

 CC-BY 4.0
CC-BY 4.0
1. Introduction
Les dernières décennies ont été une période de mutation considérable pour l’industrie du raffinage du pétrole, qui est à la fois une source d’énergie et une source de matières premières pour l’industrie chimique à l’échelle mondiale et en particulier à l’échelle de l’Europe. En effet, en cette période, les raffineurs se sont orientés vers l’optimisation de la fabrication de la matière première, en produisant des produits à forte valeur ajoutée toujours plus adaptés au marché [1]. L’Arabie Saoudite est, devant la Russie, la plus importante force de production du pétrole brut dans le monde grâce à ses réserves et ses puits pétroliers. Le pétrole est constitué d’un mélange complexe d’hydrocarbures à savoir les paraffines, les naphtènes et les aromatiques. L’obtention de produits pétroliers répondant à des spécifications bien données nécessite une séparation préalable en différentes coupes (par distillation), lesquelles doivent être purifiées, ou subir des transformations, notamment en vue de besoins pétrochimiques ultérieurs [2]. Les coupes gazoles (gasoil) proviennent de la distillation directe du pétrole brut et des procédés de conversion (vapocraquage, craquage et reformage catalytiques). Elles sont constituées essentiellement de composés aromatiques et naphténiques, avec des impuretés de composés azotés et soufrés non négligeables. Leurs caractéristiques ne respectant pas les normes internationales, elles ne peuvent pas être utilisées directement, notamment dans l’industrie automobile.
Le gazole utilisé comme carburant pour les moteurs dans l’industrie de l’automobile, doit répondre à des normes de qualité de plus en plus sévères. Il doit être adapté aux spécifications des moteurs à fort taux de compression, qui se traduisent par un indice de cétane élevé. L’indice de cétane (I.C.) représente la qualité de combustion dans un moteur à allumage commandé, il caractérise la résistance à l’auto-inflammation du carburant gasoil. Il est numériquement égal au pourcentage volumique dans un mélange de (n-cétane + α-méthylnaphtalène). Le procédé d’hydrocraquage des naphtènes conduit à la production de paraffines pouvant contribuer à améliorer l’indice de cétane et satisfaire les normes imposées pour le gazole. D’un point de vue thermodynamique, l’hydrocraquage des naphtènes est complet dans les conditions utilisées. Cinétiquement, on observe un ordre positif par rapport à l’hydrocarbure et à l’hydrogène, donc par rapport à la pression totale, une très forte énergie d’activation et une augmentation de la vitesse avec le nombre des atomes de carbone.
Notre but est d’améliorer l’indice de cétane des gazoles. A l’échelle du laboratoire, notre approche, dans cet article, est orientée vers l’étude de l’ouverture des cycles des hydrocarbures naphténiques simples, comme le méthylcyclopentane (MCP). L’ouverture de cycle de ces hydrocarbures donne lieu à la formation de paraffines contribuant à améliorer l’indice de cétane; ainsi, plus la chaine paraffinique est longue meilleur est l’indice de cétane du carburant Diesel.
Le méthylcyclopentane (MCP), de poids moléculaire 84 g⋅mol−1, est considéré comme étant une molécule très répandue et utilisée dans le domaine de l’exploitation et de l’investigation des structures des catalyseurs [3, 4, 5, 6, 7, 8]. Le MCP est très étudié dans les réactions d’ouverture du cycle [9, 10, 11] et plusieurs catalyseurs ont été utilisés pour l’étude de l’hydrogénolyse du MCP: Ni/Al2O3 [12], Rh/GNF [13], Ge-Rh [14], Rh-Ge/Al2O3 [15], Pd/SAPO-11 [16], Rh/TiO2 et Rh/SiO2 [17], Fe-KIT-6 [18], Fe-TUD-1 [19], MnW-MCM-48 [20], CoMn-MCM-48 [21], Co-KIT-6 [22, 23].
L’ouverture du cycle du MCP conduit à la formation du n-hexane (nH), 2-méthylpentane (2MP) et du 3-méthylpentane (3MP). Aussi la réaction avec l’hydrogène donne lieu, à côté de la formation du cyclohexane et du benzène comme produits d’élargissement du cycle, à des produits de craquage (C1-C5) obtenus à haute température ou en présence de sites acides de Bronsted [24]. L’étude effectuée sur des catalyseurs à base de platine a montré que sur les petites particules de métal [24], l’ouverture du cycle suit un mécanisme non sélectif (formation des MPs et du nH), tandis que sur les larges particules de platine, l’ouverture du cycle suit un mécanisme sélectif (formation des MPs seulement). La sélectivité pour le n-hexane diminue lorsque la taille des particules du métal augmente. Gault et al. [24, 25, 26] ont montré que via le mécanisme non sélectif, la production de nH, 2MP et 3MP a des proportions de 2 : 2 : 1. Les espèces diadsorbées sur les sites métalliques sont considérées comme étant les intermédiaires. Lorsque la taille des particules est plus grande que 1,8 nm, on est en face d’un mécanisme sélectif. Dans ce cas, les intermédiaires sont les espèces tétra-adsorbées sur deux sites métalliques. Un mécanisme partiellement sélectif produisant plus de nH a été proposé par Maire et al. [27] où ce mécanisme est plus important aux hautes températures. Kramer, Zuegg [28, 29] et Paal [30] ont remarqué que la sélectivité par rapport au nH augmente avec la distance interfaciale entre le Pt et le support. De là, ils ont proposé deux réactions parallèles: la première forme 2MP et 3MP et se produit sur la surface du Pt, tandis que la seconde réaction forme nH et se produit à l’interface alumine/Pt, où les sites Pt0 (métallique) et Ptδ+ (ionique) sont considérés comme étant les sites actifs [31].
Les procédés catalytiques utilisent en majorité des catalyseurs supportés à base de métaux de transition, en particulier des métaux nobles [32, 33, 34, 35, 36]. Chaque année, des quantités énormes de catalyseurs sont préparées et des millions de tonnes de catalyseurs sont régénérés, pour un coût énergétique considérable. Il serait avantageux de substituer les catalyseurs métalliques à base de métaux nobles par d’autres matériaux moins coûteux et plus écologiques tels que les oxydes [37, 38, 39, 40, 41, 42]. Les oxydes de molybdène et de tungstène sont de bons candidats car ils sont caractérisés par leur activité catalytique bifonctionnelle dans l’hydrogénolyse et l’isomérisation des alcanes (pentane, hexane et heptane) [43, 44]. Les catalyseurs à base d’oxydes de molybdène et de tungstène ont été testés dans plusieurs réactions telles que l’hydrotraitement [45], la métathèse des oléfines [46], l’hydrogénation [47], la déshydratation des alcools [48], l’oxydation des alcools [49], l’ammoxydation et l’oxydation des oléfines [50] ainsi que la réduction du NO par l’ammoniaque NH3 [51]. Les oxydes de tungstène et de molybdène MO2 (M = Mo, W) sont de possibles catalyseurs dans l’étude de la réaction d’ouverture de cycle des composés naphténiques, et cela grâce à leurs propriétés métalliques. Ces oxydes ont en commun la présence d’une structure bifonctionnelle MO2(Hx)ac sur leur surface. Le comportement catalytique de cette phase [MO2(Hx)ac] est similaire à celui observé [52] lorsque le platine est déposé sur des supports acides. L’espèce active MO2(Hx)ac est obtenue via un processus de réduction contrôlé du MO3 par l’hydrogène à des températures différentes. La réduction du MO3 en MO2 donne lieu à la formation de deux électrons libres. Les deux liaisons π et σ sont formées entre chaque atome M adjacent tout au long de l’axe C de la structure rutile déformée de MO2. La délocalisation des électrons π produit les propriétés métalliques de cette phase [53].
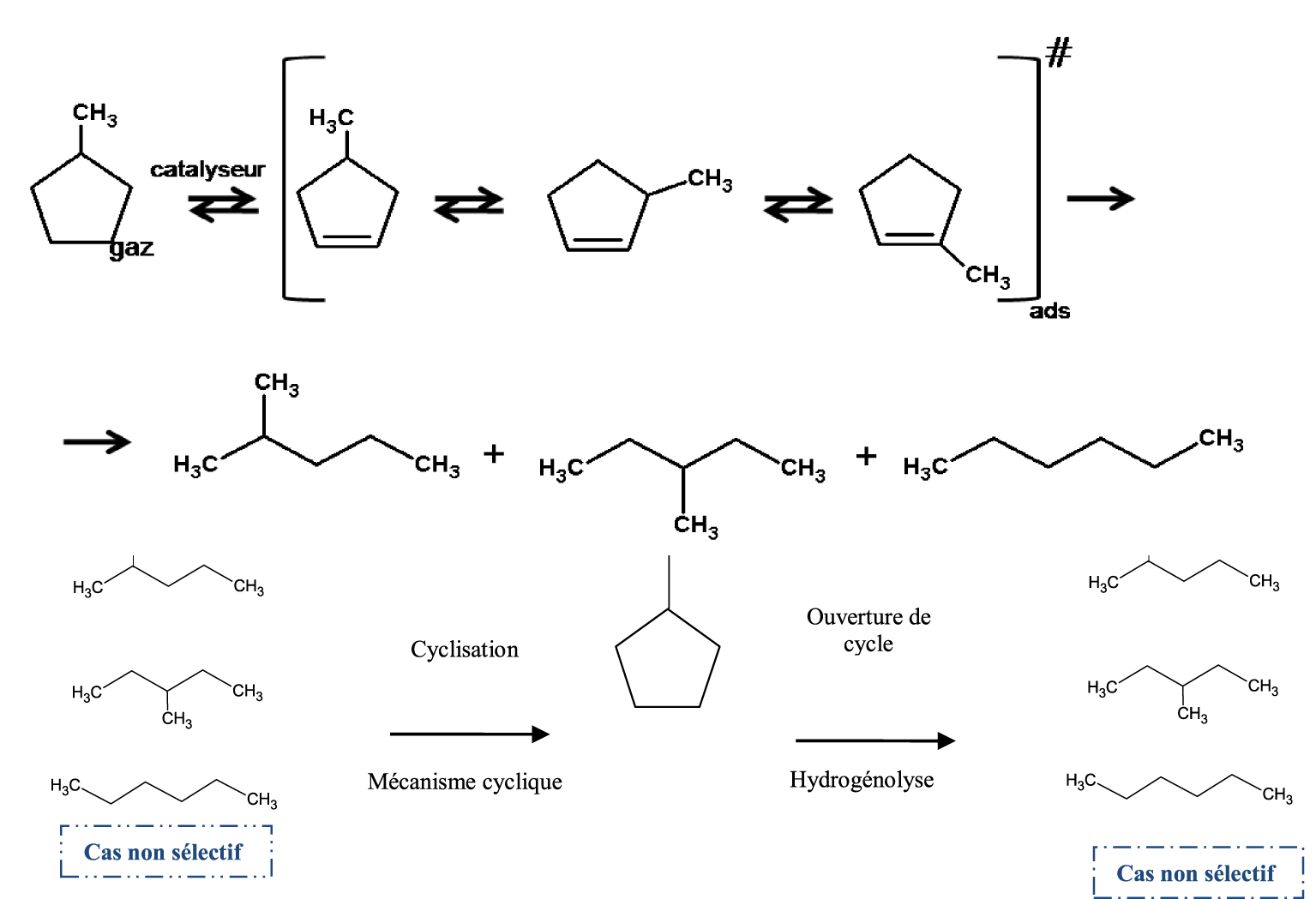
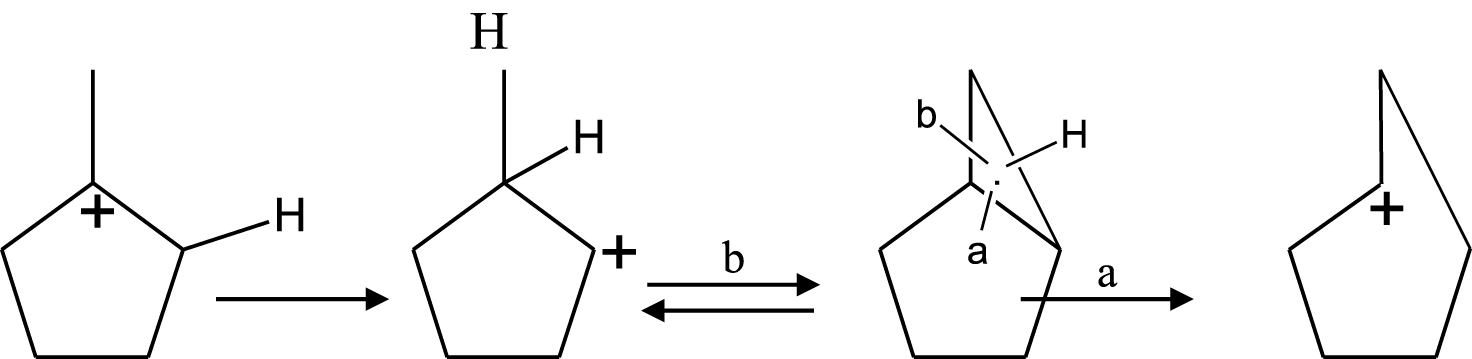
Plusieurs mécanismes réactionnels ont été proposés pour expliquer les réactions d’hydrogénolyse, de craquage, d’isomérisation et de déshydrogénation ou aromatisation des hydrocarbures. En effet, ces mécanismes vont dépendre du type de catalyseurs utilisés. Dans la catalyse métallique, à basse conversion (basse température) nous avons principalement la réaction d’ouverture du MCP. La réaction d’hydrogénolyse du MCP peut être soit sélective soit non sélective selon la nature du métal et/ou la taille de l’agrégat. Le mécanisme non sélectif (Schéma 1) a lieu généralement sur de petites particules ( <1% Pt) où la probabilité de rupture de la liaison C–C du cycle est approximativement 50%.
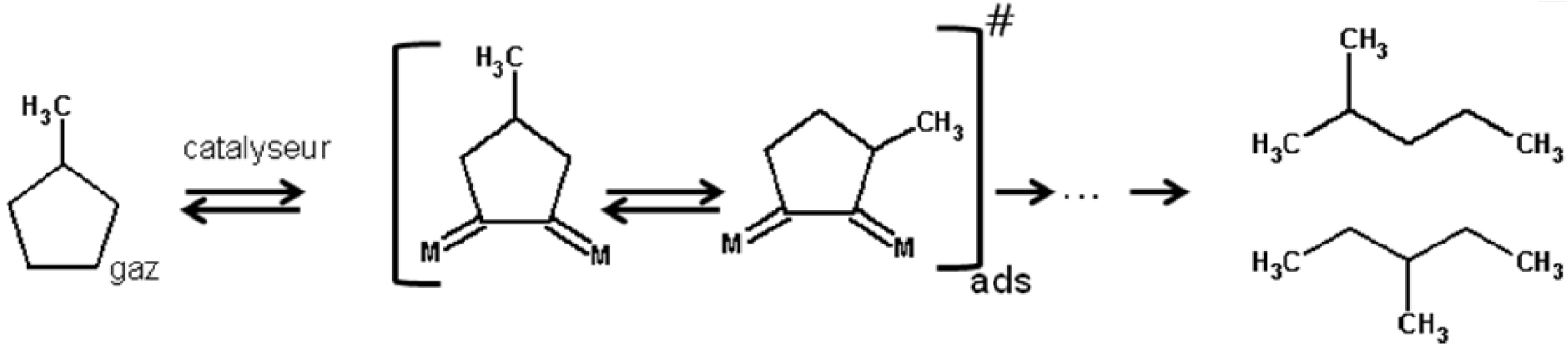
Le mécanisme sélectif il s’observe sur les grosses particules de platine ( >2% Pt) où la rupture des liaisons bisecondaires C–C a lieu, et le mécanisme proposé est est présenté dans le Schéma 2.
D’autre part, en catalyse métallique, une hydrogénolyse partiellement non sélective peut aussi se produire, par l’intervention d’un métallocyclobutène exocyclique [24]. Il s’agit d’un mécanisme qui est en compétition avec le mécanisme sélectif sur des catalyseurs à faible dispersion.
