = 580 nM). A cell-permeant MaPCa-656high acetoxymethyl ester derivative was incubated on HEK293 cells expressing nuclear localized HaloTag and, after 2 h of incubation, fluorescence microscopy images highlighted efficient HaloTag labeling of the probe under no-wash conditions with a signal-to-background ratio of 9 (Figure 5D). Moreover, the performance of the MaPCa-656high calcium indicator was evaluated in rat primary hippocampal neurons and showed efficient and homogeneous HaloTag labeling without any significant off-target signal. This work thus provides a very solid basis for the development of locally activated semisynthetic probes based on fluorogenic rhodamines, and it is possible to envision replacing BAPTA with a different receptor to target other analytes. It is worth noting that the calcium sensitivity of the MaPCa sensors (F(+Ca2+)/F(−Ca2+) between 6 and 11) is much lower than what can be attained with comparable rhodamine-based molecular calcium indicators [62]. This is another indication of the difficulty in combining the two fluorescence control mechanisms on a single molecule while maintaining their full efficiency.
The combination of an SiR fluorogen with the HaloTag protein has also been used by Emmert et al. to develop a glutathione (GSH) sensor named “TRaQ-G” based on the ring-opening process upon HaloTag binding, resulting in a bright fluorescence emission, and on the nucleophilic attack of the xanthene π-system of the silarhodamine core by the nucleophilic GSH, which quenches the fluorescence (Figure 6A) [61]. The structure containing a cyanamide nucleophile, TRaQ-G, was found to act as a GSH sensor locally activated by HaloTag. This example is particularly interesting since the unbound form of TRaQ-G is unable to react with GSH, in contrast to the previous calcium sensors where unbound indicators can bind their target but without a detectable associated signal. However TRaQ-G is based on a decrease in fluorescence, which is less practical than a turn-on effect as an absence of signal is always harder to detect without bias. The combination of HaloTag with mGold, a photostable redox-insensitive protein, was used to generate a ratiometric GSH sensor (Figure 6B). A calibration curve of the ratio of mGold/TRaQ-G fluorescence intensities to GSH concentrations confirmed the linear response of this ratiometric fluorogenic semisynthetic GSH sensor in the biologically relevant range (1–20 mM) (Figure 6C). Live-cell imaging experiments using multiple organelle-targeting sequences confirmed the efficiency and accuracy of the TRaQ-G sensor for precise detection of GSH in cells (Figure 6D). Since there is no need to wash the excess ligand, the newly expressed protein can undergo continuous labeling in prolonged experiments.
TRaQ-G dual-input glutathione sensor. (A) Principle of the TRaQ-G sensing platform. (B) Design of the ratiometric GSH sensor using TRaQ-G combined to mGold fluorescent protein. (C) Fluorescence intensity spectra of mGold–TRaQ-G ratiometric probe with varying GSH concentration. (D) Visualization of nuclear GSH concentrations during cell proliferation with H2B–TRaQ-G–mGold: images of a single HeLa nucleus showing a decrease in GSH concentration from S phase to mitosis. Image adapted from Ref. [61] under the CC BY 4.0 license.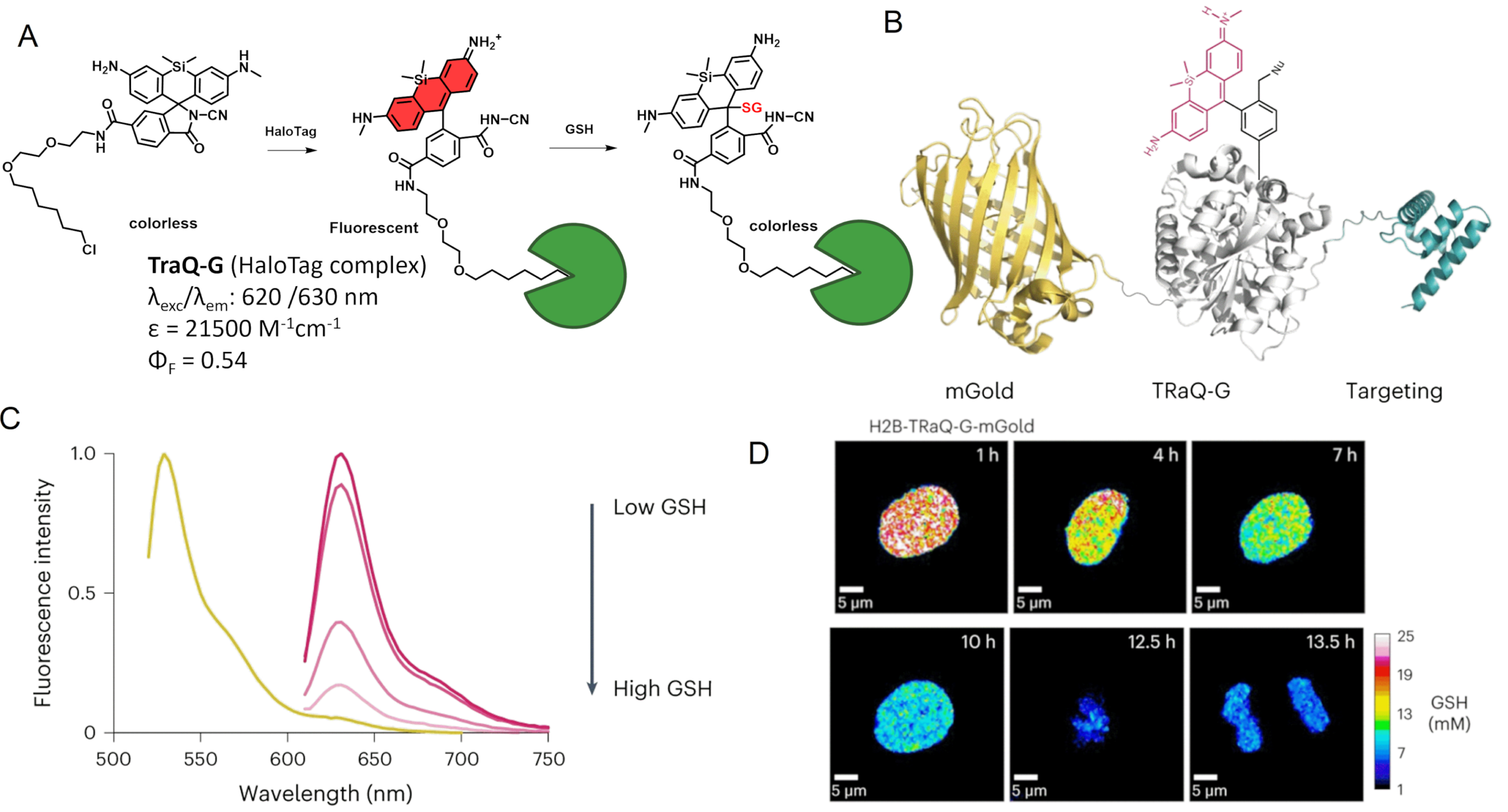
