

1 Introduction
L’augmentation de la concentration des gaz à effet de serre dans l’atmosphère (notamment le premier d’entre eux, CO2), produit essentiellement par la combustion d’énergies fossiles, est l’une des causes du réchauffement climatique [1]. Actuellement, de nombreuses recherches sont consacrées au captage et au stockage du CO2, mais peu concernent la valorisation de cette nouvelle source de carbone. En effet, l’utilisation du CO2 comme matière première pourrait contribuer au basculement de notre société vers un modèle moins dépendant des énergies fossiles. La valorisation du CO2 permettrait alors d’apporter des solutions de substitution aux produits issus de la pétrochimie, ouvrant ainsi l’opportunité de développer une chimie « verte » à partir du CO2.
À cet effet, l’une des voies de valorisation du CO2 envisagée est la transformation chimique. L’hydrogénation catalytique est considérée comme l’une des méthodes les plus performantes [2–4], car elle permet de convertir directement le CO2 et d’utiliser l’hydrogène issu de l’énergie solaire ou de l’hydroélectricité [5,6]. Par ailleurs, l’hydrogénation du CO2 permet de produire un panel de produits chimiques tels que les carburants (alcool, essence, diesel…) et les composés intermédiaires facilement valorisables (oléfines légères, aromatiques…) selon les conditions opératoires utilisées (catalyseur, température et pression). En effet, certaines études ont montré que les catalyseurs bifonctionnels hybrides oxydes/zéolithes semblent appropriés pour transformer le CO2 en hydrocarbures. Parmi celles-ci, l’hydrogénation catalytique du CO2 en méthanol sur des catalyseurs à base d’oxydes (Cu-Zn/Al2O3, Cu-Zn/ZrO2, Fe-Zn/ZrO2) dans des conditions de température de 200–350 °C et de pression de 50 bar est l’une des voies envisageables [2,7].
Toutefois, certains travaux [8–11] relevés dans la littérature ont montré la possibilité de transformer le CO2 en hydrocarbures sur des catalyseurs à base de fer avec une distribution d’Anderson–Schulz–Flory (ASF). Selon ces auteurs, cette réaction se fait en deux étapes, la première étant la réduction du CO2 en CO via la réaction inverse de la conversion du gaz à l’eau (RWGS) [réaction [1]] et la seconde la transformation du CO en hydrocarbures selon le procédé FT [réaction [2]]. Dans ce cas, le CO est l’intermédiaire réactionnel pour la synthèse des hydrocarbures.
Réaction inverse de la conversion du gaz à l’eau (reverse water gaz shift [RWGS])
| (1) |
Synthèse d’hydrocarbures selon le procédé de Fischer–Tropsch (FT)
| (2) |
Des travaux antérieurs menés dans notre laboratoire ont montré que l’ajout d’un catalyseur acide de type zéolithe à un oxyde à base de cuivre Cu–Zn–Al2O3 permet de produire des hydrocarbures avec un bon rendement, qui dépend de la masse de catalyseur acide et de la proximité oxyde–zéolithe.
L’objectif de notre travail est d’évaluer l’effet de l’ajout de la zéolithe et de la proximité entre les deux sites actifs dans le cas de catalyseur à base de fer. Pour cela, une série de catalyseurs hybrides CuO–Fe2O3–Al2O3/HZSM-5 a été préparée par mélange mécanique.
2 Partie expérimentale
2.1 Préparation des catalyseurs
Un catalyseur oxyde à base de cuivre et de fer CFA (CuO–Fe2O3–Al2O3), avec un rapport massique Cu/Fe = 2,33, a été préparé par coprécipitation. Cette méthode consiste à prendre un volume bien défini de deux solutions molaires de nitrates de cuivre (Cu(NO3)2·3 H2O), et de nitrates de fer (Fe (NO3)3·9 H2O), et de les mélanger dans un ballon tricol. Le mélange ainsi obtenu est chauffé à une température de 70 °C et coprécipité sous agitation par une solution molaire de carbonate de sodium jusqu’à pH = 7 ± 0,5. Ensuite, on introduit 10 % en masse d’alumine (γ-Al2O3, SBET = 210 m2/g, Vporeux = 0,6 cm3/g), tout en maintenant l’agitation pendant 20 min.