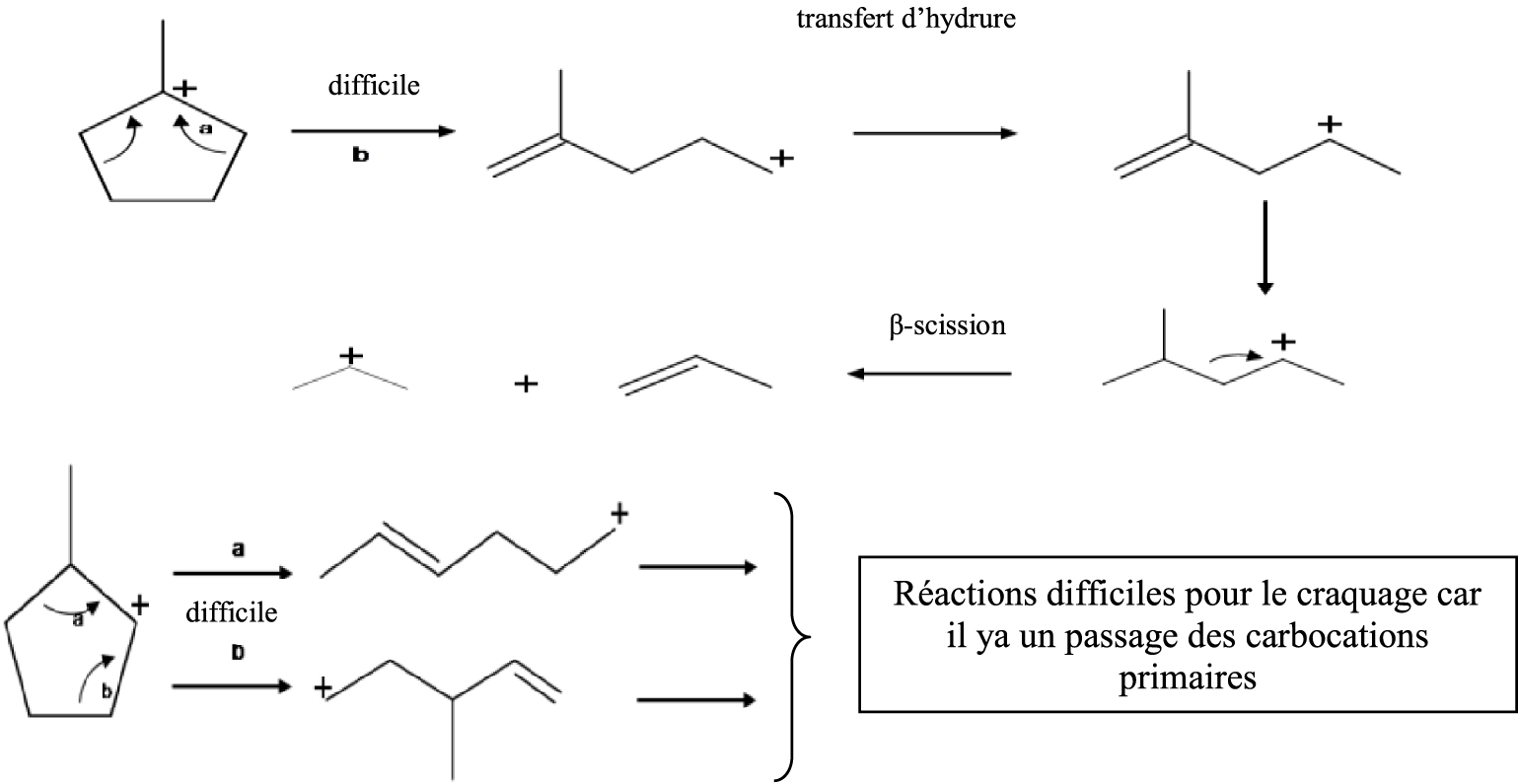
Dans la catalyse acide (Schéma 3), en général, ces catalyseurs sont moins actifs que les catalyseurs métalliques ; il faudra donc travailler à plus haute température pour obtenir de la conversion et dans ces conditions expérimentales deux réactions ont lieu simultanément : le craquage et l’élargissement du cycle.
En supposant que l’ouverture du cycle pentanique et le craquage se réalisent selon un processus de β-scission nous devons obtenir les produits présentés dans le Schéma 3.
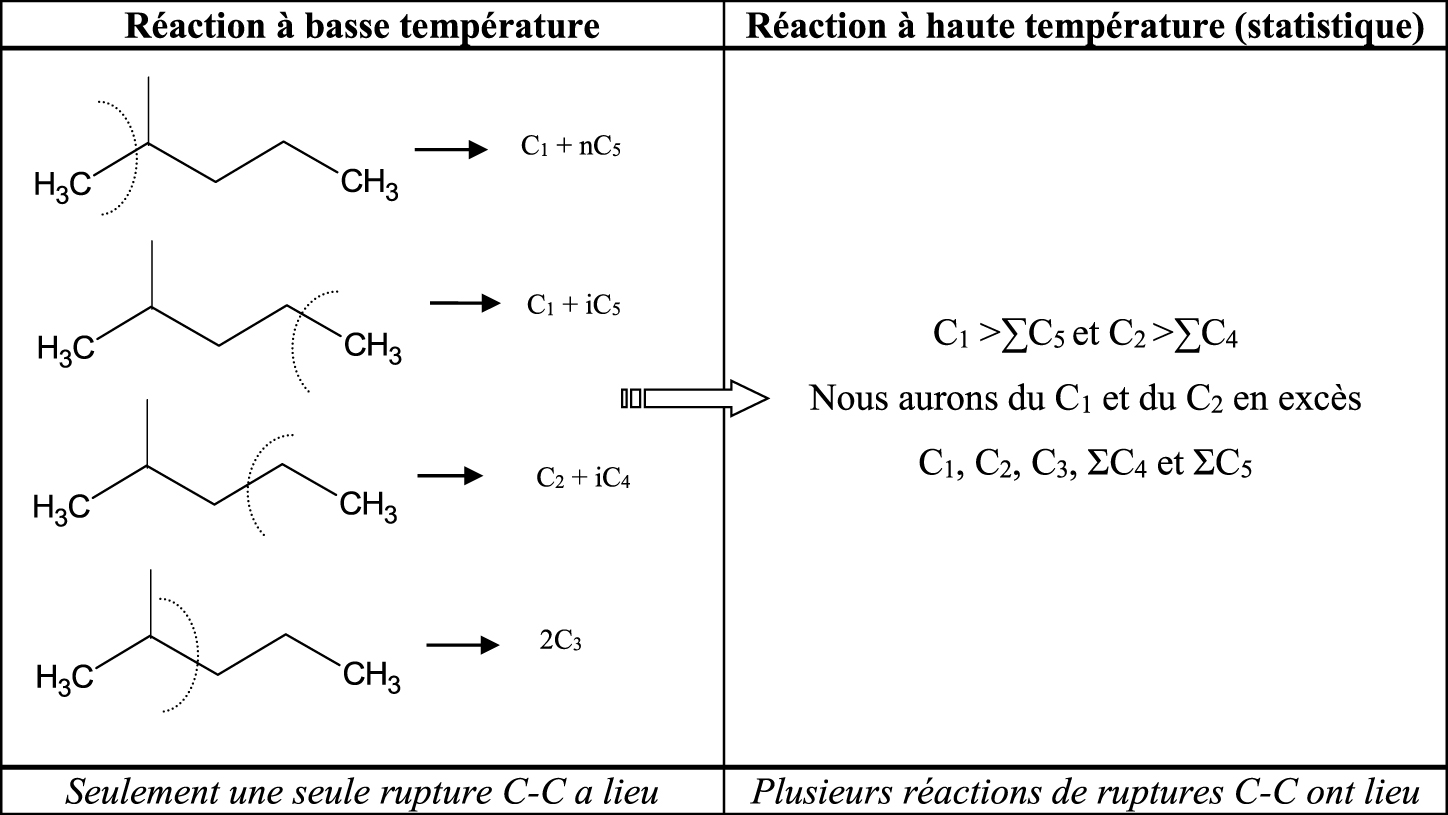
Les diverses réactions d’hydrocraquage vont dépendre de la température de réaction. A basse température, nous aurons une simple réaction de rupture C–C suivi de la désorption des produits formés. A haute température, des processus répétitifs auront lieu dans la phase adsorbée. Citons ici un exemple avec le 2-méthylpentane (Schéma 4).
Nous avons observé (Tableau 1) que la distribution des produits de craquage sur le platine est statistique à une température de réaction donnée et suit la distribution présenté dans le Tableau 1.
Bilan (%) de la distribution statistique des produits de craquage
| C1 | C2 | C3 | iC4 | nC4 | iC5 | nC5 |
|---|---|---|---|---|---|---|
| 26 | 16 | 16 | 4 | 12 | 8 | 18 |
Dans la catalyse bifonctionnelle (Schéma 5), deux sites actifs sont impliqués dans la réaction. Les sites métalliques étant les plus réactifs, les réactions d’hydrogénation-déshydrogénation et de rupture C–C se feront plus faiblement qu’en catalyse acide «pure».
La réaction d’agrandissement de cycle est favorisée en catalyse acide selon le Schéma 5.
Dans cet article, les catalyseurs WO3/TiO2 et MoO3/TiO2 ont été testés dans la réaction d’ouverture de cycle du MCP à plusieurs températures.
Spécifications des oxydes utilisés pour la préparation des catalyseurs
| Support | Spécification | Origine |
|---|---|---|
| TiO2 | 80% Anatase 20% Rutile | Degussa-Huls AG |
| WO3 | 100% | Strem Chemicals |
| MoO3 | 100% | Strem Chemicals |
2. Partie expérimentale
2.1. Préparation des catalyseurs WO3/TiO2 et MoO3/TiO2
Les catalyseurs WO3/TiO2 et MoO3/TiO2 ont été préparés par imprégnation. Les caractéristiques principales des oxydes sont regroupées dans le Tableau 2. Les supports n’ont subi aucun traitement thermique avant la procédure d’imprégnation sauf dans le cas du TiO2 qui, sous forme d’extrudés, a été lavé à l’eau distillée, puis séché à l’étuve pendant 12 h et ensuite calciné une nuit à 400 °C sous air. Le solide obtenu est broyé au mortier et passé sur un tamis de manière à obtenir des particules de diamètre comprises entre 80 et 400 μm.
Les catalyseurs à base d’oxyde supportés sur TiO2 : (MoO3/TiO2) et (WO3/TiO2) à 5 monocouches (30 wt%) ont été préparés par la méthode d’imprégnation classique du TiO2, dite de Pines. Le support est mis en contact avec la solution du sel précurseur métallique. Le mélange est maintenu pendant 2 h sous agitation à température ambiante, puis à une température de 80 °C jusqu’à l’obtention d’une pâte qui est ensuite séchée à l’étuve pendant une nuit à 120 °C. La poudre obtenue est ensuite calcinée sous flux d’air à 500 °C pendant 4 h. Pour une quantité de 5 g de TiO2 imprégnée, on a utilisé des quantités de 1,9 g et 0,2 g de (NH4)6 Mo7O24 ⋅ 4 H2O (Strem Chemicals) et (NH4)6 H2W12O40 ⋅ 6 H2O (Strem Chemicals), respectivement. Nous avons choisi de considérer un recouvrement géométrique du support en supposant qu’une molécule de WO3 occupe 23 Å2 et qu’une molécule de MoO3 occupe 20 Å2.
2.2. Caracterisation physico-chimique des catalyseurs
Les analyses chimiques des catalyseurs préparés ont été réalisées au Service Central d’Analyse Chimique à Vernaison. Les diffractogrammes de rayons X présentés ont été réalisés sur un diffractomètre à poudre, Bruker D8 Advance, utilisant une anti-cathode de cuivre (raie Kα,𝜆 = 1,5418 ). Cet appareil fonctionne sous une tension de 40 kV et une intensité de 50 mA. Les enregistrements sont sur un intervalle de valeurs d’angle 2𝜃 variant de 5 à 90°, par pas de 0,02°, avec un temps de comptage d’une seconde par pas. Quelques milligrammes d’échantillon à analyser, sous forme de poudre, sont placés sur un porte-échantillon, en veillant à sa planéité afin d’obtenir un diffractogramme optimal. L’identification des phases est réalisée par comparaison avec les fichiers standard JCPDS (Joint Committee on Powder Diffraction Standards). Les diffractogrammes de rayons X présentent un nombre de raies variable en fonction du type de matériau. La taille moyenne des cristallites est estimée à partir de l’élargissement des pics de diffraction selon la relation de Debye–Scherrer (Equation (1)) :
| (1) |
K : constante correspondant à un facteur de forme, égale dans ce cas à 0,9 ;
𝛽 : largeur angulaire à mi-hauteur de la raie de diffraction hkl (radian) ;
𝜃 : angle de Bragg (degré) ;
𝜆 : longueur d’onde du faisceau incident (Å).
L’équation (1), permet d’obtenir le diamètre moyen d’une cristallite, en considérant cette dernière comme sphérique.
L’ensemble des images MEB a été réalisé à l’IPCMS sur un appareil JEOL JSM — 6700 F avec une tension de 5 kV. Le microscope électronique à balayage consiste en l’analyse des électrons secondaires et rétrodiffusés issus du bombardement de l’échantillon par un canon à électrons. Ceux-ci sont soumis à une tension accélératrice de 10 à 20 kV. Les électrons secondaires rétrodiffusés viennent de la couche superficielle de l’échantillon. Ils sont de faible énergie et nécessitent donc d’être amplifiés avant de pouvoir être visualisés. Le phénomène de visualisation tridimensionnelle observé sur un cliché s’explique par l’accroissement du nombre d’électrons secondaires lorsque l’angle d’incidence des électrons sur la surface de l’échantillon diminue. C’est-à-dire que le nombre d’électrons réfléchis augmente sur les reliefs en arêtes et en pointes. Les échantillons sont déposés et maintenus sur un porte-échantillon en laiton grâce à du ruban adhésif double face en carbone. Les échantillons sont métallisés sous vide par pulvérisation cathodique d’une couche de carbone afin d’éliminer les effets de charge.
La porosité des catalyseurs a été déterminée grâce à la physisorption des molécules d’azote à − 196 °C. La quantité de gaz adsorbé, à température constante, est fonction de la pression relative de ce gaz. Le tracé de la droite P∕V ⋅(P0−P) en fonction de P∕P0, dans le domaine de validité de l’équation (0,05 < P∕P0 < 0,35) permet d’accéder à la valeur du volume Vm de la monocouche, qui est égale à l’inverse de la pente. La valeur de Vm est directement proportionnelle à la surface spécifique (Equation (2)) :
| (2) |
Vm : volume de la monocouche par g de solide ;
N : nombre d’Avogadro (6,02 × 1023) ;
VM : volume molaire de l’adsorbant ;
𝜎 : surface occupée par une molécule d’adsorbant (𝜎 = 16,2 × 10−20 m2 pour N2 à − 196 °C).
Les isothermes d’adsorption-désorption d’azote sont obtenues sur un appareil Micromeritics TriStar3000. Les échantillons ont été préalablement dégazés (100 mg) à 250 °C sous vide pendant 12 h. L’analyse est effectuée à la température de l’azote liquide ( − 196 °C). Les isothermes sont déterminées dans l’intervalle P∕P0 0,001–0,0995 ; le temps d’équilibre avant chaque nouvel incrément de volume est de 10 s. Le logiciel de traitement des données nous a permis de déterminer la surface spécifique BET, le volume poreux et le diamètre des pores.
Les analyses SPX sont réalisées sur un spectromètre Thermo VG Scientific opérant sous ultra-vide de 1 × 10−9 Pa. Il est constitué d’une chambre de préparation et d’une chambre d’analyse reliées par un soufflet métallique. L’échantillon, sous forme de pastille, déposé sur un porte-échantillon, est dégazé à température ambiante dans la chambre de préparation avant d’être analysé. Le rayonnement X utilisé provient de la raie Kα de l’aluminium (hν = 1253,6 eV). L’analyseur est couplé à un système informatique permettant de traiter les spectres.