These three examples show the early developments of rhodamine-based fluorogenic and chemogenetic biosensors. Although this approach benefits from the excellent photophysical properties of the rhodamine scaffold, the synthesis of the most efficient rhodamine analogues is relatively complex, especially when it comes to combining several sensing groups on the same molecule. More chemically accessible alternatives have been developed based on small viscosity-sensitive molecular rotors.
2.3. Fluorogenic semisynthetic sensors based on fluorescent molecular rotors
2.3.1. Fluorogenic molecular rotors targeting SLP tags
Another class of fluorogens are fluorescent molecular rotors (FMRs) based on fluorescent structures displaying rotation around an intramolecular single bond (Figure 7) [64, 65, 66]. These molecules exhibit viscosity-sensitive fluorescence emission related to the degree of intramolecular motion upon photoexcitation. In low-viscous media, the fast intramolecular rotation of FMRs induces nonradiative thermal relaxation of the photoexcited state, but when the viscosity of the environment increases, the immobilization of the molecule leads to a restoration of the fluorescence emission. This is a very general mechanism, and FMRs encompass a wide variety of structures, but the most commonly found are “push–pull” structures composed of electron-withdrawing and electron-donating groups conjugated through a flexible bond (Figure 7A) [65, 66]. These small fluorogens are particularly valuable tools as they are chemically accessible, combining straightforward synthesis and easily tunable photophysical properties [67, 68]. FMRs have been widely used as fluorogens to monitor viscosity in cellular compartments, but the viscosity-sensitive emission can also be used for the fluorogenic targeting of biomolecules and in particular SLP-tags. Yu and colleagues have generated a fluorescence-switchable probe using BG-CCVJ fluorogen targeting SNAP-tag with a fluorescence activation up to 170-fold (Figure 7B) [69]. In order to red-shift the emission wavelength, Zhang and colleagues developed a red Kaede-like chromophore activated by SNAP-tag binding (𝜆em = 620 nm and 90-fold enhancement upon SNAP-tag binding) (Figure 7B) [70]. The most comprehensive work on SNAP-tag fluorogens based on dipolar FMRs has led to the development of a palette of probes with emissions ranging from 485 to 700 nm by varying the electron-donating and electron-withdrawing groups [67]. As for HaloTag, Kool and coworkers developed dimethylaminostyrylpyridinium fluorogens exhibiting a viscosity-sensitive emission that is activated upon reaction with HaloTag with a 27-fold fluorescence increase (Figure 7C) [71]. Our group also strongly focuses on the development of semisynthetic fluorescent reporters based on a palette of FMRs covering the green to far-red emission range and targeted at HaloTag (Figure 7C) [68, 72].