Le précipité obtenu est lavé 10 fois à l’eau distillée, puis filtré. Le composé récupéré est séché à l’étuve à une température de 100 °C toute une nuit puis calciné sous air à 350 °C pendant 4 h. Après calcination, la poudre oxyde synthétisée est pastillée, broyée puis tamisée en fraction de grain de diamètre d = 0,2–0,4 mm utilisés en tests catalytiques.
Les catalyseurs hybrides sont préparés par mélange physique de deux familles de catalyseurs, un oxyde et un catalyseur acide de type zéolithe. Le catalyseur acide utilisé est une zéolithe de type MFI (HZSM-5, Si/Al = 40, SBET = 452 m2/g et Vporeux = 0,3 cm3/g).
Afin de déterminer l’effet de la proximité oxyde–zéolithe sur les performances catalytiques, des catalyseurs hybrides constitués de 25 % en poids d’oxyde et de 75 % en poids de zéolithe sont préparés. Deux dispositions du lit catalytique ont été utilisées, ainsi que le montre la Fig. 1.
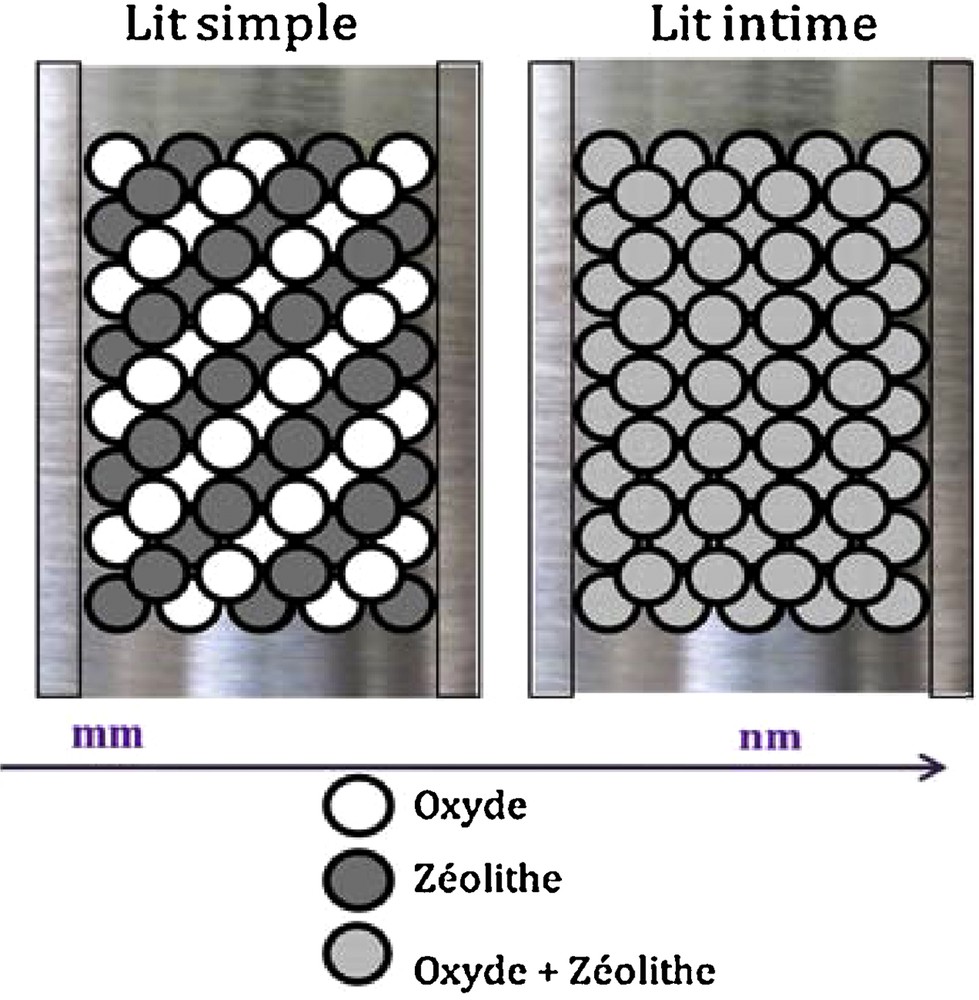
Disposition du lit catalytique.
2.1.1 Lit simple
Ce dispositif a été préparé de la façon suivante : nous avons mis en contact l’oxyde et la zéolithe dans un rapport massique de 1/3. Avant cela, l’oxyde et la zéolithe sont pastillés, broyés et tamisés séparément ; enfin, une fraction de grain de diamètre 0,2–0,4 mm est récupérée pour chacun d’eux.