2.3. Mesure de la réactivité catalytique
2.3.1. Le montage pilote, pour la conversion du MCP, à pression atmosphérique
Les tests catalytiques ont été réalisés dans un montage à pression atmosphérique (Figure 1). Avant la réaction, une purge à l’azote est nécessaire pour chasser l’air de l’ensemble du système. Une entrée d’H2 est ouverte, la pression étant fixée, le débit du gaz est commandé par un débitmètre. Le gaz circule dans tout le système et passe par le bas du réacteur (chargé en catalyseur). La programmation de la température (ainsi que le gradient de la température et la sécurité) du four se fait sur le tableau de bord du bâti. La réaction se fait par injection de l’hydrocarbure dans le piège en U.
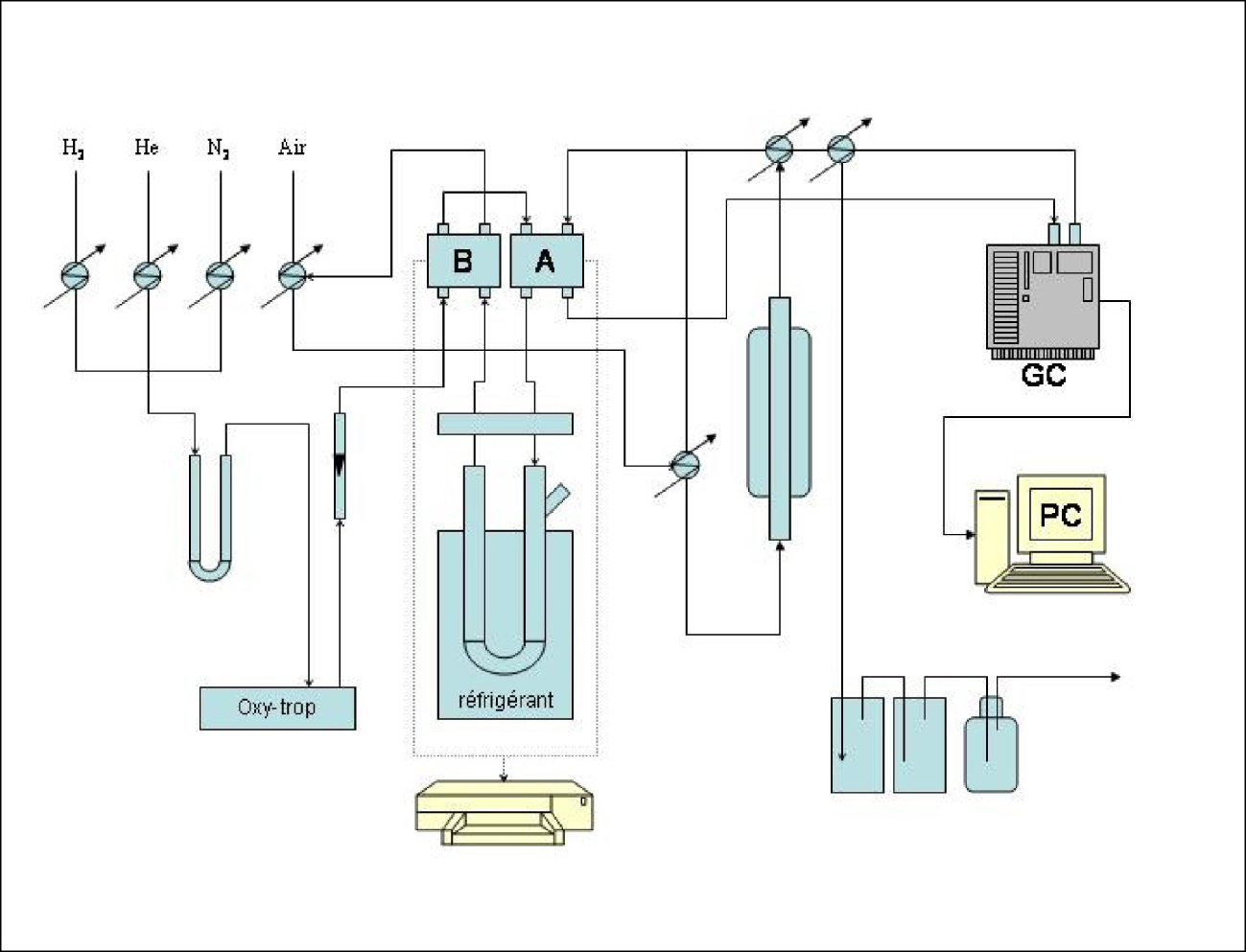
Schéma du montage pour la conversion du MCP à pression atmosphérique.
Le tube en U est plongé dans un mélange réfrigérant (anisole — azote liquide). L’hydrogène entraîne la vapeur de l’hydrocarbure jusqu’au réacteur tout en passant par les catharomètres. Les vapeurs (produits de la réaction) sont détectées sur le chromatographe où un réglage préalable de la température de la colonne, de l’injecteur et du détecteur a eu lieu. Les produits obtenus de la réaction sont visualisés sur l’écran de l’ordinateur. Les quantités de catalyseur sont choisies de manière à éliminer l’influence du phénomène de diffusion. Nous avons utilisé 200 mg de catalyseur « frais » pour chaque expérience. Ces catalyseurs sont, préalablement à leur utilisation, réduits sous hydrogène in situ pendant une durée bien déterminée de 12 h à 500 °C. Le débit d’hydrogène est de 40 mL⋅min−1. Des pulses de 5 μL d’hydrocarbure sont envoyés sur les surfaces continuellement balayées par l’hydrogène. Un test à blanc a permis de vérifier qu’aucune réactivité thermique ne se manifestait en l’absence du catalyseur.
Les tests catalytiques ont été réalisés à des températures de réaction variant entre 180 et 500 °C. Le réacteur utilisé pour le déroulement de la réaction catalytique est un réacteur catalytique pulsé en ligne avec un chromatographe en phase gazeuse Varian 3300 (Figure 1). L’analyse des produits de réaction a été effectuée par chromatographie gazeuse à l’aide d’un détecteur à ionisation de flamme (FID). La colonne chromatographique utilisée (de type 5CB WCOT Fused Silica) avait une longueur de 50 m, un diamètre intérieur de 0,53 mm, un diamètre extérieur de 0,70 mm et une épaisseur de 5 μm. La phase stationnaire de la colonne capillaire est CP-Sil. L’identification des produits de réaction a été réalisée par comparaison avec les temps de rétention observés pour des substances étalons. Les signaux de chromatographie en phase gazeuse (convenablement corrigés pour tenir compte de la sensibilité variable du détecteur) ont été employés pour calculer le nombre total de moles de chaque produit dans le courant gazeux de sortie. Par la méthode de normalisation des aires (appelée en anglais « area normalization »), les pourcentages en poids de chaque composant sont obtenus en divisant les surfaces individuelles par la somme des surfaces de tous les pics, multipliée par 100. Les surfaces correspondant aux différents composés ne sont pas directement proportionnelles aux teneurs dans le mélange. Il est en fait nécessaire de déterminer des facteurs de correction. Ces facteurs de correction de Dietz sont déterminés pour les détecteurs FID et sont présentés dans le Tableau 3 avec les temps de rétention correspondants.
Coefficients de Dietz et temps de rétention relatif aux différents hydrocarbures obtenus lors de la conversion du méthylcyclopentane
| Numéro | Hydrocarbure | Abréviation | Facteur de Dietz | Temps de rétention (s) |
|---|---|---|---|---|
| 1 | Méthane | C1 | 0,97 | 182 |
| 2 | Ethane | C2 | 0,97 | 188 |
| 3 | Propane | C3 | 0,98 | 199 |
| 4 | Isobutane | iC4 | 1,09 | 210 |
| 5 | Butane | C4 | 1,09 | 220 |
| 6 | Iso-Pentane | iC5 | 1,05 | 247 |
| 7 | Pentane | C5 | 1,04 | 259 |
| 8 | 2-Méthylpentane | 2MP | 1,05 | 307 |
| 9 | 3-Méthylpentane | 3MP | 1,04 | 322 |
| 10 | n-Hexane | nH | 1,03 | 336 |
| 12 | Méthylcyclopentane | MCP | 1,01 | 380 |
| 15 | Benzène | Bz | 1,12 | 419 |
| 16 | Cyclohexane | C6 | 1,01 | 434 |
2.3.2. Détermination de la conversion 𝛼 (%)
La conversion (𝛼) représente le nombre de moles d’hydrocarbure injecté qui a réagi et est exprimée en pourcentage.
| (3) |
2.3.3. Détermination de la sélectivité (%)
La sélectivité Si d’un produit i donné est définie par la quantité ni de produit i sur la quantité totale de produit obtenu nréagi.
| (4) |
2.3.4. Le calcul du flux de l’hydrocarbure (HC)
| (5) |
d : masse volumique de l’HC (g⋅cm−3) ;
m : masse molaire de l’HC (g⋅mol−1) ;
t : temps de passage de l’HC sur le catalyseur (s) ;
F = (5 × 10−3 × 0,784)∕(84 × 5 × 60)
F = 1,53 × 10−7 mol⋅s−1
2.3.5. Détermination de la vitesse de réaction
Habituellement, l’activité d’un catalyseur est définie par la vitesse de réaction, exprimée par gramme de catalyseur et par unité de temps.
Nous présenterons un exemple de calcul de la vitesse spécifique, en considérant la réaction
| (6) |
- Dans le cas d’un réacteur différentiel opérant sous flux molaire constant F, nous pouvons considérer la vitesse constante en tout point.
- Une fine section, de masse
- Dans un point x du réacteur, nous avons : C = (FA0 − FA)∕FA0, avec FA0 flux molaire de A à l’entrée du réacteur et FA le flux molaire de A au point x et dC = −dFA∕FA0. Ce qui implique − dFA = FA0⋅d𝜔 = F⋅dC et par conséquent la vitesse de la transformation V sera alors V ⋅d𝜔 = F⋅dC. On obtient d𝜔∕F = dC∕V .
- Pour des conversions faibles : 𝜔∕F = C∕V .
- Entre l’entrée et la sortie du réacteur, l’équation s’écrit : 𝜔∕F = ∫dC∕V .
- Pour une réaction d’ordre 1, on a V = k⋅PA.
- En tout point du réacteur on a : FA = F⋅(1 − C) et FB = F × C. Mais, le nombre de moles étant constant au cours de la réaction implique FA + FB = F = Cte.
- Les pressions partielles s’expriment pour A par PA = P⋅FA∕(FA + FB) = P⋅(1 − C) et pour B par PB = P × C.
La vitesse (μmol⋅g−1⋅s) s’écrit alors :
| (7) |
F : flux d’hydrocarbure (μmol⋅s−1) ;
𝜔 : masse de catalyseur (g) ;
C : conversion de A (%).
2.3.6. Détermination des énergies apparentes d’activation
Nos études de réactivité sur ces différents systèmes sont réalisées en fonction de la température, ceci nous a donc permis de déterminer les énergies apparentes d’activation de la réaction de contact du MCP. Nous allons expliquer maintenant cette donnée cinétique.
- Sans faire d’hypothèse particulière sur les mécanismes de réaction nous pouvons écrire la vitesse de réaction
- Nos expériences sont réalisées avec PHC = Cte et PH2 = Cte. Nous pouvons donc écrire v = k⋅Cte dans un faible domaine de température.
Si maintenant nous faisons l’hypothèse que nous avons un mécanisme de type Langmuir–Hinshelwood, la vitesse de réaction sera proportionnelle aux fractions de surface recouvertes par HC et H2, respectivement 𝜃HC et 𝜃H2.
| (8) |
| (9) |
| (10) |
| (11) |
| (12) |
Et nous avions vu r = 𝛼F∕m à faible conversion et dans notre cas F∕m = Cte
F : flux de l’hydrocarbure HC ;
𝜔: masse du catalyseur ;
𝛼 : conversion ;
d’où : r ∼𝛼⋅ Cte′′∼ k⋅Cte
Dans les tableaux 𝛼F∕m est VG exprimée en [106 s−1].
- La constante de vitesse k de la réaction de transformation de l’hydrocarbure dépend de la température selon l’équation d’Arrhenius :
| (13) |
k : constante de vitesse ;
A : facteur préexponentiel ;
R : constante des gaz parfaits ;
T : température de la réaction (K) ;
- Pour la détermination expérimentale de l’énergie apparente d’activation, les tests catalytiques sont réalisés à différentes températures. Ainsi on a tracé le graphe Ln(k) = f(1∕T). La pente de la droite obtenue est égale à (−Ea∕R). Cette valeur est déterminée comme étant l’énergie apparente d’activation.
- Il est nécessaire de déterminer le facteur préexponentiel A. Les valeurs de Ln(V ) (Ln vitesse globale de réaction) en fonction de l’inverse de la température sont portées dans les tableaux (11 à 14). Elles ont été calculées à partir des courbes Ln(k) = −Ea∕RT + Ln(A) où nous avons porté Ln(k) = f(1∕T) ; l’ordonnée à l’origine donnant la valeur de A.
3. Resultats et discussions
3.1. Caractérisation physico-chimique
3.1.1. Catalyseurs supportés MoO3 /TiO2
Les photographies en microscopie électronique à balayage correspondant au MoO3 non calciné et MoO3 réduit, sont présentées dans la Figure 2. La morphologie est variable, consistant en agrégats de différentes formes géométriques avec prépondérance des plaquettes de différentes dimensions. Les photographies en microscopie électronique à balayage correspondant au support TiO2 P25 calciné, sont présentées dans la Figure 3. La morphologie est caractéristique aux cristaux d’oxyde de titane commercial. La taille moyenne des cristaux d’oxyde de titane est de l’ordre de 40 nm. Les échantillons apparaissent cristallisés sans traces d’oxydes amorphes.
Les photographies en microscopie électronique à balayage correspondant aux catalyseurs MoO3/TiO2 calcinés et réduits sont présentées sur la Figure 4. Ainsi, l’ajout d’oxyde de molybdène sur le support TiO2 détermine une perte d’identité morphologique du MoO3 et par contre, la morphologie du support TiO2 est inchangée. La taille des grains est comprise entre 50 nm et 100 nm. La présence de plaquettes dans les catalyseurs calcinés et réduits à été observée.

Photographies en microscopie électronique à balayage du MoO3 non réduit (a) et réduit 12 h à 400 °C (b).

Photographie en microscopie électronique à balayage du support TiO2 calciné.

Photographies en microscopie électronique à balayage du MoO3/TiO2 calciné 4 h à 500 °C (a) et réduit 12 h à 400 °C (b).
Le diffractogramme de rayons X présenté sur la Figure 5a et correspondant au support de MoO3 non calciné, ne présente que des pics caractéristiques de MoO3 de structure orthorhombique (JCPDS 03-065-2421). Le traitement réducteur de 12 h à 400 °C détermine un changement structural (Figure 5b), qui se traduit par la disparition des raies de diffraction de MoO3 et par l’apparition d’un seul oxyde de MoO2 (JCPDS 00-032-0671). De plus, aucun sous-oxyde n’a été détecté. Les résultats sont différents de ceux rapportés dans la littérature, puisque certains auteurs citent l’apparition de sous oxydes Mo4O11 et Mo18O52 comme intermédiaires dans la réduction de MoO3 en MoO2 [54]. Dans les mêmes conditions, Belatel [55] a rapporté également la formation de molybdène métallique. Le diamètre des cristaux d’oxyde de molybdène a été calculé à l’aide de l’équation de Scherrer à partir des positions des pics principaux à 2𝜃 = 27,49° pour l’échantillon de MoO3 non calciné et à 2𝜃 = 26,09° pour MoO3 réduit sous hydrogène. Le calcul donne 26 nm pour le MoO3 non calciné et 23 nm pour le MoO3 réduit, indiquant que le traitement réducteur présente un effet positif sur la taille des cristaux, en favorisant la formation des petites particules dans le cas d’oxyde de molybdène.