(A) Principle of fluorescence activation mechanism of FMR-based semisynthetic probes upon SLP binding. An EDG is π-conjugated to an EWG. Examples of some FMRs for (B) SNAP-tag labeling and (C) HaloTag labeling.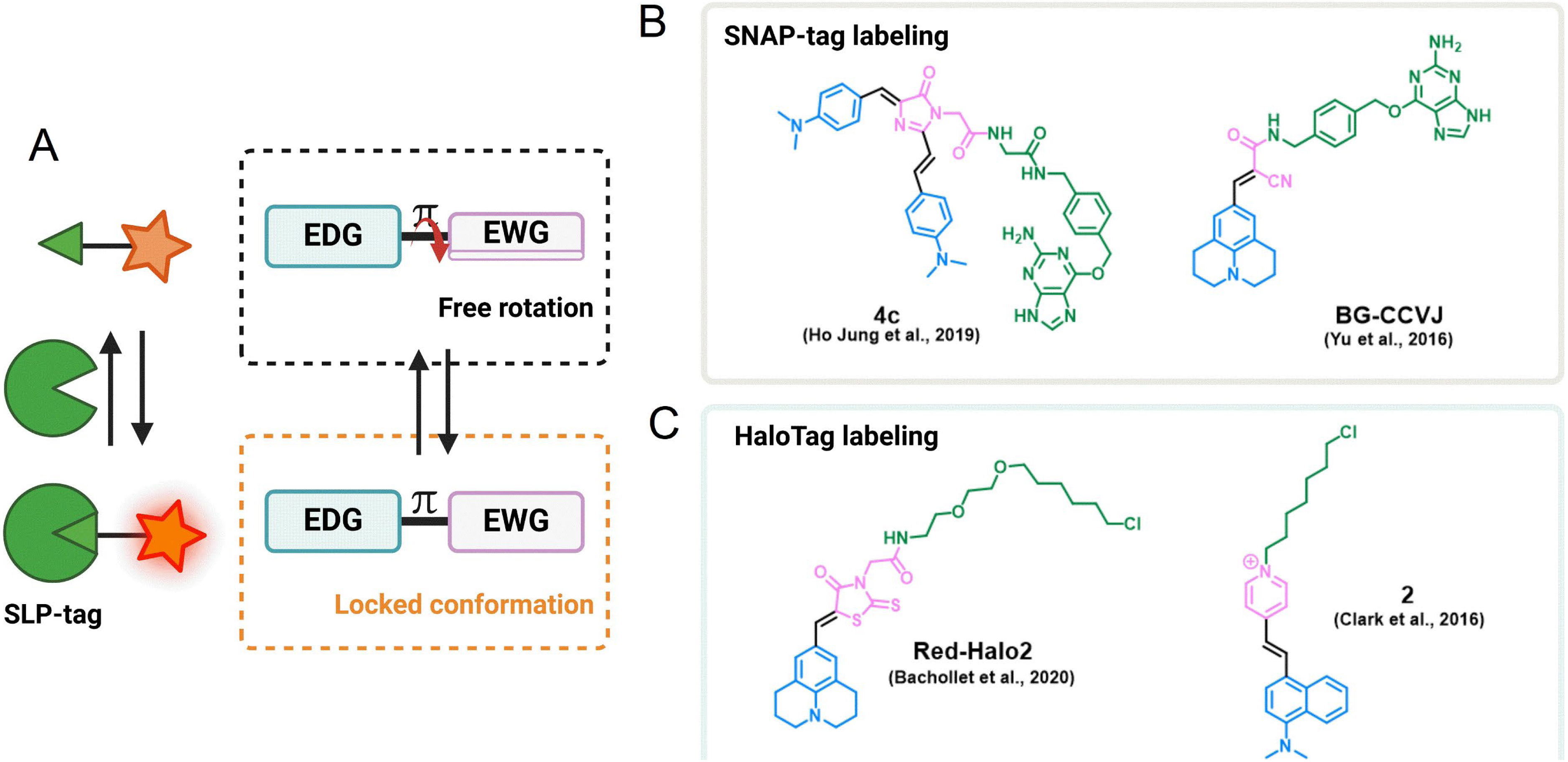
Noncovalent semisynthetic fluorogenic probes involving FMRs have also been reported for noncovalent genetically encoded strategies such as 4-hydroxybenzylidene rhodanine derivatives combined to the FAST protein [73, 74] or 3,5-difluoro-4-hydroxybenzylidene imidazolinone (DFHBI) derivatives that have been extensively engineered as fluorogens, enabling fluorescence emission upon binding to RNA aptamers [3, 75, 76]. Noncovalent fluorogen-activating protein (FAP) labeling has also been used to selectively bind FMR-based fluorogens such as malachite green and thiazole orange, and activate their fluorescence for wash-free bioimaging of subcellular organelles [77, 78].
2.3.2. FRET-based semisynthetic pH sensors
Using FAP labeling, Bruchez and coworkers reported semisynthetic fluorogenic pH probes based on FRET ratiometric fluorescence activation [63, 81]. These pH probes rely on a tandem of a pH-sensitive cyanine dye and a molecular rotor activated by a FAP that is either the donor [81] or the acceptor (Figure 8A) [63]. The variation in pH modulates the ICT in the cyanine structures and thus the emission properties of the donor cyanine3 (Cy3) dye, which in turn impacts the energy transfer (Figures 3C and 8A,B). Local activation of the fluorogen moiety by FAP binding thus creates a ratiometric pH sensor that can be used under wash-free conditions. The second design, with a fluorogenic acceptor, ensures better selectivity since the nonemissive acceptor also quenches the fluorescence of the pH-sensitive donor by energy transfer for unbound molecules (Figure 8) [63]. The resulting cell-impermeant probe was used to monitor membrane GPCR trafficking but cannot be used to image intracellular events. Additionally, the dual probe design uses most of the visible spectrum and precludes the use of other fluorescent probes for multiplexing.