2.1.2 Lit intime
Les deux poudres oxyde et zéolithe sont mélangées intimement dans un mortier en porcelaine. Le mélange obtenu est pastillé, broyé puis tamisé en fraction de grain (d = 0,2–0,4 mm).
2.2 Caractérisations des catalyseurs
Afin de déterminer les teneurs massiques en éléments dans les catalyseurs, la spectrométrie plasma à couplage inductif a été utilisée. Le dosage est effectué sur un spectromètre PerkinElmer OPTIMA 2000 DV. Le principe de la méthode consiste à vaporiser à l’aide d’un plasma la solution dans laquelle a été dissout le catalyseur (1 volume de HNO3 [1 M] + 3 volumes de HCl [1 M]) et mesurer l’intensité d’émission d’une radiation caractéristique de l’élément à doser.
La mesure de la surface spécifique des différents catalyseurs a été réalisée en utilisant l’adsorption–désorption d’azote à −196 °C à l’aide de deux appareils Micromeritics ASAP 2000 et Tristar. Les échantillons sont prétraités sous vide primaire à 90 °C pendant 1 h, puis sous vide secondaire à 350 °C pendant 8 h pour la zéolithe et à 250 °C pendant 4 h pour l’oxyde. Les calculs de surface BET sont effectués à partir des logiciels ASAP 2000 et Tristar.
La diffraction des rayons X in situ a été utilisée dans le but d’identifier la nature des phases obtenues et de vérifier la bonne cristallinité des catalyseurs. Ces mesures ont été effectuées sur un diffractomètre de type D8 ADVANCE utilisant la radiation Kα du cuivre (λ = 1,5406 Å), pour une tension de 40 kV et une intensité de 30 mA. Avant chaque analyse, l’échantillon est purgé sous hélium pendant 1 h afin d’éliminer l’air contenu dans la chambre d’analyse, puis sous un flux 3 % H2/He pour homogénéiser le mélange réactionnel. Les analyses sont réalisés pour des températures allant de 30 °C jusqu’à 350 °C, avec un pas de 5 °C/min.
L’échantillon est maintenu à chaque température pendant 1 h avant acquisition. L’acquisition des données se fait pour un angle 2θ compris entre 20° et 90°, avec un pas de 0,024° et un temps d’acquisition de 1 s. Les positions et les intensités des raies de diffraction observées sont comparées à celles données par les fiches de références Joint Committee on Powder Diffraction Standards (JCPDS).
Pour évaluer la réductibilité des catalyseurs, des mesures de réduction en température programmée ont été réalisées dans les mêmes conditions afin de pouvoir les comparer. Une masse de 100 mg de l’échantillon (poudre ou grain) introduite dans un tube en quartz en forme de U entre deux pelotes de laine de quartz est soumise à un flux gazeux constitué d’un mélange 10 % H2/Ar (30 mL/min). La réduction en température programmée est réalisée de la température ambiante jusqu’à 900 °C, avec une rampe de 10 °C/min. Le catalyseur est maintenu à cette température pendant 20 min, puis refroidi à température ambiante avec l’azote liquide. La consommation de l’hydrogène est suivie à l’aide d’un détecteur à conductivité thermique (TCD). L’eau formée au cours de la réduction est piégée dans une trappe contenant de l’isopropanol et de l’azote liquide placée avant le détecteur.
Afin de déterminer la quantité d’hydrogène adsorbée sur la surface des catalyseurs, une masse bien définie est réduite pendant 3 h à 350 °C sous flux d’hydrogène (30 mL/min), avec une rampe de température de 10 °C/min. Ensuite, un dégazage à la même température pendant 1 h est réalisé sous un flux gazeux (Ar) à 30 mL/min. Après le prétraitement in situ, le réacteur est refroidi sous un flux d’argon à température ambiante avant les mesures. Les mesures d’adsorption sont réalisés par injection de pulses, 10 % H2/Ar jusqu’à saturation (C1x). On dégaze ensuite pendant 10 min sous argon afin d’évacuer l’hydrogène réversiblement adsorbé (C2X), puis on lance une deuxième série de pulses d’injection. La quantité d’hydrogène chimisorbée (CX) est obtenue par différence entre C1X et C2X :
2.3 Réaction catalytique : conversion du CO2
L’hydrogénation catalytique du CO2 a été réalisée dans un réacteur en acier inoxydable dans les conditions suivantes : une pression de 30 bars, un rapport H2/CO2 = 3 et une température de 350 °C. La masse de lit catalytique a été fixée à 5 g, dont 0,5 à 2 g de catalyseur dilué dans du carborundum (SiC). Avant réaction, le catalyseur est réduit à pression atmosphérique pendant 4 h sous hydrogène pur (1,8 L/h) à 350 °C, selon une rampe de 5 °C/min.