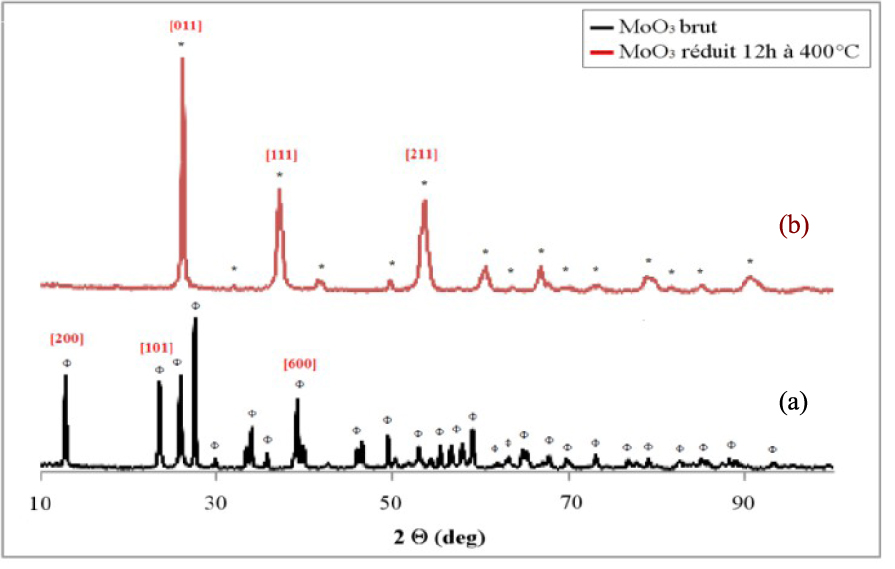
Diffractogrammes de rayons X du MoO3 non réduit (a) et réduit 12 h à 400 °C (b) ; (𝛷) MoO3, (*) MoO2.
Les diffractogrammes du catalyseur MoO3/TiO2 calciné 4 h à 500 °C et réduit 12 h à 400 °C sont présentés sur la Figure 6. Le catalyseur MoO3/TiO2 calciné à 500 °C pendant 4 h (Figure 6a), contient en plus des raies de diffraction caractéristiques du TiO2 (80% anatase et 20% rutile), des raies de diffraction à 2𝜃 : 12,91° et 23,40°, 30,01°, 33.51° et 46,12° correspondant au MoO3 orthorhombique (JCPDS 00-001-0706). Ce résultat montre la présence d’agrégats de MoO3 dans le catalyseur calciné, indiquant une faible dispersion de l’oxyde de molybdène sur le support de TiO2 et l’impossibilité de la formation d’une monophase catalytique. Le diamètre des cristaux d’anatase est de 20 nm, de rutile 25 nm et de MoO3 22 nm. Le traitement réducteur du catalyseur MoO3/TiO2 à 400 °C pour une durée de 12 h, provoque la disparition des raies de diffraction du MoO3 et l’apparition des raies de diffraction à 2𝜃 = 26,5° correspondant à la phase MoO2 qui coexistent avec les raies du TiO2 (Figure 6b). Dans ce cas, le diamètre des cristaux d’anatase est de 19 nm, de rutile 25 nm et de MoO2 45 nm. Aucune raie caractéristique d’un composé mixte entre le TiO2 et le MoO3 ou le MoO2 n’est observable.
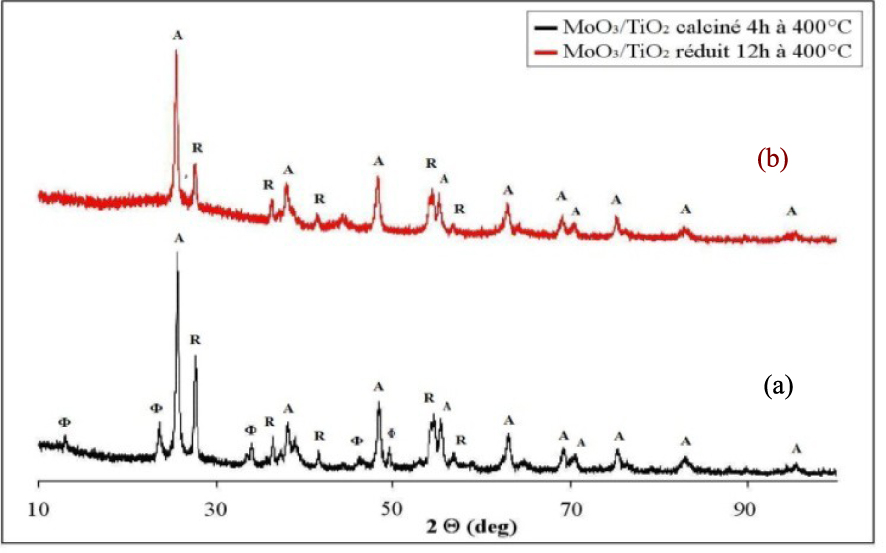
Diffractogrammes de rayons X du MoO3/TiO2 calciné (a) et réduit (b) ; (𝛷) MoO3, (*) MoO2, (A) Anatase, (R) Rutile.
Les valeurs de la surface spécifique BET correspondant aux MoO3 non réduit et réduit sont de 1,0 m2⋅g−1 et 6 m2⋅g−1. Ces résultats montrent une augmentation considérable, d’un facteur 6, de la surface spécifique du trioxyde de molybdène après une réduction de 12 h sous hydrogène à 400 °C contrairement à ce qui a été observé dans le cas du MoO2 où on a constaté une faible augmentation de la surface spécifique après le traitement réducteur. L’augmentation de la surface spécifique du MoO3, après la réduction, pourrait être attribuée à un effondrement de la structure cristalline, conduisant ainsi à la formation de pores et de canaux. Les résultats obtenus en sorption d’azote sont en concordance avec les résultats de microscopie électronique à balayage, qui montrent clairement la formation de pores ou de fissures au niveau des grains après le traitement réducteur.
La surface spécifique BET du catalyseur MoO3/TiO2 calciné est de 25 m2⋅g−1 mais elle augmente à 29 m2⋅g−1 après le traitement réducteur. Le volume poreux du catalyseur MoO3/TiO2 calciné est de 0,23 cm3⋅g−1 avant le traitement réducteur et 0,22 cm3⋅g−1 après. Le volume poreux du MoO3/TiO2 reste faible et ne varie pratiquement pas avec le traitement réducteur.
Les spectres de photoémission SPX de la raie 3d du molybdène du catalyseur MoO3/TiO2 calciné non réduit et réduit à 400 °C sont représentés respectivement sur les Figures 7 et 8. Pour le catalyseur MoO3/TiO2 calciné (Figure 7) nous avons observé la formation d’un doublet électronique, dont les niveaux d’énergie (232,8 ± 0,2 eV et 235,8 ± 0,2 eV), sont caractéristiques du MoO3. Ce résultat confirme que pour une température de calcination de 500 °C, le molybdène (Mo6+) existant à la surface de l’échantillon et provenant du sel précurseur, se trouve sous forme de MoO3. L’exposition de l’échantillon à l’hydrogène pendant 12 h à 400 °C (Figure 8) entraîne une réduction du MoO3 de surface en MoO2. Au cours de la réduction à 400 °C, la contribution des espèces Mo6+ diminue au profit des espèces Mo5+ et Mo4+.
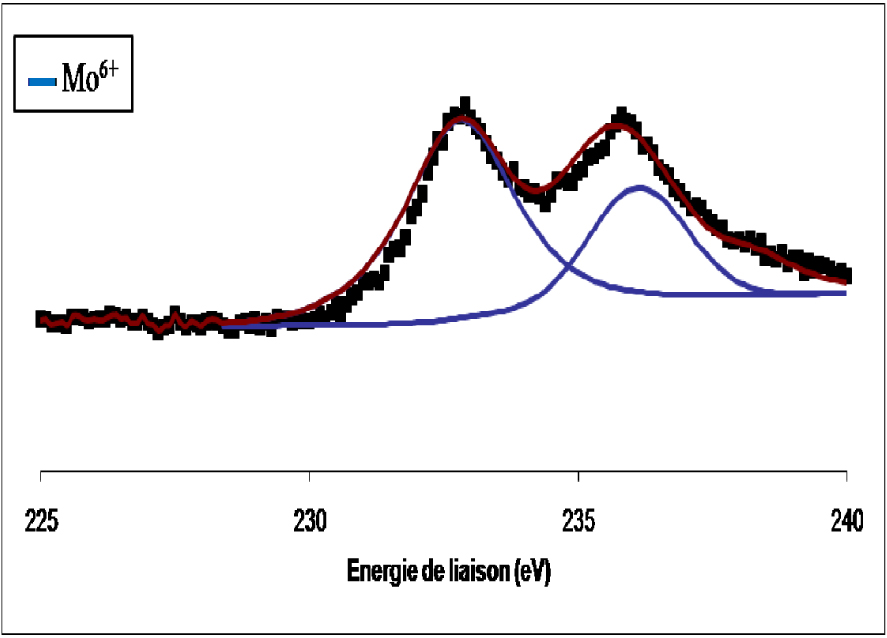
Spectre de photoémission de la raie Mo3d du MoO3/TiO2calciné.
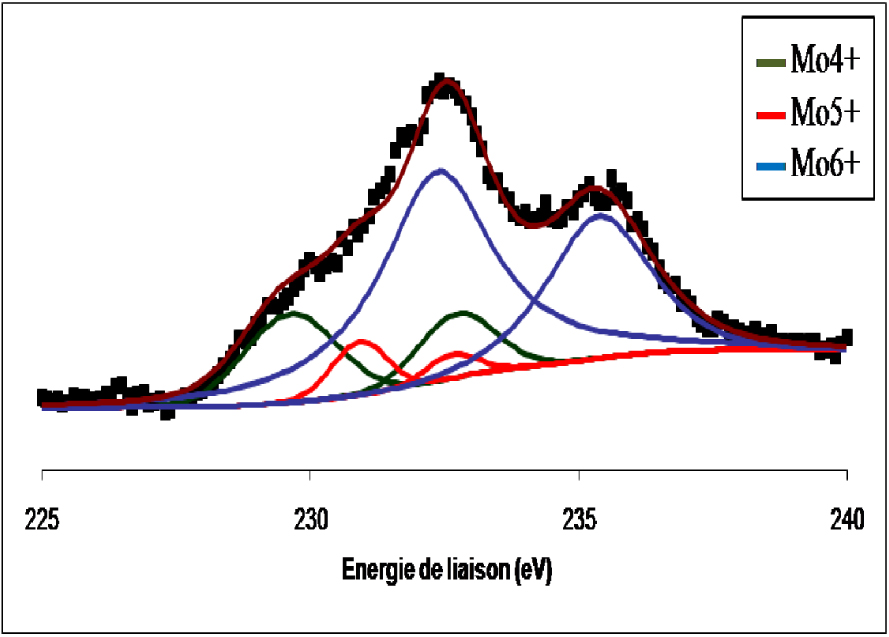
Spectre de photoémission de la raie Mo3d du MoO3/TiO2 réduit 12 h à 400 °C.
Les spectres de photoémission de la raie 1s de l’oxygène de l’échantillon du catalyseur MoO3/TiO2 calciné et réduit sont présentés sur les Figures 9 et 10.
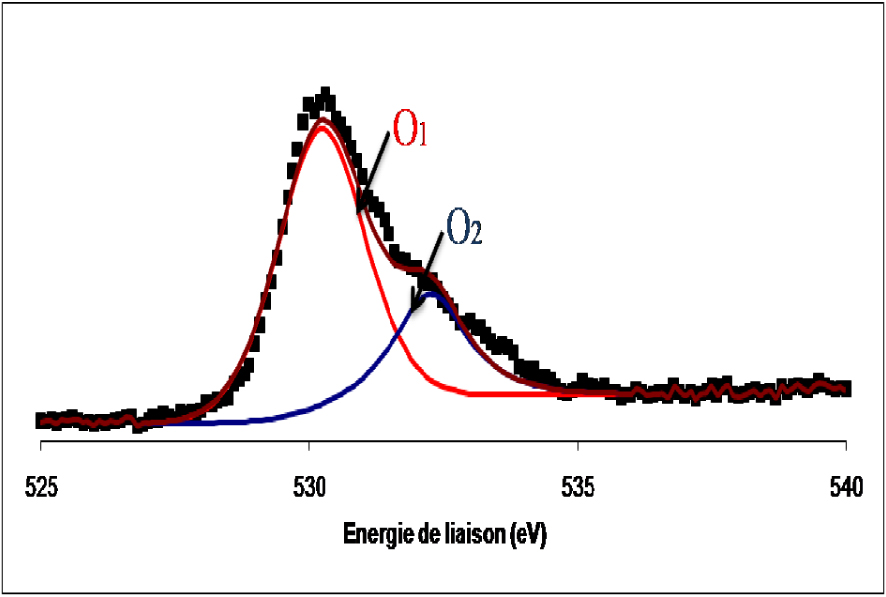
Spectre de photoémission de la raie 1s de l’oxygène dans le catalyseur de MoO3/TiO2 calciné.
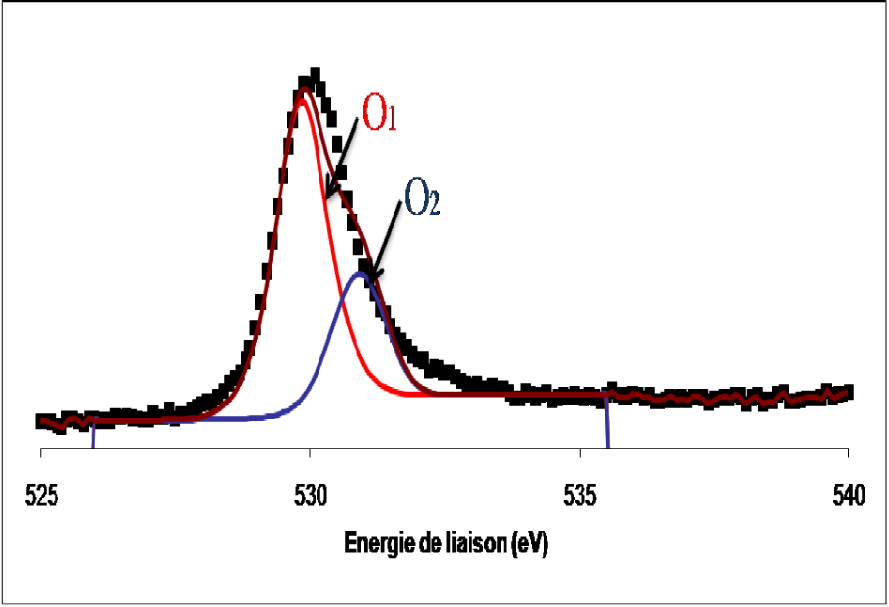
Spectre de photoémission de la raie 1s de l’oxygène dans le catalyseur de MoO3/TiO2 réduit 12 h à 400 °C.
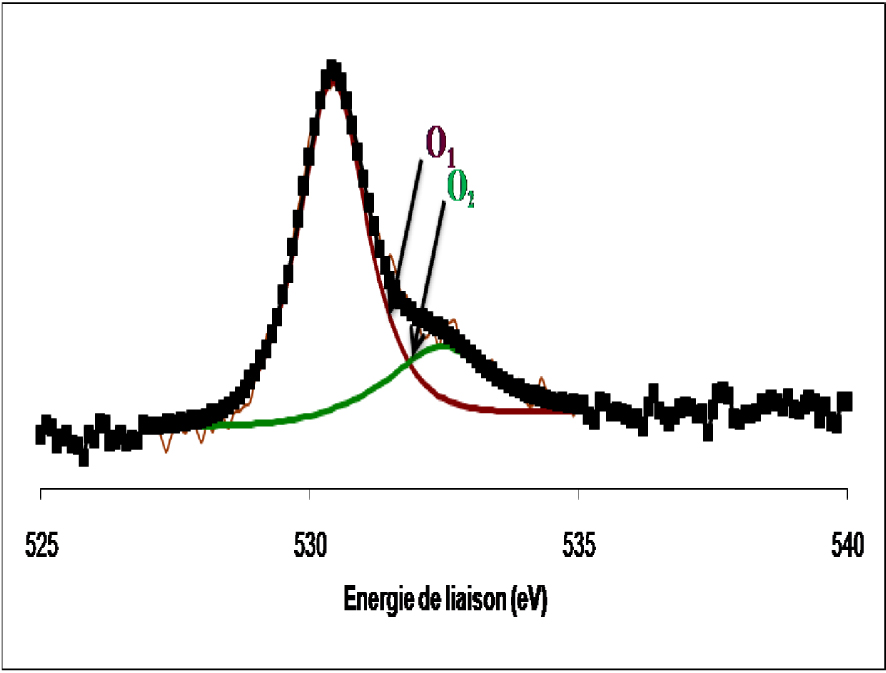
Les spectres de l’oxygène dans les deux cas se présentent sous forme d’une raie asymétrique constituée de deux composantes, la première située à 530,3 ± 0,2 eV attribuée à l’oxygène de réseau des oxydes de métaux de transition et une seconde détectée à 532,5 ± 0,2 eV, attribuée à l’oxygène adsorbé à la surface de l’oxyde sous forme de groupes OH.