(A) Structure of the FRET-based locally activated pH indicator targeting FAP. (B) Fluorescence emission of the tandem, showing the shift in emission of the Cy3 moiety due to pH and the FRET with MG. (C) Ratiometric pH calibration curve. Adapted with permission from Ref. [63]. Copyright 2018 American Chemical Society.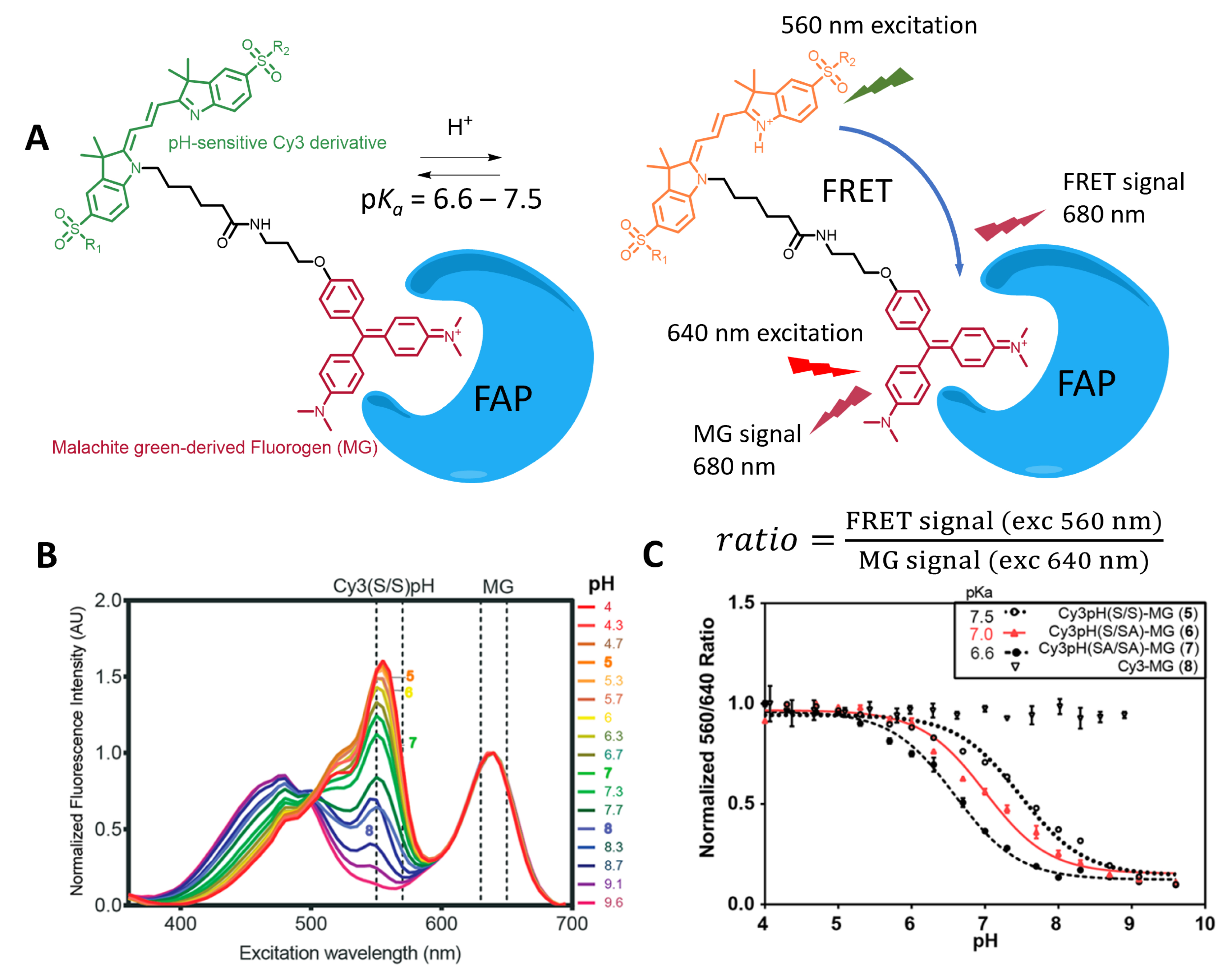
2.3.3. PeT- and ICT-based fluorogenic sensors
More versatile semisynthetic sensors based on a single fluorogen scaffold have been recently developed. Our group reported a dual-input calcium probe by combining a viscosity-sensitive red-emitting molecular rotor targeting HaloTag with a BAPTA group that controls the fluorescence by PeT (Ca-DIP, Figure 9) [79]. Similarly to previous MaPCa indicators based on SiR fluorogens, Ca-DIP exhibits a locally activated red-emitting fluorescence signal upon both HaloTag binding and Ca2+ chelation. More precisely, in the absence of HaloTag protein, the intramolecular rotation of Ca-DIP quenches the fluorescence emission, resulting in a very weak signal even in the presence of calcium. When bound to HaloTag, Ca-DIP becomes fluorescent (25-fold fluorescence activation) with good calcium sensitivity (ΔF/F0 = 5.4). Although the calcium sensitivity is on par with that of the MaPCa indicators, its brightness is much lower owing to a partial activation of the fluorescence by HaloTag.