Les produits de réaction sont analysés avec un système analytique composé de quatre chromatographes de type Varian muni de quatre détecteurs (microTCD, TCD et 2FID) placés en série.
Les gaz comme CO et CO2 sont analysés à l’aide d’un détecteur à conductivité thermique (TCD) en utilisant une colonne remplie Porapack Q (15 m, 80/100) et l’hydrogène (H2) par microTCD en utilisant une colonne remplie Carbosieve.
Les produits comme CH3OH et CH3OCH3 sont analysés à l’aide d’un FID sur une colonne capillaire AT Aquawax (30 m, 0,25 mm) et les hydrocarbures (C1–C6) sur un FID muni d’une colonne capillaire HP PLOT Q (30 m, 0,32 mm).
La conversion du CO2 est donnée par l’expression suivante :
Le rendement molaire des produits :
RX, rendement en produit X (X = CO, MeOH ou DME),
nX formé, nombre de moles de produit formé ; nCO2 initial : nombre de moles de CO2 initial
Rendement hydrocarbures = XCO2 − (RCO + RMeOH + RDME), en C-mol % où RCO, RMeOH et RDME représentent respectivement le rendement molaire en CO, méthanol et diméthyléther.
3 Résultats et discussion
3.1 Caractérisation des catalyseurs
3.1.1 Effet de la masse de la zéolithe
Les différentes caractérisations physicochimiques des différents catalyseurs sont rapportées dans le Tableau 1. Les teneurs massiques en cuivre et en fer déterminées par ICP pour le catalyseur oxyde CFA sont de 48,4 % et 18,5 %, respectivement. Ces valeurs sont en accord avec les teneurs théoriques, validant ainsi la méthode de synthèse par coprécipitation.
Caractéristiques physicochimiques des catalyseurs : ICP, SBET, DRX.
| Catalyseurs | Teneurs (% massique) | SBET (m2/gCat) | dhkl (nm)a | ||||
| Cu | Fe | Al | Avant réduction | Après réduction | |||
| CuO | Cu | Fe3O4 | |||||
| HZSM-5 | – | – | – | 452 | – | – | – |
| CFA | 48,4 | 18,5 | 4,4 | 52 | 30 | 42 | 24 |
| CFA-HZSM-5(1/1) | 24,2 | 9,3 | – | 238 | 30 | 42 | 24 |
| CFA-HZSM-5(1/3) | 12,1 | 4,6 | – | 346 | 30 | 42 | 24 |
a Calculs effectués en utilisant le pic le plus intense (111) de CuO, Cu et (220) pour Fe3O4.
Les diffractogrammes réalisés sous flux d’hydrogène avec une montée en température entre 30 et 350 °C dans le cas du catalyseur oxyde CFA et catalyseurs hybrides CFA-HZSM-5 sont présentés sur les Fig. 2–4.
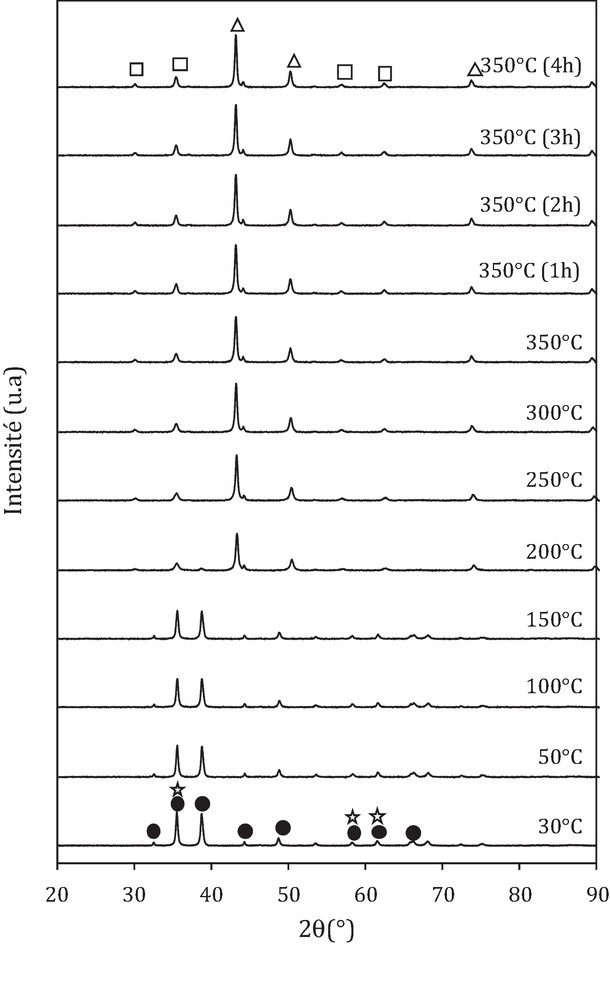
Diffractogrammes diffraction des rayons X (DRX) sous flux 3 %H2/He de CFA en fonction de la température. • : CuO ; : Fe2O3 (hématite) ; Δ : Cu ; □ : Fe3O4 (magnétite).