L’analyse des spectres SPX du Ti (2p) de l’oxyde de molybdène supporté sur l’oxyde de titane calciné 4 h à 500 °C d’une part et celui réduit 12 h à 400 °C d’autre part sont présentés sur la Figure 11.
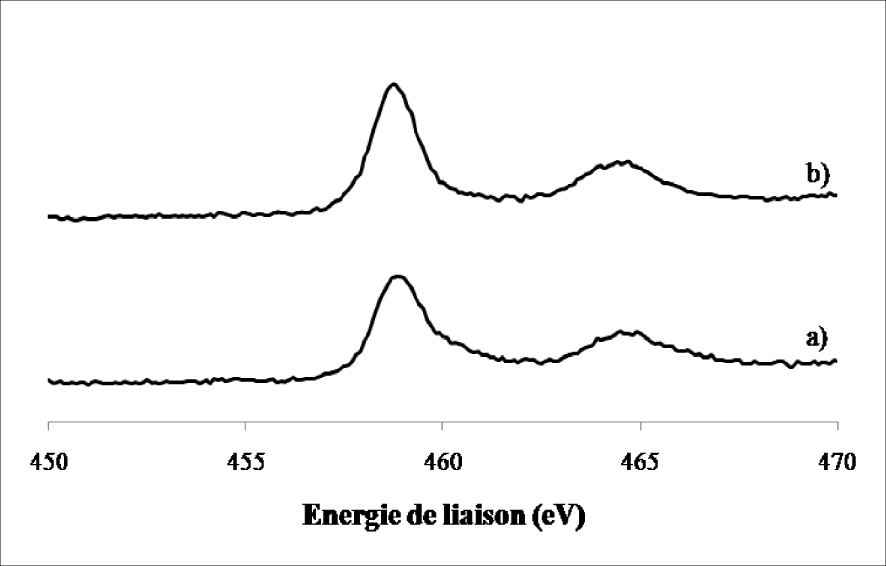
Spectres de photoémission X du Ti (2p) dans le MoO3/TiO2 calciné 4 h à 500 °C (a) et réduit 12 h à 400 °C (b).
Nous avons observé la présence de deux lignes spectrales à 459 ± 0,2 eV et 464,8 ± 0,2 eV caractéristiques du TiO2 aussi bien sur l’échantillon calciné que sur l’échantillon réduit. Aucun autre changement n’a été détecté au niveau du support (TiO2) après réduction, ce qui confirme les résultats obtenus par DRX concernant la stabilité de la phase anatase et rutile après le traitement réducteur.
3.1.2. Catalyseurs supportés WO3/TiO2
Les photographies en microscopie électronique à balayage correspondant respectivement au WO3 commercial non réduit et réduit sont présentées dans la Figure 12. Dans tous les cas, la morphologie consiste en des particules agglomérées de type plaquettes distinctes et assez irrégulières. La taille des agglomérats est comprise entre 100 nm et 700 nm pour WO3 brut, entre 100 nm et 500 nm pour le WO3 réduit. Après la réduction sous hydrogène, la taille des particules diminue et il apparaît clairement la formation de pores ou de fissures au niveau des grains dans le WO3 réduit. En outre, les échantillons apparaissent cristallisés sans traces d’oxydes amorphes.
Les photographies en microscopie électronique à balayage correspondant aux catalyseurs WO3/TiO2 calcinés et réduits sont présentées dans la Figure 13. La morphologie est caractéristique des cristaux d’oxyde de titane commercial (Figure 3) avec des cristaux de taille comprise entre 10 et 40 nm. Ainsi, l’ajout d’oxyde de tungstène sur le support TiO2 par imprégnation entraîne une perte d’identité morphologique du WO3 mais ne semble pas affecter la morphologie du support TiO2.

Photographies en microscopie électronique à balayage du WO3 non réduit (a) et réduit (b).

Photographies en microscopie électronique à balayage du WO3/TiO2 calciné (a) et réduit (b).
Le diffractogramme de rayons X présenté sur la Figure 14a et correspondant au WO3 non réduit, ne présente que des pics caractéristiques de WO3 de structure monoclinique (JCPDS 00-005-0363). Après un traitement réducteur de 12 h à 500 °C, nous avons constaté un changement structural, qui se traduit par la disparition des raies de diffraction de WO3 et par l’apparition de la phase métallique de type W3O. Le diffractogramme correspondant à l’échantillon de WO3 réduit est présenté sur la Figure 14b.
Le diamètre des cristaux du W3O est de 9 nm et a été calculé à l’aide de l’équation de Scherrer à partir de la position du pic principal à 2𝜃 = 39,97° correspondant à une distance interplanaire de 2,25 nm. Le diamètre des cristaux, dans le WO3 non réduit est de 27 nm et a été calculé d’après le pic correspondant à l’indice de Miller (200). Le traitement réducteur favorise donc la formation des petites particules dans le cas d’oxyde de tungstène.
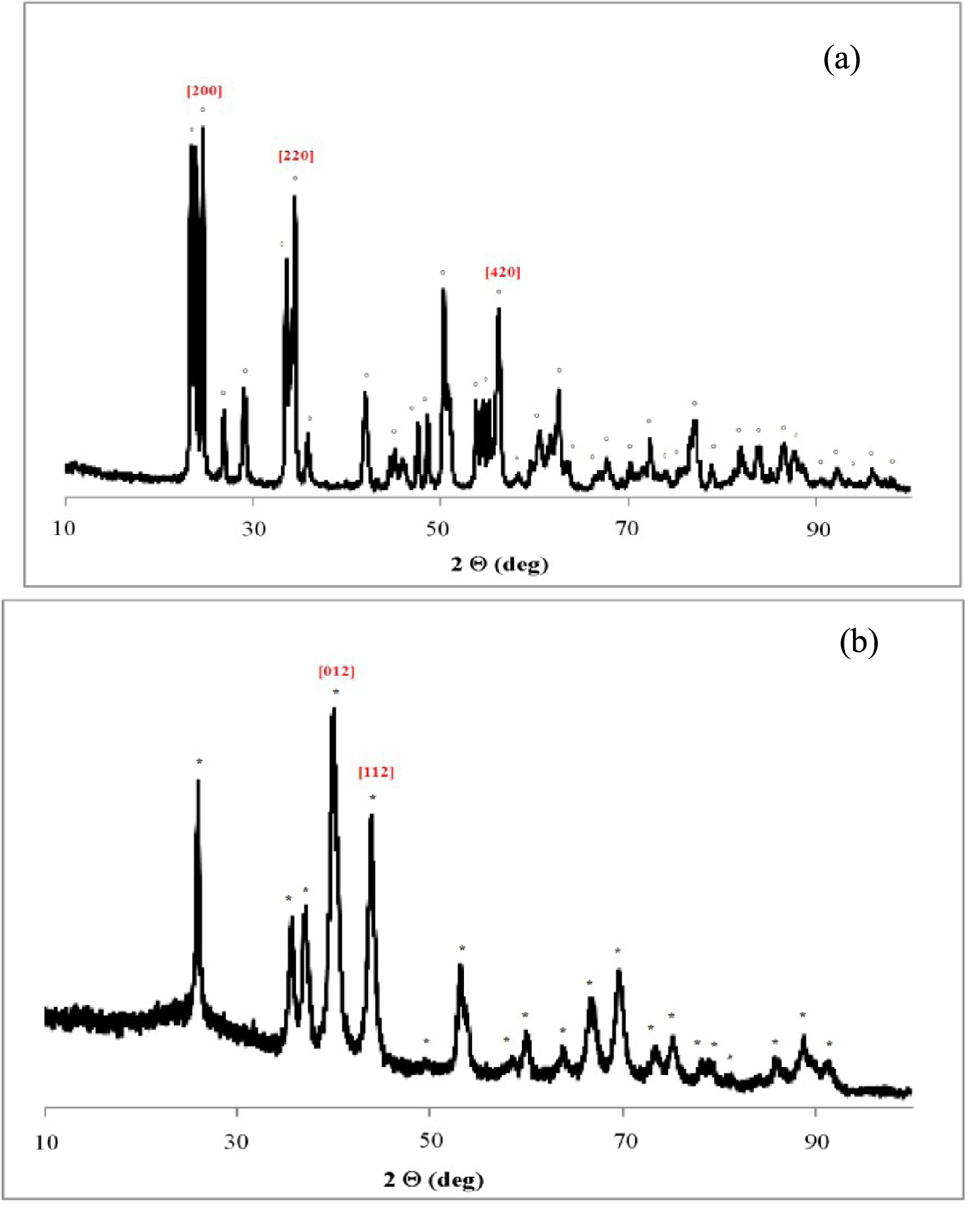
Diffractogrammes de rayons X du WO3 non réduit (a) et du WO3 réduit 12 h à 500 °C (b) ; (°) WO3 (*) W3O.
Les diffractogrammes du catalyseur WO3/TiO2 calciné et réduit 12 h à 500 °C sont présentés sur la Figure 15. Le catalyseur WO3/TiO2 calciné à 500 °C pendant 4 h (Figure 15a), contient en plus des raies de diffraction caractéristiques du TiO2 (80% anatase et 20% rutile), des raies de diffraction correspondant au WO3 monoclinique (JCPDS 00-043-1035). Ce résultat montre la présence des agrégats de WO3 dans le catalyseur calciné, indiquant une faible dispersion de l’oxyde de tungstène sur le support de TiO2 et l’impossibilité de la formation d’une monophase catalytique dans ces conditions. Le diamètre des cristaux d’anatase est de 20 nm, de rutile 26 nm et de WO3 de 40 nm.
Le traitement réducteur du catalyseur WO3/TiO2 à 500 °C pendant 12 h (Figure 15b), conduit à la disparition des raies de diffraction du WO3 et à l’apparition des raies de diffraction à 2𝜃 = 37°, 53°, 60° correspondant à la phase métallique W3O qui coexiste avec les raies de diffraction du TiO2. Dans ce cas, le diamètre des cristaux d’anatase est de 19 nm, de rutile 23 nm et de WO2 de 11 nm.
Nous mentionnons qu’aucune raie correspondante à la formation de composés mixtes entre l’oxyde de tungstène et TiO2 n’a été observée.
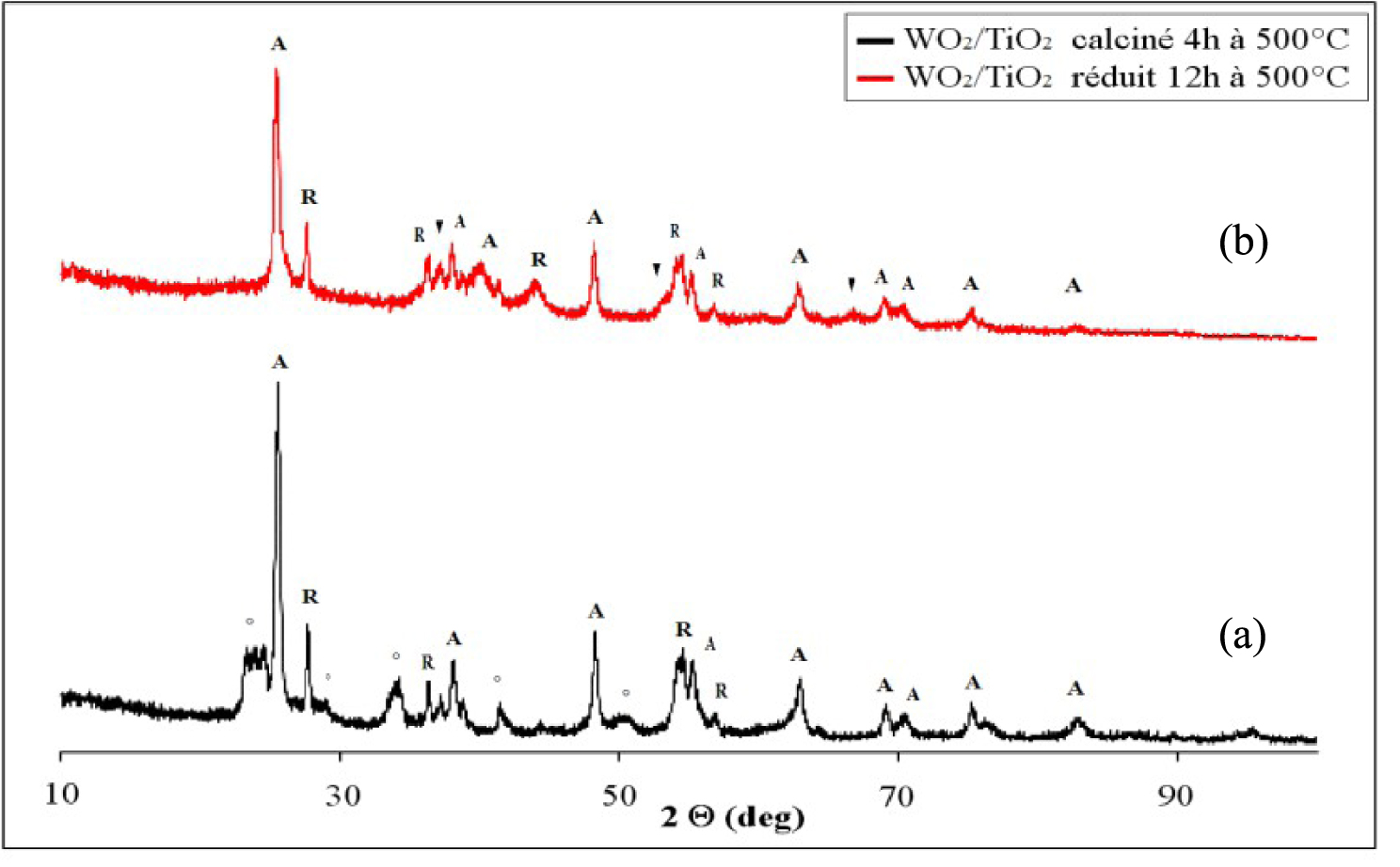
Diffractogrammes de rayons X du catalyseur de WO3/TiO2 calciné (a) et réduit 12 h à 500 °C ; (°) WO3, (▾) WO2, (A) TiO2 anatase, (R), TiO2 rutile.
Les valeurs de la surface spécifique BET correspondant aux supports de TiO2 et de WO3 sont 44 m2⋅g−1 et 1,1 m2⋅g−1. Après une réduction de 12 h sous hydrogène à 500 °C, la surface spécifique BET du TiO2 est quasiment identique, autour de 45 m2⋅g−1, mais la surface spécifique BET du WO3 augmente à 24 m2⋅g−1. La surface spécifique BET du catalyseur WO3/TiO2 calciné est de 31 m2⋅g−1, mais après le traitement réducteur elle augmente à 42 m2⋅g−1. Le volume poreux du catalyseur WO3/TiO2 calciné est de 0,13 cm3⋅g−1 et après le traitement réducteur il atteint 0,17 cm3⋅g−1. Les résultats obtenus en sorption d’azote sont en concordance avec les résultats de microscopie électronique à balayage, qui montrent clairement la formation de pores ou de fissures au niveau des grains après le traitement réducteur.