Structure and properties of the Ca-DIP sensor. (A) Structure of Ca-DIP. (B) Absorption and emission properties in the four possible states of the system with and without calcium and HaloTag. (C) Fluorimetric titration. (D) Dual-input fluorescence activation. (E) Genetically targeted calcium imaging in HeLa cells expressing HaloTag–NLS protein: a selective signal sensitive to calcium can be recorded in cell nuclei under wash-free conditions. Adapted with permission from Ref. [79]. Copyright 2022 Royal Society of Chemistry.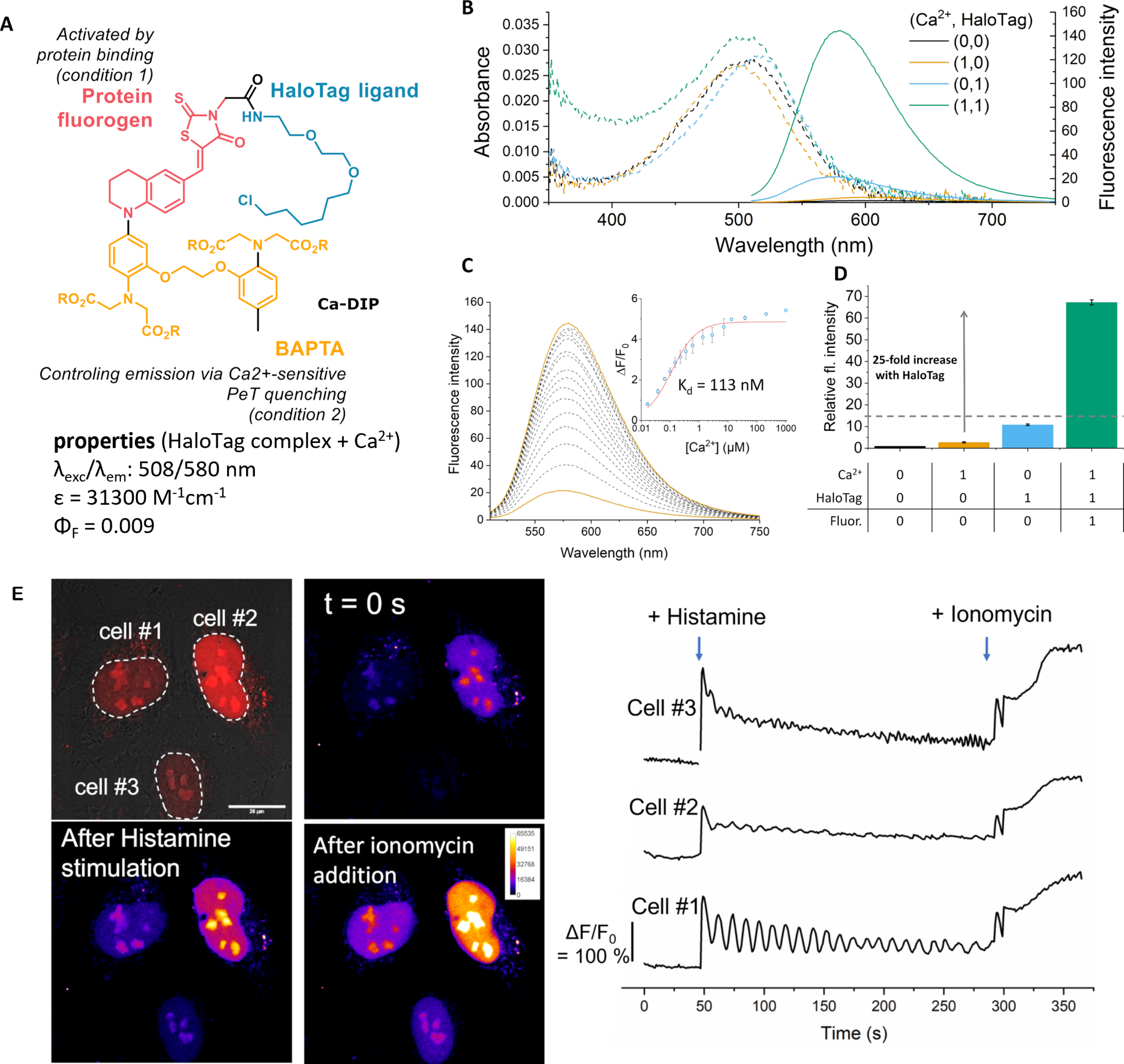
Following this work on a calcium probe, we have used a similar idea to design a dual-input pH probe and develop a locally activated chemogenetic sensor to image protein exocytosis [80]. By using a phenol electron-donating group, we obtained the pH-sensitive HaloTag ligand pHluo-Halo-1 (Figure 10A). Thanks to an adjacent amide function, the pKa of the phenol was lowered to 6.3, a value adapted to measure the transition from acidic vesicles (pH 5.5) to the neutral environment (pH 7.4) during protein exocytosis (Figure 10D). As for Ca-DIP, only the HaloTag-bound form is fluorescent with 21-fold activation due to the protein, and pH modulates the ICT from the phenol with a ratiometric absorption and only one fluorescence emission originating from the phenolate form (Figure 10B). In addition to tuning the pKa, the amide function was used to introduce a hydrophilic diethylene glycol chain in order to limit the nonspecific activation in lipophilic organelles. In HeLa cells, pHluo-Halo-1 indeed exhibits remarkable labeling selectivity, with virtually no nonspecific signal under wash-free conditions (Figure 10C). It was applied to image the exocytosis of a CD63-HaloTag protein using TIRF microscopy. Moreover, comparison with the pH biosensors super-ecliptic pHluorin and pHuji showed that our hybrid sensor reported correctly on single fusion events (Figure 10D) [80].