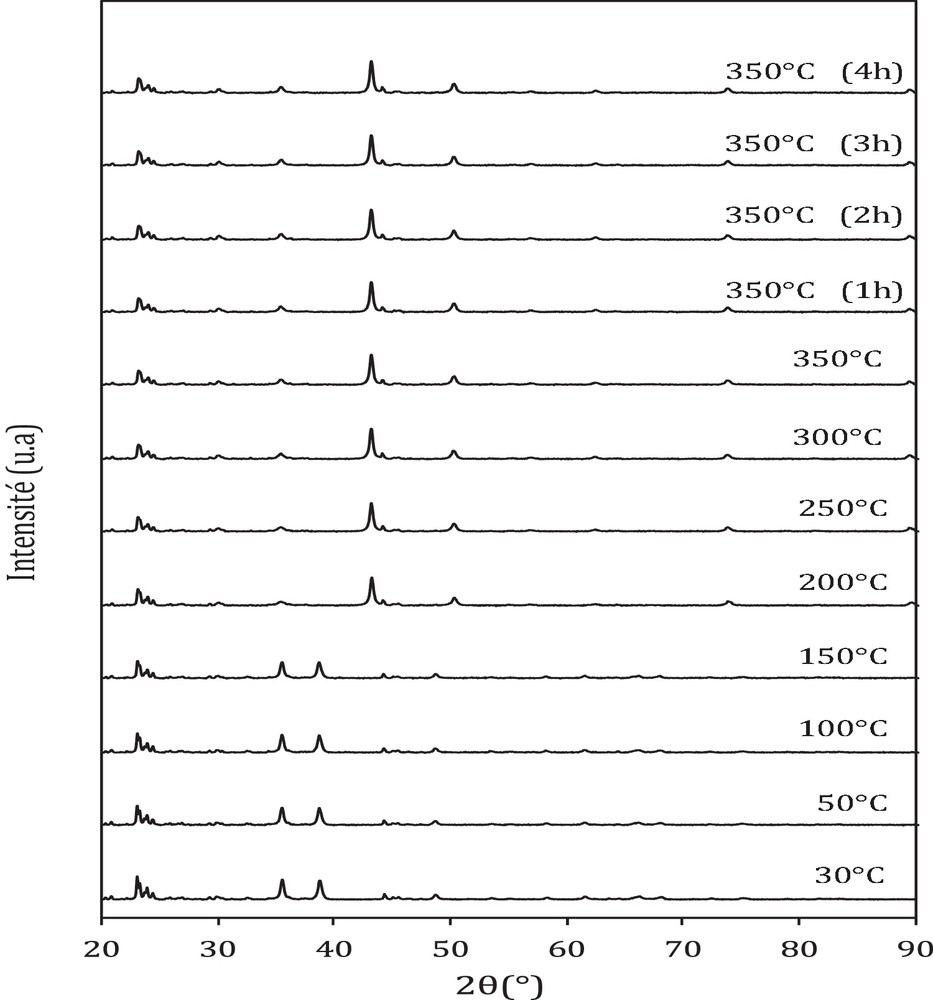
Diffractogrammes diffraction des rayons X (DRX) de CFA + HZSM5 (1/1) en fonction de la température sous flux 3 %H2/He.