Les spectres de photoémission des raies W4f du WO3 non réduit et du WO3 réduit 12 h à 500 °C ex situ sont représentés sur les Figures 16 et 17.
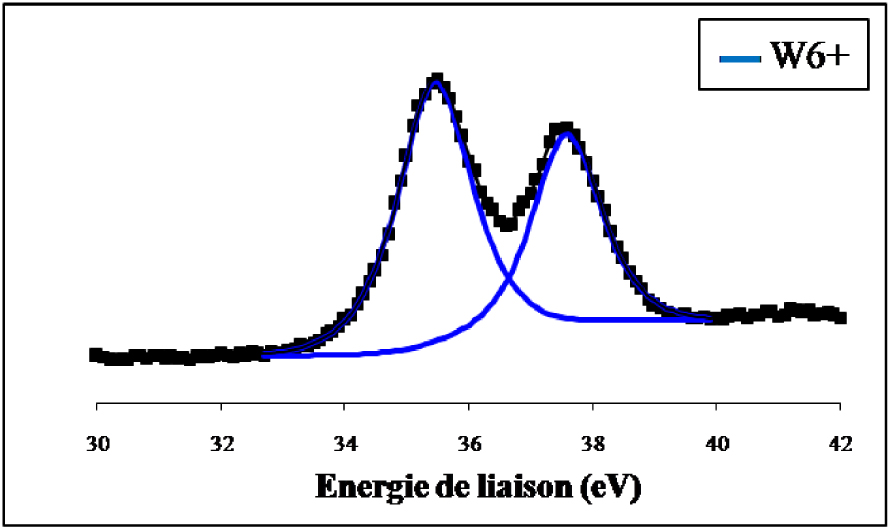
Spectre de photoémission des raies W4f du WO3 non réduit.
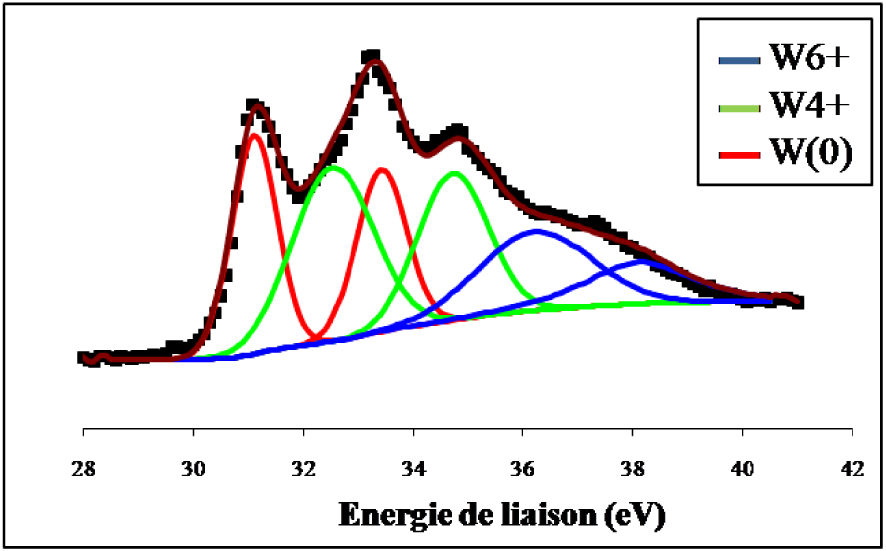
Spectre de photoémission des raies W4f du WO3 réduit 12 h à 500 °C.
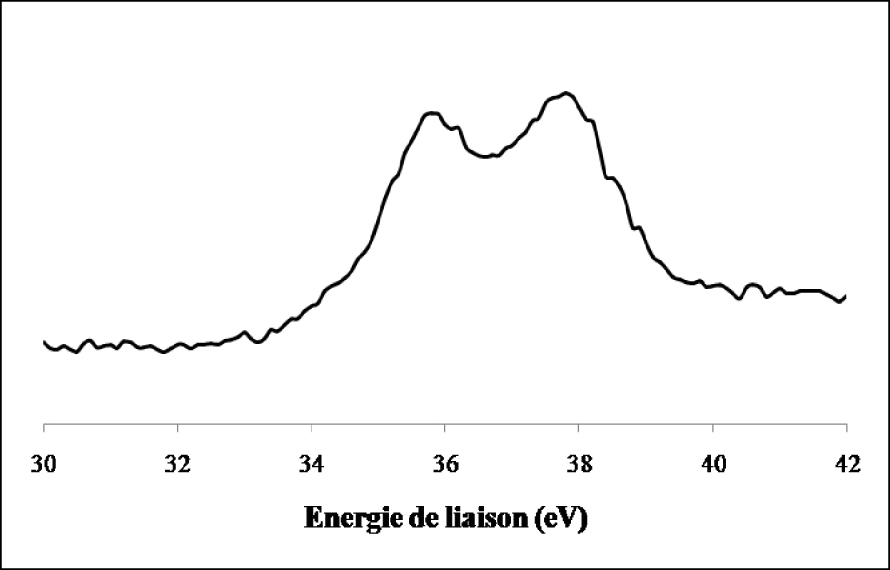
Nous constatons que le signal du tungstène de l’échantillon non réduit se compose de deux lignes spectrales bien définies à 37,5 ± 0,2 eV et à 35,4 ± 0,2 eV qui sont les niveaux d’énergie caractéristique de WO3 dont le tungstène présente un degré d’oxydation +6. La réduction ex situ de l’échantillon par l’hydrogène pendant 12 h à 500 °C (Figure 17) indique en plus du doublet correspondant à l’espèce W6+ et qui représente 24,2% du signal, la présence de raies supplémentaires attribuées à W4+ pour 43,3% et W(0) pour 32,5%. Les spectres de photoémission des raies W4f du WO3/TiO2 calciné et du WO3/TiO2 calciné et réduit 12 h à 500 °C ex situ sont représentés sur les Figures 18 et 19. Sur l’échantillon du catalyseur WO3/TiO2 calciné (Figure 18) nous avons observé la formation d’un doublet électronique, aux niveaux d’énergie 35,8 ± 0,2 eV et 37,7 ± 0,2 eV, caractéristiques du tungstène au degré d’oxydation +6, correspondant ainsi à la phase WO3. Ce résultat confirme que pour une température de calcination de 500 °C, le tungstène provenant du sel précurseur, existe à la surface de TiO2 sous forme de grains de WO3.
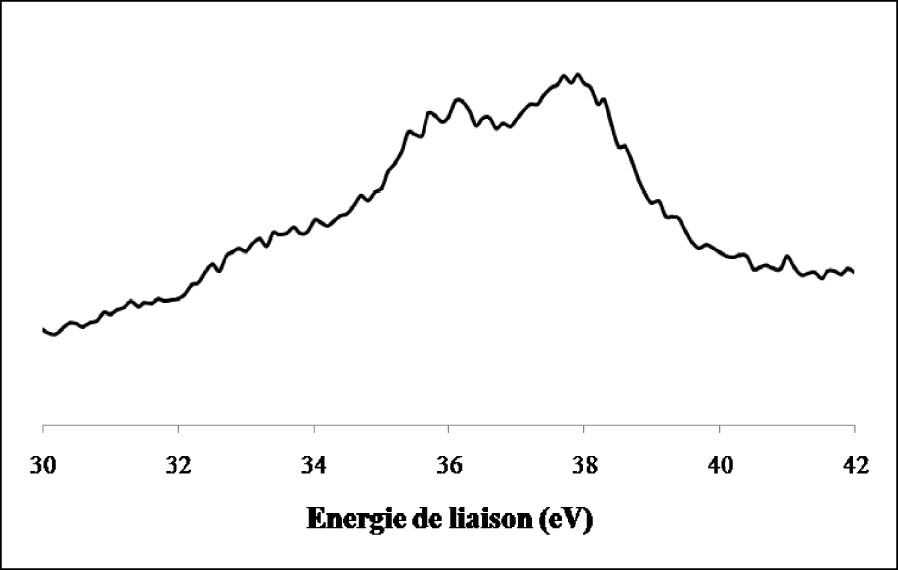
La réduction ex situ de l’échantillon de WO3/TiO2 calciné par l’hydrogène pendant 12 h à 500 °C (Figure 19) entraîne en plus du doublet correspondant à l’espèce W6+ l’apparition de raies supplémentaires attribuées à W4+ et W(0).
Les résultats corroborent ceux de diffraction des rayons X qui ont montré la réduction du WO3 en WO2 et W3O (état métallique).
Les spectres de photoémission de la raie 1s de l’oxygène de l’échantillon du WO3 calciné et réduit sont présentés sur les Figures 20 et 21.
Spectre de photoémission des raies W4f du WO3/TiO2 calciné.
Spectre de photoémission des raies W4f du WO3/TiO2 réduit.
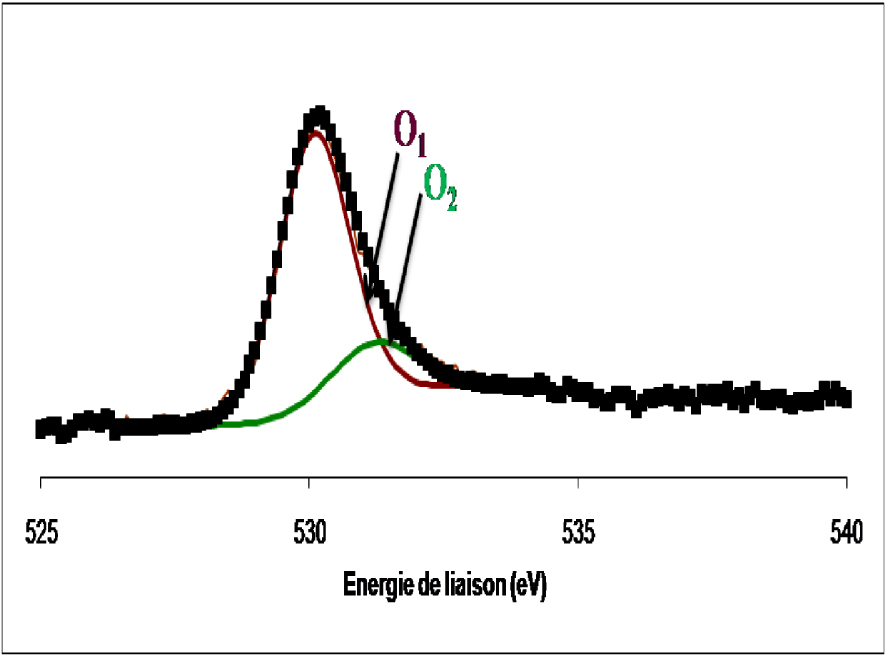
Spectre de photoémission de la raie 1s de l’oxygène dans le WO3 non réduit.
Spectre de photoémission de la raie 1s de l’oxygène dans le WO3 réduit 12 h à 500 °C.
Dans l’échantillon WO3 non réduit, l’oxygène se présente sous forme de deux composantes. La première composante, à 530,1 ± 0,2 eV, est rapportée comme étant l’oxygène de réseau des oxydes de métaux de transition. La deuxième composante, à 531,2 ± 0,2 eV, est attribuée à l’oxygène adsorbé à la surface de l’oxyde sous forme de groupes OH. Pour le WO3 réduit, l’oxygène se présente sous forme d’une raie asymétrique formée de deux composantes. Le spectre est identique à celui de l’échantillon non réduit. Les deux contributions sont relatives à l’oxygène du réseau et à l’oxygène des groupes OH formés au cours de la réduction. Dans le Tableau 4 sont mentionnées les quantités des différents types d’oxygène à la surface du WO3 dans les deux cas, non réduit et réduit. L’analyse des spectres SPX du Ti (2p), de l’oxyde de tungstène supporté sur l’oxyde de titane calciné 4 h à 500 °C et celui réduit 12 h à 400 °C, sont présentés sur la Figure 22.
Spectres de photoémission X du Ti (2p) dans le WO3/TiO2 calciné 4 h à 500 °C (a) et réduit 12 h à 400 °C (b).
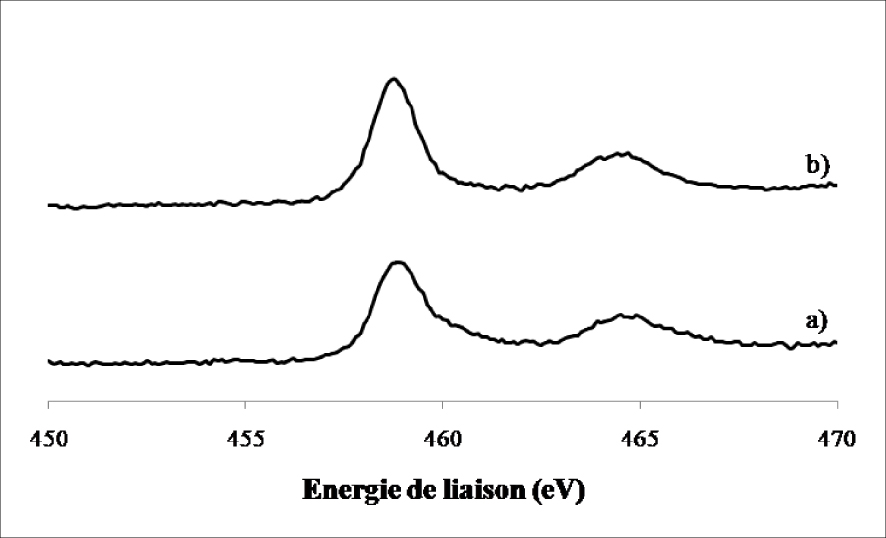
Sur l’échantillon calciné, nous avons observé la présence de deux lignes spectrales à 459 ± 0,2 eV et 464,7 ± 0,2 eV caractéristiques du TiO2. L’échantillon réduit montre aussi l’apparition de ces deux lignes à 459 ± 0,2 eV et 464,8 ± 0,2 eV. Aucun autre changement n’a été détecté au niveau du support (TiO2) après réduction ce qui confirme les résultats obtenus par DRX concernant la stabilité de la phase anatase et rutile après le traitement réducteur.
3.2. Etude de la réactivite catalytique des catalyseurs à base d’oxydes supportés MoO3, MoO3/TiO2, WO3, WO3/TiO2
3.2.1. Etude de la réactivité catalytique des catalyseurs à base d’oxydes supportés MoO3, MoO3/TiO2
La distribution des produits de réaction formés dans la réaction du MCP en présence de MoO3 et MoO3/TiO2, en fonction de la température de réaction, est répertoriée dans les Tableaux 5 et 6.
Contributions des différents types d’oxygène à la surface du WO3 non réduit et réduit
| Echantillon | % O1 | % O2 |
|---|---|---|
| WO3 non réduit | 82,93 | 17,07 |
| WO3 réduit 12 h à 500 °C | 76,57 | 23,42 |
O1 : oxygène du réseau, O2 : oxygène formé lors de la réduction.
Distribution des produits de réaction lors de la conversion du MCP en présence du MoO3 réduit 12 h à 400 °C : influence de la température de réaction
| Température de réaction (°C) | C1 | C2 | C3 | iC4 | nC4 | iC5 | nC5 | 2MP | 3MP | nH | C6 | Bz |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 340 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1,3 | 3,3 |
| 400 | 5,5 | 0,7 | 0,6 | 0,8 | 1 | 0,7 | 0,2 | 2,7 | 0,7 | 0,7 | 0,2 | 17,3 |
| 58 | 7 | 6 | 8,5 | 10,5 | 7 | 2 | - | - | - | - | - | |
| Distribution statistique | 26 | 16 | 16 | 4 | 12 | 8 | 18 | - | - | - | - | - |
En gras : Produits de craquage normalisés à 100.En italique : Distribution statistique des produits de craquage.