Semisynthetic fluorogenic sensor to monitor protein exocytosis. (A) Structure and dual-input activation of pHluo-Halo-1. (B) Absorption and emission spectra at various pH values. (C) Labeling selectivity of pHluo-Halo-1 in HeLa cells expressing a nuclear HaloTag–NLS under wash-free conditions. Turn-on number represents the median value for 130 cells. (D) General principle of exocytosis imaging with pHluo-Halo-1. (E) TIRF microscopy images of HeLa cell expressing CD63-HaloTag labeled with pHluo-Halo-1 undergoing fusion: normalized intensity trace for a selected fusion event corresponding to the orange arrow on the image. Adapted with permission from Ref. [80]. Copyright 2024 American Chemical Society.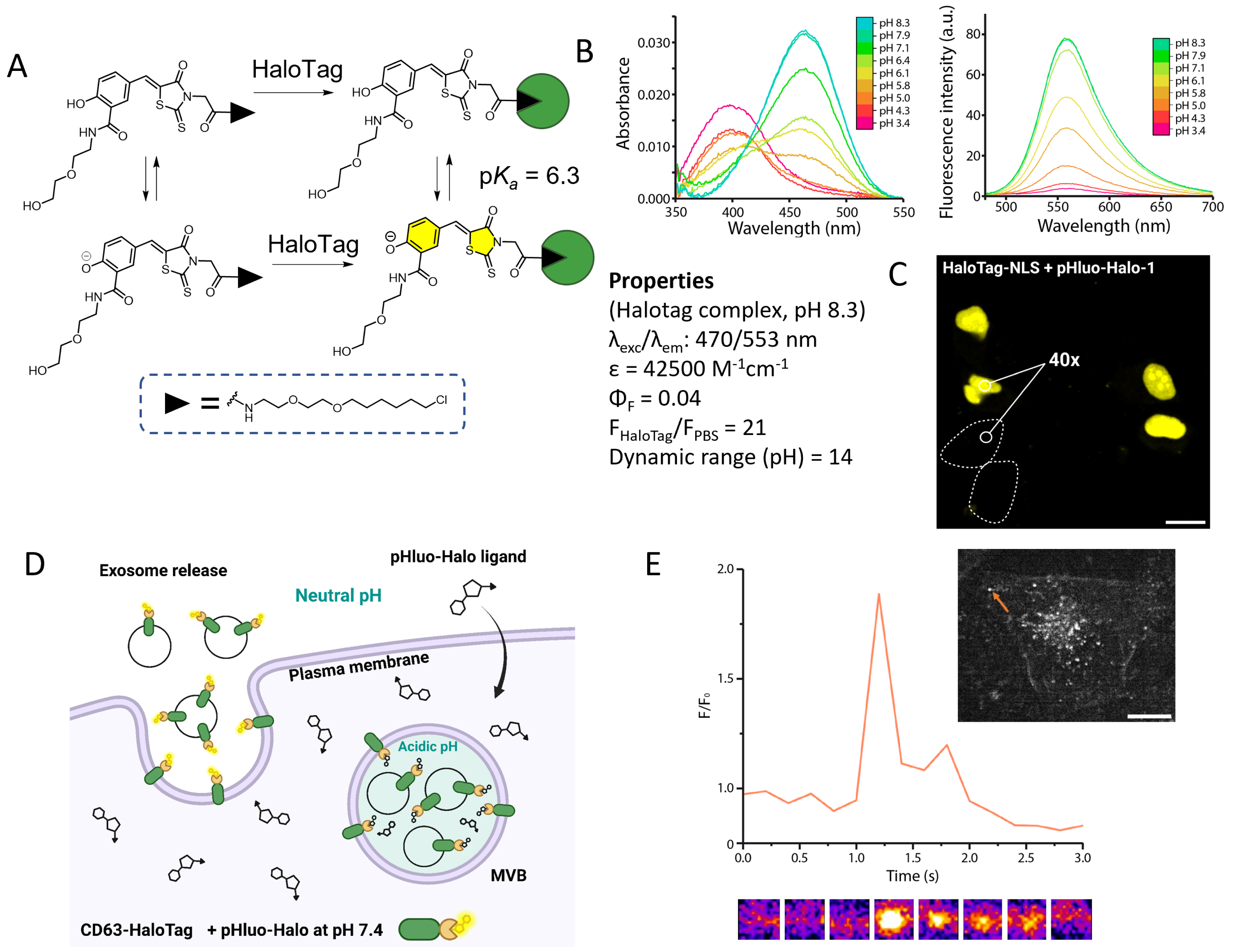
As for Ca-DIP, the fluorescence brightness of pHluo-Halo-1 remains low. Dipolar FMRs are inherently less bright than symmetrical fluorophores like rhodamines and cyanines, but they still can attain much better properties and HaloTag does not fully activate the fluorogen. HaloTag was indeed optimized for the rhodamine scaffold and does not necessarily activate efficiently the fluorescence of FMRs. Recent findings however suggests that protein engineering can improve this aspect [83].
The Baker group used existing molecular rotor DFHBI derivatives to develop a dual-input fluorogenic semisynthetic platform for intracellular pH monitoring [82]. This strategy is based on the binding of pH-sensitive DFHBI to mini fluorogen-activating proteins (mFAPs; Figure 11A). The moderate size, photostability, and fast binding kinetics of mFAPs make them attractive candidates for the development of hybrid chemogenetic biosensors [84]. The challenge was about the ability of the mFAP to bind both phenolic and phenolate states of DFHBI. Hence, to generate an effective pH-responsive semisynthetic probe using a DFHBI–mFAP labeling strategy, the authors successfully designed with a de novo computational strategy an mFAP mutant, mFAP_pH, capable of binding these two states with equivalent affinities (Figure 11A). Fluorimetric pH titration showed a suitable pKa of 5.4, corresponding to the pKa of free, unbound DFHBI in solution [85]. Upon stabilization of the cis-planar conformation by mFAP_pH, the deprotonation of the phenol moiety at neutral pH increases the ICT, which creates a bathochromic shift of the excitation spectra and activates the fluorescence, thus creating interesting ratiometric properties for quantitative pH sensing in the biological range (Figure 11B). It is worth noting that in this paper, the authors also report split reporters for protein/protein interactions and an mFAP-based calcium sensor, two features that are slightly out of the scope of this study. Although it reports on a similar type of chemogenetic sensor with a molecule combining sensitivity to pH and fluorogenic activation by a genetically encoded protein, this work presents a key difference compared to the other articles reviewed herein. It is indeed not based on small-molecule engineering, since they use a previously reported pH-sensitive fluorogenic probe, but solely on protein engineering to adapt the binding properties of mFAP to this molecule. While conventional protein engineering methods rely on the mutation of existing proteins, the de novo design used in this example consists in the generation of entirely new protein structures and functions [86].