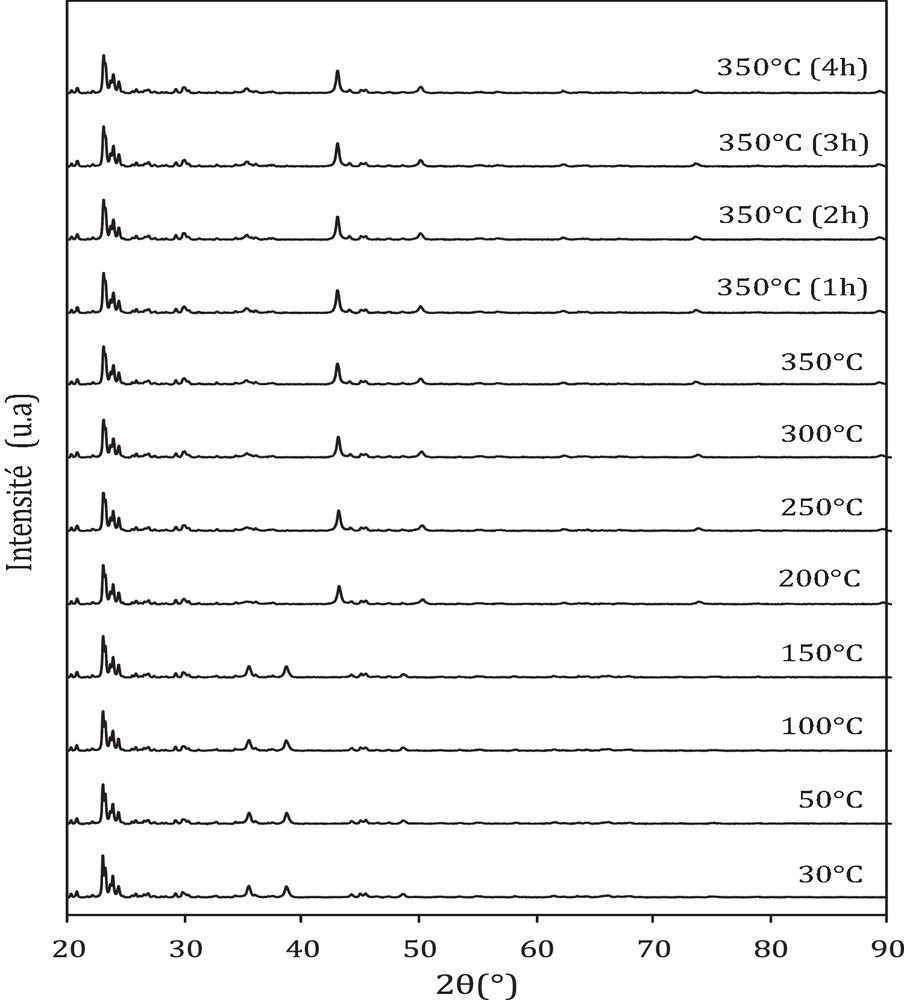
Diffractogrammes diffraction des rayons X (DRX) de CFA + HZSM5 (1/3) en fonction de la température sous flux 3 %H2/He.
L’analyse des diffractogrammes de la Fig. 1 montre que, pour des températures inférieures à 200 °C, on décèle la présence de deux phases CuO et Fe2O3. Les pics les plus intenses caractéristiques de CuO sont observés vers 2θ = 35,5° et 38,7° (fiche JCPDS 00-045-0937). Par ailleurs, les pics caractéristiques de Fe2O3 sont confondus avec ceux de CuO (fiches JCPDS), ce qui indique que ces deux phases (CuO et Fe2O3) diffractent pour des valeurs de 2θ identiques. Le pic le plus intense relatif à Fe2O3 est observé vers 2θ = 35,9° (fiche JCPDS 01-084-0311).
Après réduction sous hydrogène pour des températures supérieures à 200 °C, on décèle la disparition des pics caractéristiques de CuO et Fe2O3. Toutefois, des pics relatifs au cuivre métallique sont observés à 2θ = 43,3°, 50,4° et 74,1° (fiche JCPDS 01-089-2838), indiquant la réduction complète de CuO en Cu. On observe aussi des pics à 2θ = 30°, 35° et 62,2°, attribués à la formation de la phase cristalline Fe3O4 (fiche JCPDS no 01-089-0951). Cette dernière provient de la réduction des particules Fe2O3. Un pic caractéristique de l’alumine est observé dans le catalyseur oxyde avant et après réduction vers 2θ = 44,3°.