Distribution des produits de réaction lors de la conversion du MCP en présence du MoO3/TiO2 réduit 12 h à 400 °C : influence de la température de réaction
| Température de réaction (°C) | C1 | C2 | C3 | iC4 | nC4 | iC5 | nC5 | 2MP | 3MP | nH | C6 | Bz |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 180 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| 220 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1,4 |
| 260 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 2,5 |
| 300 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0,7 | 0,3 | 0,04 | 1 | 3,7 |
| 340 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1,9 | 1 | 1,8 | 6,5 | 6,6 |
| 400 | 4,7 | 1,4 | 2,2 | 0 | 0 | 0 | 0 | 2,3 | 0,7 | 1,6 | 3,6 | 28,1 |
L’analyse des produits de réaction obtenus pour tous les catalyseurs a mis en évidence une large gamme d’hydrocarbures : C1–C5, 2MP, 3MP, nH, C6, Bz. Ceci montre que le processus de conversion du méthylcyclopentane est très complexe, la réaction principale d’ouverture de cycle du MCP (2MP, 3MP, nH) étant accompagnée de la réaction d’élargissement de cycle (C6, Bz) et des réactions secondaires de craquage (C1–C5). Nous notons que des produits insaturés, les oléfines, n’ont pas été détectés dans nos conditions de travail. Le Tableau 5 fournit également la distribution statistique des produits de craquage à 440 °C sur le catalyseur MoO3. L’activité catalytique de ces catalyseurs est caractérisée par les pourcentages de conversion totale et de sélectivité correspondant à chaque réaction.
Dans le Tableau 7 sont répertoriées les valeurs de la conversion totale (𝛼%), les sélectivités dans les réactions de craquage (SC%), d’ouverture de cycle (SO%), d’élargissement de cycle (SE%), les vitesses spécifiques globales VG (μmol⋅g−1⋅s−1 et 106 s−1) et les vitesses d’ouverture VO (μmol⋅g−1⋅s−1) de cycle en fonction de la température de réaction.
3.2.1.1. Etude de l’évolution de l’activité catalytique en fonction de la température de réaction
L’évolution de l’activité catalytique exprimée par la conversion totale (𝛼) du MCP sur les catalyseurs MoO3 et MoO3/TiO2 réduits 12 h à 400 °C, en fonction de la température de réaction (180 °C–500 °C), est présentée dans le Tableau 7.
Les données de ce tableau montrent que MoO3 ne présente pas d’activité catalytique en dessous de 340 °C. A 340 °C, il convertit 4,6% de MCP. Au fur et à mesure que la température augmente jusqu’à 400 °C, le MoO3 développe une activité catalytique importante, car il convertit 31,0% de MCP.
Nous notons que la vitesse globale de réaction varie de 0,3 (μmol⋅g−1⋅s−1) à 2,2 (μmol⋅g−1⋅s−1) quand la température de réaction augmente de 340 °C à 400 °C. Ce résultat, concernant la vitesse globale de réaction, est identique avec celui obtenu sur le MoO2 réduit et corrobore les résultats DRX qui ont montré l’apparition de la phase MoO2 dans le MoO3 réduit. L’énergie apparente d’activation entre 340 et 500 °C est de 25,3 ± 2,5 kcal⋅mol−1. Le catalyseur MoO3/TiO2 se distingue par son activité catalytique dès 220 °C: il convertit 1,4% de MCP. Pour les deux catalyseurs, MoO3 et MoO3/TiO2, l’accroissement de la température de réaction conduit, logiquement, à l’augmentation de la conversion totale du MCP.
La réactivité des catalyseurs dans la réaction de conversion du MCP décroît dans l’ordre suivant :
On constate que le catalyseur le plus actif est MoO3/TiO2 quelle que soit la température de réaction.
Nous notons que la vitesse globale de réaction (
Performances des catalyseurs MoO3 réduits 12 h à 400 °C et MoO3/TiO2 réduit 12 h à 400 °C (les tests catalytiques ont été réalisés dans tous les cas entre 180 °C et 500 °C, mais dans les tableaux sont répertoriées seulement les valeurs de la température pour laquelle se manifeste l’activité catalytique)
| Température de réaction (°C) | 𝛼 (%) | SC (%) | SO (%) | SE (%) | 2MP / 3MP | 3MP / nH | VG (μmol⋅g−1⋅s−1) | VG (106 s−1) | VO (μmol⋅g−1⋅s−1) |
|---|---|---|---|---|---|---|---|---|---|
| MoO3 réduit | |||||||||
| 340 | 4,6 | 0 | 0 | 100 | 0 | 0 | 0,3 | 25,2 | 0 |
| 400 | 31 | 27,7 | 13,5 | 56,4 | 3,7 | 1 | 2,2 | 184,8 | 0,9 |
| MoO3/TiO2 | |||||||||
| 220 | 1,4 | 0 | 0 | 100 | 0 | 0 | 0,1 | 8,4 | 0 |
| 260 | 2,5 | 0 | 0 | 100 | 0 | 0 | 0,2 | 16,8 | 0 |
| 300 | 5,7 | 0 | 18,5 | 81,4 | 3,7 | 2,8 | 0,4 | 33,6 | 0,07 |
| 340 | 17,8 | 0 | 26,3 | 73,7 | 1,9 | 0,6 | 1,3 | 109,2 | 0,34 |
| 400 | 44,5 | 18,5 | 10,2 | 71,3 | 3,1 | 0,5 | 3,2 | 268,8 | 0,31 |
3.2.1.2. Etude de l’évolution des sélectivités en fonction de la température de réaction
Les valeurs de la sélectivité pour les réactions secondaires de craquage (SC) (avec la formation des produits C1–C5) sur les catalyseurs MoO3 et MoO3/TiO2, en fonction de la température de réaction, sont répertoriées dans le Tableau 7. On constate que la surface de ces catalyseurs est inactive vis-à-vis de la réaction de craquage en dessous de 400 °C. A 400 °C, ils présentent une sélectivité pour la réaction de craquage, respectivement 27,7% (MoO3) et 18,5% (MoO3/TiO2).
L’analyse de la distribution des produits de craquage montre que le MoO3 favorise la formation de C1–C5 à 400 °C (SC = 27,7%). Il est important de noter que le pourcentage en produit C1 est le plus important, mettant en évidence une fonction métallique du MoO3 réduit. Ce comportement est dû probablement à des nombreux processus répétitifs, favorisant ainsi la réaction de déméthylation. Sur le catalyseur MoO3/TiO2, le produit C1 est prédominant à 400 °C (C1 > C3 > C2).
Ces résultats montrent que la réaction de déméthylation est favorisée sur les deux catalyseurs, mais la dispersion d’oxyde de molybdène sur l’oxyde de titane conduit à la diminution de l’importance de la réaction de craquage.
Les valeurs de la sélectivité vers la réaction principale d’ouverture de cycle du MCP, avec la formation de 2MP, 3MP et nH, en présence des catalyseurs MoO3 et MoO3/TiO2, en fonction de la température de réaction, sont répertoriées dans le Tableau 7.
Distribution des produits de réaction lors de la conversion du MCP en présence du WO3 réduit 12 h à 500 °C : influence de la température de réaction
| Température de réaction (°C) | C1 | C2 | C3 | iC4 | nC4 | iC5 | nC5 | C6 | Bz |
|---|---|---|---|---|---|---|---|---|---|
| 400 | 4,6 | 4,9 | 4,6 | 1,7 | 2,2 | 0,1 | 0,6 | 1,5 | 0 |
| 440 | 25,8 | 11,1 | 9 | 1,8 | 3,6 | 1,4 | 1,1 | 1,1 | 1,1 |
| 470 | 53 | 14,4 | 8,4 | 1,3 | 2,4 | 0,7 | 0,7 | 0 | 2,7 |
| 500 | 82,4 | 12,1 | 3,2 | 0,04 | 0,2 | 0 | 0 | 0 | 1,8 |
Les données de ce tableau montrent que le catalyseur le plus sélectif dans la réaction d’ouverture de cycle du MCP est le MoO3/TiO2. Il présente une sélectivité en ouverture de cycle du MCP, SO = 18,5% pour une conversion (𝛼) de 5,7% à 300 °C. Les valeurs du rapport 2MP/3MP de 3,7 et du rapport 3MP/nH de 2,8, signifient une affinité du catalyseur MoO3/TiO2 pour l’ouverture de cycle selon un mécanisme de type sélectif. L’accroissement de la température de réaction jusqu’à 400 °C conduit à la diminution de la sélectivité vers l’ouverture de cycle (
Une sélectivité en ouverture de cycle du MCP de 13,5%, pour une conversion du MCP de 31%, a été observée sur le catalyseur MoO3 seulement à 400 °C. Les valeurs du rapport 2MP/3MP de 3,7 et du rapport 3MP∕nH de 1, signifient une affinité du catalyseur MoO3 pour l’ouverture de cycle selon un mécanisme de type non sélectif.
Nous notons que la vitesse d’ouverture de cycle (VO) (Tableau 7) sur le catalyseur MoO3 est de 0,9 à 400 °C et varie sur le MoO3/TiO2 entre 0,07 et 0,31 quand la température augmente de 300 à 400 °C.
Le Tableau 7 présente les valeurs de la sélectivité pour la réaction d’élargissement (SE) de cycle (avec la formation du benzène et du cyclohexane), sur les catalyseurs MoO3 et MoO3/TiO2, en fonction de la température de réaction.
Les données de ce tableau montrent que le MoO3 présente une sélectivité en élargissement de cycle de 100%, pour une conversion (𝛼) de 4,6% à 340 °C. Les produits d’élargissement de cycle formés sont le cyclohexane et le benzène. L’augmentation de la température jusqu’à 400 °C conduit à la diminution de la sélectivité en élargissement de cycle à SE = 56,4%. La distribution des produits d’élargissement montre que le produit majoritaire est le benzène et le cyclohexane est formé seulement en traces.
Le catalyseur MoO3/TiO2 présente une sélectivité en élargissement de cycle de 100%, à 220 °C et 260 °C pour conversion (𝛼) de 1,4% et 2,5 respectivement. Le seul produit formé dans la réaction d’élargissement de cycle est le produit de déshydrogénation : le benzène. L’augmentation de la température de réaction jusqu’à 400 °C, conduit à une diminution de la sélectivité en élargissement de cycle à 71,3% (𝛼 = 44,5). Dans ce cas, les produits d’élargissement de cycle formés sont le cyclohexane et majoritairement le benzène.
3.2.2. Etude de la réactivité catalytique des catalyseurs à base d’oxydes supportés WO3, WO3/TiO2
La distribution des produits formés dans la réaction du MCP en présence de WO3 et WO3/TiO2, en fonction de la température de réaction, est répertoriée dans les Tableaux 8 et 9. Elle met en évidence une large gamme d’hydrocarbures : C1–C5 et C6, Bz. Ceci montre que le processus de conversion du méthylcyclopentane est très complexe, la réaction principale d’ouverture de cycle du MCP (2MP, 3MP, nH) n’a pas lieu sur ces catalyseurs, seulement la réaction d’élargissement de cycle (C6, Bz) et des réactions secondaires de craquage (C1–C5). Nous notons que des produits insaturés, les oléfines, n’ont pas été détectés dans nos conditions de travail.
Distribution des produits de réaction lors de la conversion du MCP en présence du WO3/TiO2 réduit 12 h à 500 °C : influence de la température de réaction
| Température de réaction (°C) | C1 | C2 | C3 | iC4 | nC4 | iC5 | nC5 | C6 | Bz |
|---|---|---|---|---|---|---|---|---|---|
| 340 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1,9 |
| 400 | 2,3 | 2,8 | 1,1 | 1,2 | 0,4 | 0,5 | 0,9 | 0 | 2 |
| 440 | 11,5 | 4,9 | 8,5 | 0,4 | 1,8 | 0 | 0 | 0 | 4,3 |
| 470 | 34,1 | 10,1 | 5,3 | 0,2 | 1,3 | 0 | 0 | 0 | 4,3 |
| 500 | 78,5 | 10,8 | 2,5 | 0,1 | 0,2 | 0 | 0 | 0 | 3 |
L’activité catalytique de ces catalyseurs est caractérisée par les pourcentages de conversion totale et de sélectivité correspondant à chaque réaction. Les notations utilisées sont similaires à celles utilisées pour les catalyseurs supportés sur l’alumine.
Dans le Tableau 10 sont répertoriées les valeurs de la conversion totale (𝛼%), les sélectivités dans les réactions de craquage (SC%), d’élargissement de cycle (SE%), les vitesses spécifiques globales VG (μmol⋅g−1⋅s−1 et 106 s−1) et les vitesses d’ouverture VO (μmol⋅g−1⋅s−1) de cycle en fonction de la température de réaction.
3.2.2.1. Etude de l’évolution de l’activité catalytique en fonction de la température de réaction
L’évolution de l’activité catalytique exprimée par la conversion totale (𝛼) du MCP sur les catalyseurs WO3 et WO3/TiO2 réduits 12 h à 500 °C, en fonction de la température de réaction (180 °C–500 °C), est présentée dans le Tableau 10. Les données de ce tableau montrent que le WO3 est actif catalytiquement dans une gamme de température comprise entre 400 et 500 °C. A 400 °C il convertit 20% de MCP. Ce pourcentage augmente de façon importante avec la température, jusque 99,6% à 500 °C.
Nous notons que la vitesse globale de réaction varie de 1,6 à 7,6 quand la température de réaction augmente de 400 °C à 500 °C. Il est important de mentionner que les vitesses de réaction entre 440 et 470 °C sont similaires aux vitesses de réaction obtenues sur WO2. L’énergie apparente d’activation calculée entre 400 et 440 °C est de 24,3 ± 2,4 kcal⋅mol−1.
Le catalyseur WO3/TiO2 se distingue par son activité catalytique dès 340 °C : il convertit 1,9% de MCP. Ce pourcentage augmente de façon importante avec la température, jusque 95,0% à 500 °C. La réactivité des catalyseurs dans la réaction de conversion du MCP décroît dans l’ordre suivant :
On constate que le catalyseur le plus actif est WO3/TiO2 à basse température et le WO3 à haute température.
Nous notons que la vitesse globale de réaction (
Performances des catalyseurs WO3 réduits 12 h à 500 °C et WO3/TiO2 réduit 12 h à 500 °C (les tests catalytiques ont été réalisés dans tous les cas entre 180 °C et 500 °C, mais dans les tableaux sont répertoriées seulement les valeurs de la température pour laquelle se manifeste l’activité catalytique)
| Température de réaction (°C) | 𝛼 (%) | SC (%) | SE (%) | VG (μmol⋅g−1⋅s−1) | VG (106 s−1) |
|---|---|---|---|---|---|
| WO3 réduit | |||||
| 400 | 20,3 | 92,4 | 7,6 | 1,6 | 134,4 |
| 440 | 56 | 96,2 | 3,8 | 4,3 | 361,2 |
| 470 | 83,1 | 96,8 | 3,2 | 6,4 | 537,6 |
| 500 | 99,6 | 98,2 | 1,8 | 7,6 | 638,4 |
| WO3/TiO2 | |||||
| 340 | 1,9 | 0 | 100 | 0,1 | 8,4 |
| 400 | 11,3 | 82 | 18 | 0,8 | 67,2 |
| 440 | 31,5 | 86,3 | 13,7 | 2,3 | 193,2 |
| 470 | 55,3 | 92,3 | 7,7 | 4,1 | 344,4 |
| 500 | 95 | 96,8 | 3,2 | 7 | 588 |
3.2.2.2. Etude de l’évolution de la sélectivité dans la réaction de craquage (SC) en fonction de la température de réaction
Les valeurs de la sélectivité pour les réactions secondaires de craquage (SC) (avec la formation des produits C1–C5) sur les catalyseurs WO3 et WO3/TiO2, en fonction de la température de réaction, sont répertoriées dans le Tableau 10. On constate que la surface de ces catalyseurs est inactive vis-à-vis de la réaction de craquage en dessous de 400 °C pour les deux catalyseurs. A 400 °C, ils présentent une sélectivité pour la réaction de craquage, respectivement 92,4% (WO3) et 82% (WO3/TiO2). L’analyse de la distribution des produits de craquage (Tableau 10) montre que le WO3 favorise la formation de C1–C5 à 400 °C (SC = 92,4%), avec C2, C3 et C1 majoritaires. A 500 °C, le produit majoritaire est le C1. Sur le catalyseur WO3/TiO2, parmi les produits formés, le produit C2 est prédominant à 400 °C et le C1 entre 440 et 500 °C (Tableau 10). Ces résultats montrent que les deux catalyseurs, WO3 et WO3/TiO2 présentent le même comportement dans les réactions de craquage : la réaction de dééthylation est favorisée à 400 °C et la réaction de déméthylation est prépondérante entre 440 et 500 °C. La différence entre ces catalyseurs consiste en une sensible diminution de la sélectivité de craquage sur le WO3/TiO2.