Fluorogenic chemogenetic pH sensor based on the de novo design of mFAPs. (A) Principle of DFHBI–mFAP_pH complex formation for pH monitoring. (B) Normalized fluorescence excitation spectra of the mFAP_pH–DFHBI complex for a pH titration between pH 3.6 and pH 8.4 (left) and normalized emission spectra at pH 3.6 and 8.4 (right). Image adapted from Ref. [82] under the CC BY 4.0 license.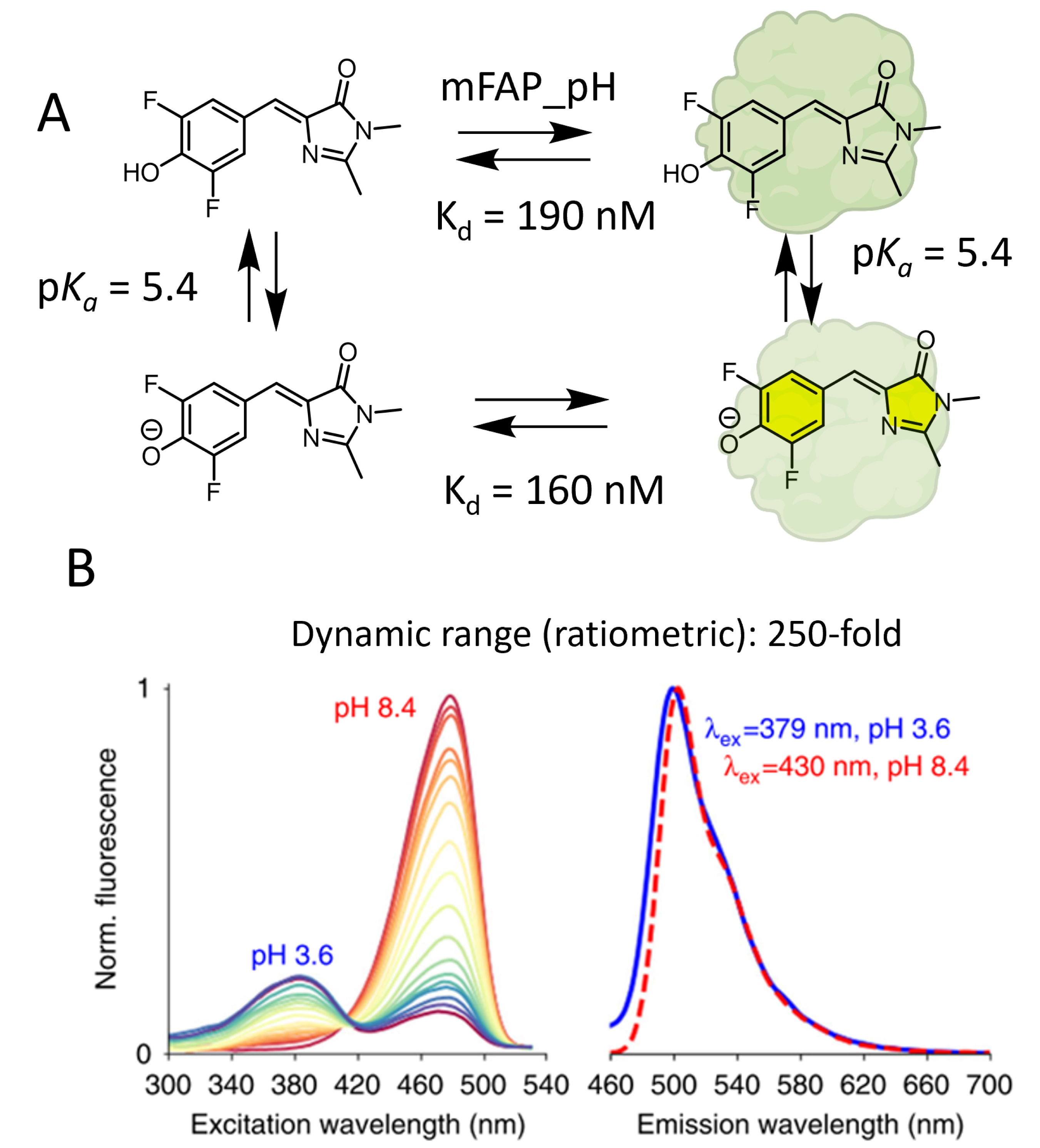
2.4. Another example of locally activated fluorogenic semisynthetic biosensor for pH
Xu and coworkers developed a semisynthetic pH probe based on the combination of 1,8-naphthalimide fluorogens (BG-NDM) targeted at SNAP-tag and incorporating an amine group that quenches the fluorescence via PeT (Scheme 2) [87]. In this design, the SNAP-tag protein activates BG-NDM by preventing fluorophore aggregation, and protonation of the amine at low pH inhibits the PeT. It is worth noting that the residual fluorescence of BG-NDM is high, and consequently, the fluorescence enhancement due to the reaction with SNAP-tag is moderate. This dual-input locally activated pH probe displays a pKa of 8.0, which is slightly high for the range of biological pH values albeit with a good dynamic response. So far this is the only example of a dual-input probe with an activation based on the aggregation ability of naphthalimide. Although it is not mentioned in this study, PeT between the benzylguanine ligand of SNAP-tag and a naphthalimide has been reported and may also contribute to the fluorogenicity of this probe and constitutes a possible strategy to design dual-input probes [88].