Dans le cas des catalyseurs hybrides, on observe la présence des phases CuO et Fe2O3 avant réduction et des phases Cu et Fe3O4 après réduction (Fig. 3 et 4). La présence de ces phases a été rapportée par certains auteurs avec des catalyseurs oxydes cuivre-fer [12,13]. Par ailleurs, un pic correspondant à l’alumine est détecté vers 2θ = 44,3° (fiche JCPDS no 34-0493) dans tous les catalyseurs, quelle que soit leur composition. Ce pic est très faible en intensité, ce qui indique que l’alumine se présente sous forme de petites cristallites dans les catalyseurs [14] ; on constate aussi, pour tous les catalyseurs, des pics situés entre 23 et 25°, caractéristiques de la zéolithe ZSM-5 [15].
La taille des particules avant et après réduction calculée par la relation de Debye Scherrer est reportée dans le Tableau 1. Il ressort de ces résultats que tous les catalyseurs (oxyde et hybrides) possèdent la même taille des particules CuO (30 nm), Cu (42 nm) et Fe3O4 (24 nm) avant et après réduction. Ceci indique que l’ajout de la zéolithe n’a aucun effet sur la taille des particules.
La réduction de l’oxyde de cuivre (CuO) par l’hydrogène est décrite dans la littérature [16–18] par la réaction [3].
| (3) |
Par ailleurs, la réductibilité de l’oxyde de fer (Fe2O3) est donnée par les différentes réactions présentées ci-dessous [19–21] :
| (4) |
| (5) |
| (6) |
| (7) |
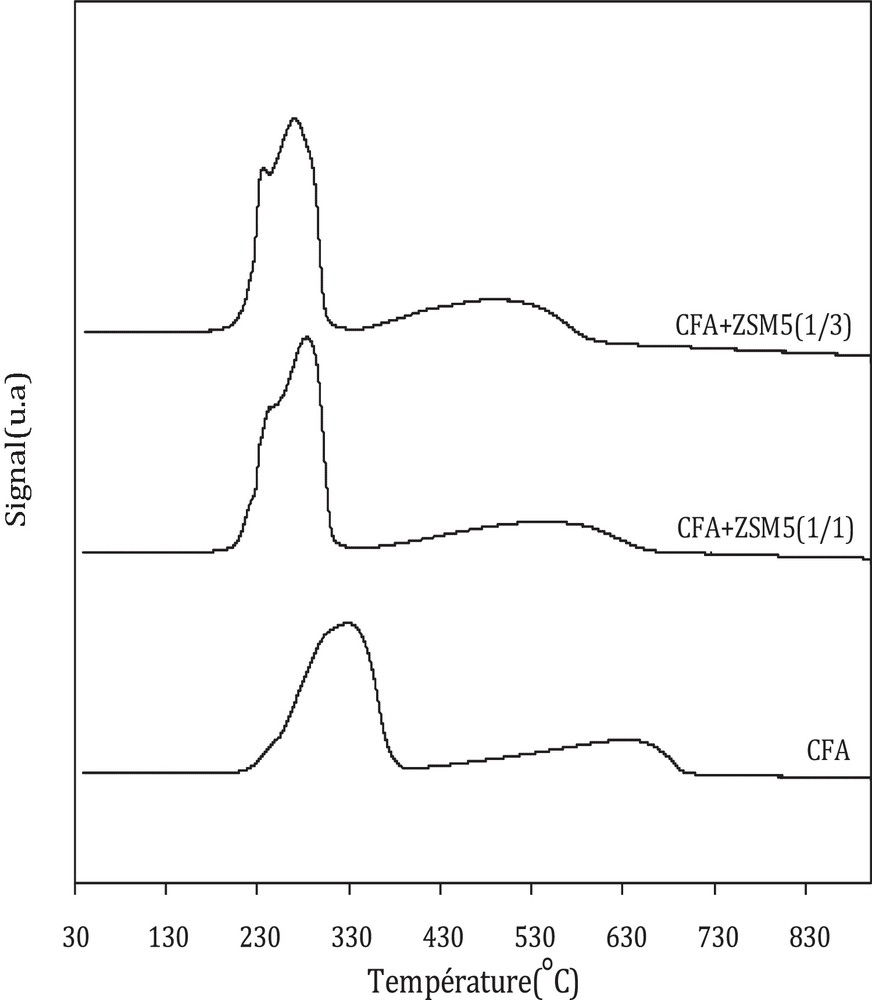
La Fig. 5 présente l’effet de l’ajout de la zéolithe sur la réductibilité du catalyseur oxyde CFA dans la gamme de température comprise entre 30 et 900 °C (profils normalisés à la masse de l’oxyde CFA).