3.2.2.3. Etude de l’évolution de la sélectivité dans la réaction d’élargissement de cycle (SE) en fonction de la température de réaction
Le Tableau 10 présente les valeurs de la sélectivité pour la réaction d’élargissement (SE) de cycle (avec la formation du benzène et du cyclohexane), sur les catalyseurs WO3 et WO3/TiO2, en fonction de la température de réaction.
WO3 présente une sélectivité en élargissement de cycle de SE = 7,6%, pour une conversion (𝛼) de 20,3% à 400 °C. Le seul produit formé dans la réaction d’élargissement de cycle est le produit d’isomérisation du MCP : le cyclohexane. L’augmentation de la température de réaction conduit à la diminution de la sélectivité en élargissement de cycle. Ainsi, à 440 °C la valeur de la SE est 3,8% (𝛼= 56%) et les produits d’élargissement de cycle formés sont le cyclohexane et le benzène en quantités équimolaires. Entre 470 et 500 °C, les valeurs de la sélectivité en élargissement de cycle sont respectivement de 3,2 (𝛼= 83,1%) et 1,8% (𝛼 = 99,6%). Les produits d’élargissement de cycle formés sont le cyclohexane et le benzène.
Le catalyseur WO3/TiO2 présente une sélectivité en élargissement de cycle de 100%, à 340 °C pour une conversion (𝛼) de 1,9%. L’augmentation de la température de réaction jusqu’à 500 °C, conduit à une diminution de la sélectivité en élargissement de cycle qui descend à 3,2% (𝛼= 95,0) à 500 °C. Pour toutes les valeurs de température étudiées, le seul produit formé dans la réaction d’élargissement de cycle est le produit de déshydrogénation : le benzène.
Ces résultats montrent que le WO3/TiO2 est plus sélectif par rapport au WO3 dans la réaction d’élargissement de cycle à basse température. Pour les deux catalyseurs, l’augmentation de la température de réaction diminue l’importance de la réaction d’élargissement.
3.2.3. Corrélation des résultats obtenus avec ceux obtenus en littérature
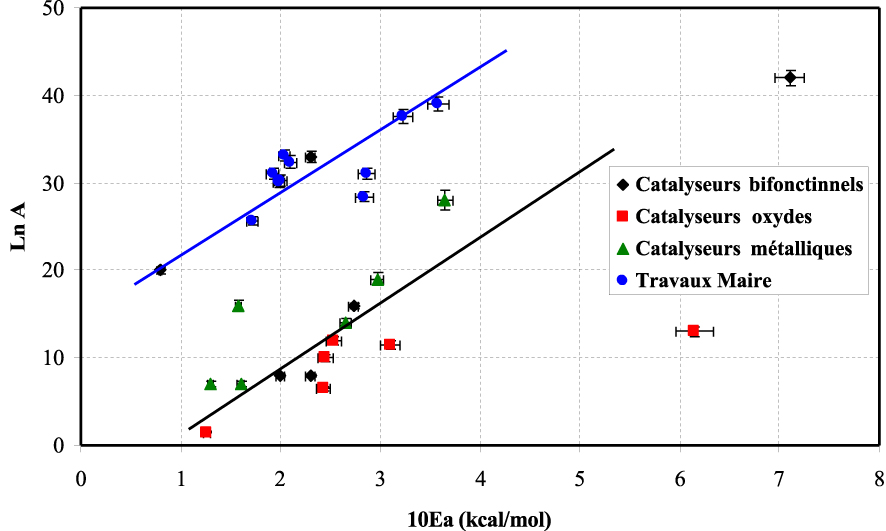
Nous avons comparé les résultats obtenus dans la réaction de conversion du MCP avec les résultats obtenus dans la littérature [6, 7, 11, 56, 57, 58] (Figure 23).
Les valeurs de Ln(V) (avec V vitesse globale de réaction) en fonction de l’inverse de la température sont presentées dans les Tableaux 11 à 14.
Nous avons ensuite tracé Ln(A) = mEa + c, où m et c sont des constantes, pour la réaction d’hydrogénolyse du MCP et nous avons comparé nos résultats à ceux obtenus dans les travaux de Maire [56]. L’ensemble des résultats obtenus sur les différents catalyseurs sont portés dans le Tableau 15.
Variation du Ln de la vitesse globale de réaction en fonction de l’inverse de la température de réaction sur un catalyseur MoO3
| T (°C) | T (K) | 103∕T (K) | V | Ln(V) |
|---|---|---|---|---|
| 180 | 453 | 2,2 | - | - |
| 220 | 493 | 2,02 | - | - |
| 260 | 533 | 1,87 | - | - |
| 300 | 573 | 1,74 | - | - |
| 340 | 613 | 1,63 | 0,32 | −1,14 |
| 400 | 673 | 1,48 | 2,17 | 0,77 |
Comparaison entre les courbes obtenues lors de ce travail et les résultats obtenus lors des travaux de Maire [56].
Variation du Ln de la vitesse globale de réaction en fonction de l’inverse de la température de réaction sur un catalyseur MoO3/TiO2
| T (°C) | T (K) | 103∕T (K) | V | Ln(V) |
|---|---|---|---|---|
| 180 | 453 | 2,2 | - | - |
| 220 | 493 | 2,02 | 0,1 | −2,3 |
| 260 | 533 | 1,87 | 0,18 | −1,7 |
| 300 | 573 | 1,74 | 0,41 | −0,89 |
| 340 | 613 | 1,63 | 1,29 | 0,25 |
| 400 | 673 | 1,48 | 3,22 | 1,17 |
Variation du Ln de la vitesse globale de réaction en fonction de l’inverse de la température de réaction sur un catalyseur WO3
| T (°C) | T (K) | 103∕T (K) | V | Ln(V) |
|---|---|---|---|---|
| 180 | 453 | 2,2 | - | - |
| 220 | 493 | 2,02 | - | - |
| 260 | 533 | 1,87 | - | - |
| 300 | 573 | 1,74 | - | - |
| 340 | 613 | 1,63 | - | - |
| 400 | 673 | 1,48 | 1,55 | 0,44 |
| 440 | 713 | 1,4 | 4,29 | 1,46 |
| 470 | 743 | 1,34 | 6,35 | 1,85 |
| 500 | 773 | 1,29 | 7,62 | 2,03 |
Les résultats obtenus ont montré l’obtention de deux droites parallèles. L’évolution de ces droites montre qu’à la plus forte valeur d’énergie apparente d’activation correspond la valeur la plus élevée du facteur préexponentiel.
On traduit cette évolution par l’apparition d’un effet de compensation existant pour cette réaction et ayant lieu sur différents catalyseurs comme trouvé dans le cas des travaux de Maire [56].
L’interprétation de cet effet n’est pas encore bien élucidée. Conner [59] a expliqué ce phénomène par la présence d’un effet de compensation entre l’entropie et l’enthalpie de l’état de transition. Il relia la variation d’entropie de transition à un changement des niveaux d’énergie de l’état de transition et la réduction de l’énergie d’activation à une modification de l’état de transition. Par ailleurs Rooney [60] a focalisé ces recherches sur les phénomènes de catalyse hétérogène et d’adsorption.
Variation du Ln de la vitesse globale de réaction en fonction de l’inverse de la température de réaction sur un catalyseur WO3/TiO2
| T (°C) | T (K) | 103∕T(K) | V | Ln(V) |
|---|---|---|---|---|
| 180 | 453 | 2,2 | - | - |
| 220 | 493 | 2,02 | - | - |
| 260 | 533 | 1,87 | - | - |
| 300 | 573 | 1,74 | - | - |
| 340 | 613 | 1,63 | 0,14 | −1,97 |
| 400 | 673 | 1,48 | 0,83 | −0,19 |
| 440 | 713 | 1,4 | 2,33 | 0,85 |
| 470 | 743 | 1,34 | 4,09 | 1,41 |
| 500 | 773 | 1,29 | 7,02 | 1,95 |
Valeurs des énergies apparentes d’activation et du facteur exponentiel obtenu sur les différents systèmes catalytiques étudiés
| Catalyseurs | Ea (kcal/mol) | Ln(A) | A (s−1) |
|---|---|---|---|
| MoO3 | 25,3 | 12 | 1,6 × 105 |
| MoO3/TiO2 | 12,6 | 1,5 | 4,5 |
| WO3 | 24,3 | 6,5 | 6,0 × 102 |
| WO3/TiO2 | 24,5 | 10 | 2,2 × 104 |
4. Conclusions
Les catalyseurs à base des oxydes (MoO3 et WO3) ont été préparés par la méthode d’imprégnation, seulement sur le support faiblement réductible, TiO2 : MoO3/TiO2 et WO3/TiO2. Par ailleurs, les paramètres de maille de ces trois oxydes sont voisins, ce qui pourrait induire une réactivité particulière. De plus ces oxydes sont des semi-conducteurs qui pourraient présenter des interactions électroniques aux interfaces « oxyde ». Les mesures des surfaces spécifiques ont mis en évidence l’augmentation de la surface spécifique des oxydes de tungstène massiques et supportés (WO3, WO2 et WO3/TiO2), suite au traitement réducteur de 12 h à 500 °C. Les analyses par DRX sur un échantillon de WO3/TiO2 calciné 4 h à 500 °C ont montré la présence des cristallites du trioxyde de tungstène. Après réduction, le trioxyde s’est complètement réduit en dioxyde (WO2) et tungstène métallique (W3O). L’évolution de la surface des oxydes de molybdène, suivie par SPX avant et après traitement réducteur à 400 °C, a montré la présence de trois espèces de molybdène : Mo6+, Mo5+ et Mo4+. Les espèces Mo6+ et Mo4+ représentent respectivement le trioxyde et le dioxyde de molybdène, l’espèce Mo5+ correspondrait, dans ce cas, aux sous-oxydes intermédiaires entre MoO3 et MoO2. Cette espèce représenterait donc les atomes du molybdène situés sur les plans de cisaillement. L’évolution de la surface des oxydes de tungstène suivie par SPX avant le traitement réducteur a mis en évidence la présence de W6+ correspondant à la phase WO3. Après traitement réducteur à 500 °C, la surface est décorée avec des espèces W6+ et W4+, avec W6+ à la surface et W4+ en volume.
Les oxydes de molybdène et de tungstène possèdent une fonction de type « métal » sur le MoO2 (WO2) et une fonction de type « acide » par les groupements OH associée à une acidité de Bronsted. Ces systèmes peuvent être considérés comme bifonctionnels MoO2(OHx)ac (WO2(OHx)ac)(« ac=acide »).
Le catalyseur MoO3réduit 12 h à 400 °C a été testé dans la réaction de conversion du MCP dans la gamme de température de 180 −−400 °C. Il est inactif catalytiquement pour l’ouverture du cycle avant 340 °C mais il présente une sélectivité de 100% pour la réaction d’élargissement de cycle. A 400 °C, il présente une activité catalytique pour la réaction d’ouverture de cycle selon un mécanisme de type sélectif. A basse température (340 °C) le MoO3 réduit favorise exclusivement la réaction d’élargissement de cycle. A haute température (400 °C), le MoO3 réduit est actif pour la réaction d’ouverture de cycle, d’élargissement de cycle et de craquage. L’ordre de sélectivité pour chaque réaction est la suivante : SE (56%) > SC (27,7%) > SO (13,5%). MoO3 réduit favorise la réaction de déméthylation (produit C1) par un processus répétitif et la réaction de déshydrogénation, avec la formation exclusive du benzène.
Le catalyseur MoO3∕TiO2réduit 12 h à 400 °C a été testé dans la réaction de conversion du MCP dans la gamme de température 180 −−400 °C. Il est inactif catalytiquement avant 220 °C. A 220 °C, il présente une sélectivité de 100% pour la réaction d’élargissement de cycle. A 400 °C, il présente une activité catalytique pour la réaction d’ouverture de cycle selon un mécanisme de type sélectif. Aux basses températures (220 −−260 °C), le MoO3/TiO2 réduit favorise en exclusivité la réaction d’élargissement de cycle avec la formation du benzène. A températures moyennes (300 −−340 °C), le MoO3/TiO2 réduit favorise les réactions d’ouverture de cycle du MCP et d’élargissement de cycle. A haute température (400 °C) le MoO3/TiO2 réduit est actif pour la réaction d’ouverture de cycle, d’élargissement de cycle et de craquage. L’ordre de sélectivité pour chaque réaction est la suivante : SE (71%) > SC (18,5%) > SO (10,2%)
Le catalyseur WO3réduit 12 h à 500 °C a été testé dans la réaction de conversion du MCP dans la gamme de température de 180 −−500 °C. Il est inactif catalytiquement avant 400 °C. A partir de 400 °C, il présente une activité catalytique dominée par la réaction de craquage (92%) et secondairement la réaction d’élargissement de cycle du MCP (8%). L’augmentation de la température jusqu’à 500 °C, favorise encore la réaction de craquage. A basse température (400 °C), le WO3 réduit favorise les réactions de dééthylation (produit C2), de déméthylation (produit C1) et de dépropylation (produit C3) par un processus répétitif et la réaction d’isomérisation du MCP, avec la formation exclusive du cyclohexane. A haute température (500 °C), le WO3 réduit favorise la réaction de déméthylation (produit C1) par un processus répétitif et la réaction de déshydrogénation, avec la formation exclusive du benzène. D’un point de vue thermodynamique, on attend 100 fois plus de benzène à 500 °C qu’à 400 °C.
Le catalyseur WO3∕TiO2réduit 12 h à 500 °C a été testé dans la réaction de conversion du MCP dans la gamme de température de 180 −−500 °C. Il est inactif catalytiquement avant 340 °C. A partir de 340 °C, il présente une activité catalytique, dominée par la réaction d’élargissement de cycle (100%) avec la formation exclusive du benzène. L’augmentation de la température jusqu’à 500 °C favorise la réaction de craquage au détriment de la réaction d’élargissement de cycle. A basse température (340 °C), le WO3/TiO2 réduit, favorise la réaction de déshydrogénation avec la formation exclusive du benzène. A haute température (500 °C), le WO3/TiO2 réduit, favorise la réaction de déméthylation (produit C1) et la réaction de déshydrogénation, avec la formation exclusive du benzène. Le catalyseur WO3/TiO2 présente une activité catalytique supérieure au catalyseur WO3, mais il est inactif par rapport à la réaction d’ouverture de cycle. Seules les réactions d’élargissement de cycle et de craquage ont lieu. L’oxyde supporté WO3/TiO2 réduit est plus actif que le WO3 réduit dans les mêmes conditions de réaction. Dans les deux cas, le caractère acide est mis en évidence via la réaction d’élargissement de cycle du MCP à basse température. Le caractère métallique apparaît à plus haute température via la réaction de craquage.
Les résultats obtenus sur les oxydes et les oxydes supportés prouvent que la fonction acide est très active conduisant en général aux produits issus de la réaction d’élargissement de cycle et de craquage. Les produits d’ouverture de cycle sont formés dans une faible proportion mais uniquement sur les oxydes de molybdène. Les oxydes étudiés ne possèdent pas l’aptitude à faire désorber les molécules ayant subi une réaction de rupture de la liaison C–C endocyclique du MCP.
En catalyse acide, sur les supports réductibles, la fonction acide est active à basse température pour la réaction d’élargissement de cycle et la fonction métallique est active à haute température pour la réaction d’ouverture et d’élargissement de cycle. Cette constatation est valable uniquement pour le MoO3 et MoO3/TiO2. Les sites actifs présents sur les oxydes de tungstène ne sont pas réactifs pour l’ouverture de cycle du MCP.