Structure of the naphthalimide-based dual-input pH probe BG-NDM.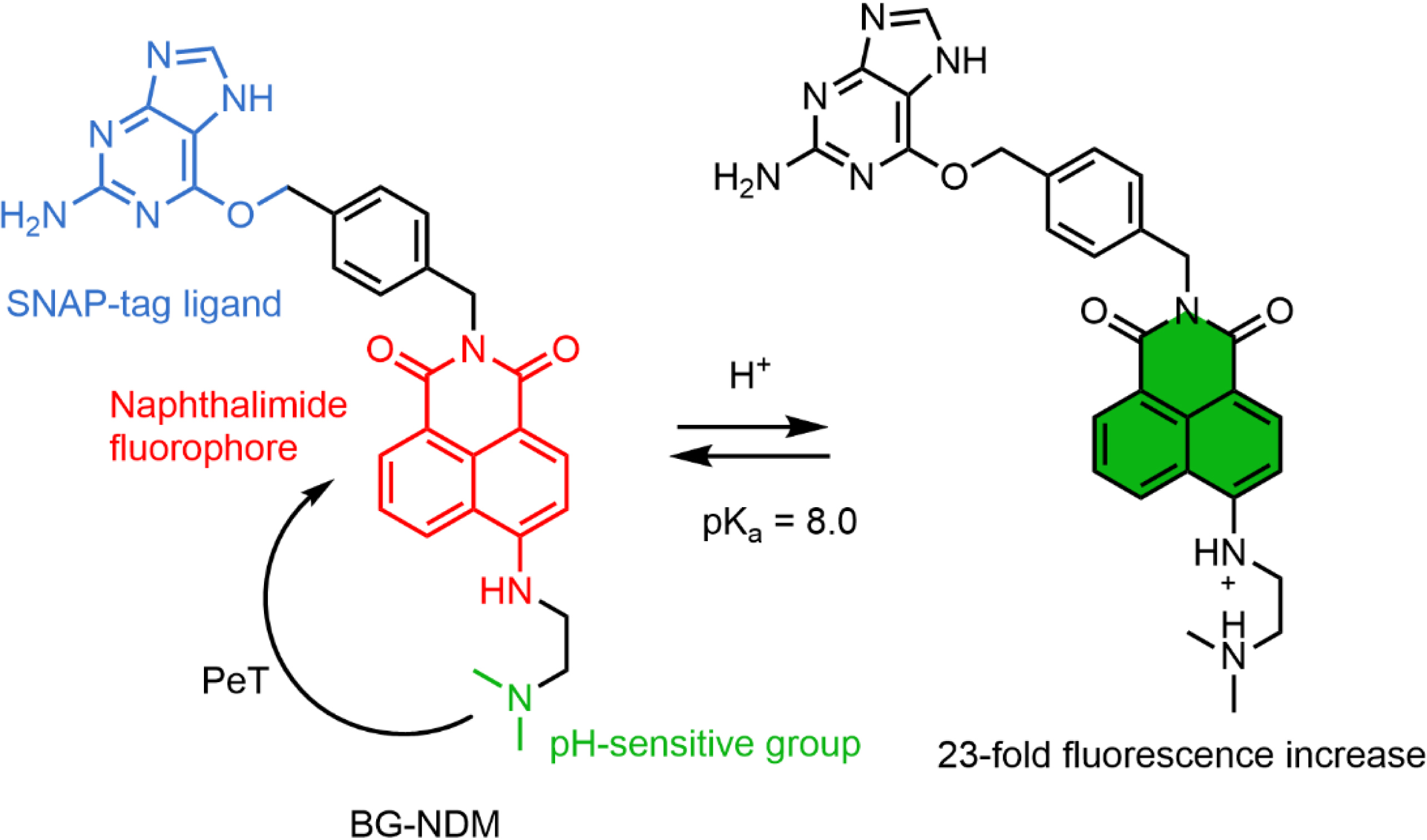
3. Conclusion
There have been so far only few examples of locally activated semisynthetic biosensors, likely owing not only to the recent nature of the field but also to the design and synthetic challenges of efficiently combining two fluorescence-activating mechanisms on a single molecule. Nonetheless, this approach certainly holds great potential for developing efficient sensors that benefit not only from the high versatility of small molecular fluorophores and sensing groups but also from the selectivity of genetic encoding of the recombinant protein. Although FRET-based examples have interesting properties such as ratiometric emissions that enable quantitative measurements, the most versatile design is the one based on a single fluorophore. The development of biocompatible fluorogens targeting SLP-tags with low cytotoxicity, cell permeability, high photostability, and a wide range of emission wavelengths provides molecular platforms that can be combined with a variety of sensing groups to engineer dual-input probes. Due to the small number of examples, only a few analytes have been targeted (pH, Ca2+ and Mg2+), but the dual-input probes described herein should be adjustable to other targets using the wide repertoire of chemical receptors as has been done with the FlAsH derivative to shift from a calcium to a magnesium indicator (Scheme 1). Measurements of the redox potential of fluorogens and receptors or density functional theory calculations can help predict the PeT process between two moieties to help design dual-input probes targeting new analytes with good sensitivity. Although remarkable results have been achieved, researchers have also encountered limitations such as limited fluorogenicity and brightness. Even though it may be convenient and versatile to use well-established SLP-tags that can bind a variety of small molecular probes, the future of the field will likely rely on the combination of molecular and protein engineering to ensure the adequation between the two partners and in particular the efficient activation of fluorescence. Protein engineering can be performed by rational design, directed evolution, or more recent computational de novo design methods. A recent example of the incorporation of a molecular calcium chelator (BAPTA) in a fusion between a GFP and HaloTag also underlines other possibilities that combine chemical and genetic approaches to developing hybrid sensors with unique properties [89].
Declaration of interests
The authors do not work for, advise, own shares in, or receive funds from any organization that could benefit from this article, and have declared no affiliations other than their research organizations.
Funding
JC is supported by a PhD fellowship funded by the “interface pour le vivant” (IPV) initiative of Sorbonne Université.