Profils de réduction en température programmée des catalyseurs.
Les profils de réduction en température programmée des trois catalyseurs sont caractérisés par deux pics principaux. Un premier pic de consommation d’hydrogène est observé pour des températures comprises entre 150 et 400 °C et un deuxième pic entre 400 et 800 °C.
Dans le cas du catalyseur oxyde CFA, le premier pic observé vers les basses températures est précédé par un épaulement entre 150–250 °C, avec un pic principal centré vers 328 °C. Le deuxième pic, observé vers les hautes températures, est centré vers 638 °C. Le pic observé vers les basses températures est attribué à la réduction combinée de CuO en Cu [réaction [3]] et de Fe2O3 en Fe3O4 [réaction [4]], alors que le pic décelé vers les hautes températures est assigné à la réduction de Fe3O4 en Fe métallique [réaction [5]] [12,22–24].
Dans le cas des catalyseurs hybrides, l’ajout de la zéolithe induit un changement de réductibilité. En effet, un décalage des pics vers les basses températures est observé. Ce phénomène est amplifié avec l’augmentation de la masse de la zéolithe. En effet, les deux pics principaux sont décalés de 45 °C et de 88 °C dans le cas du catalyseur CFA-HZSM-5(1/1) et de 57 °C et de 143 °C pour le catalyseur CFA-HZSM-5(1/3).
Le premier pic obtenu sur les catalyseurs hybrides a été attribué dans la littérature [16,25] à la réduction de CuO en Cu et de Fe2O3 en Fe3O4 dans la gamme de température 200–350 °C, alors que le deuxième pic observé entre 350 et 700 °C l’était à la réduction de Fe3O4 en Fe. Erena et al. [26] et Cheon et al. [27] ont attribué la présence de plusieurs épaulements dans le pic observé vers les basses températures à une distribution hétérogène des particules oxydes avec différentes réductibilité, attribué aux interactions de l’oxyde métallique avec la zéolithe et/ou avec des particules métalliques environnantes.
Le décalage des pics vers les basses températures (amélioration de la réductibilité) est souvent assigné, soit à la diminution de la taille des particules [14,26–28], soit à un effet de synergie issu des interactions solide–solide entre l’oxyde et la zéolithe [29–31]. Par ailleurs, un épandage d’hydrogène est aussi utilisé pour expliquer cette amélioration de la réductibilité en présence de la zéolithe [32–35]. Afin de vérifier cette dernière hypothèse, une adsorption d’hydrogène à température ambiante a été réalisée sur les catalyseurs près-réduits.
Les résultats obtenus après adsorption d’hydrogène sont présentés dans le Tableau 2 (quantités d’hydrogène adsorbées et d’hydrogène épandu (hydrogène spillover) ont été normalisées à la masse du catalyseur oxyde). Les valeurs de Hspillover sont calculées par la différence entre la quantité d’hydrogène adsorbée sur les catalyseurs hybrides et celle adsorbée sur le catalyseur oxyde.
Quantité d’hydrogène adsorbée et d’hydrogène épandu sur les différents catalyseurs.
| Catalyseurs | Hads (μmol/gcat) | Hads (μmol/gCFA) | Hspillovera (μmol/gCFA) |
| HZSM-5 | 0 | – | – |
| CFA | 0,6 | 0,6 | 0 |
| CFA-HZSM-5(1/1) | 0,7 | 1,5 | 0,9 |
| CFA-HZSM-5(1/3) | 0,7 | 2,9 | 2,3 |
a Hspillover = Hads (μmol/gcat) − Hads (μmol/gCFA).
Aucune adsorption d’hydrogène n’est observée en présence de la zéolithe seule ; en revanche, une adsorption de 0,6 μmol/gCFA est décelée dans le cas de l’oxyde seul. Dans le cas des catalyseurs hybrides, les quantités d’hydrogène adsorbées sont respectivement de 1,5 et de 2,9 μmol/gCFA. L’augmentation de la quantité d’hydrogène adsorbée en présence de la zéolithe peut être expliquée par l’hydrogène épandu (taille des particules Cu inchangée en présence de la zéolithe, Tableau 1). En effet, la valeur (Hspillover, Tableau 2) augmente avec la masse de la zéolithe, favorisant ainsi une meilleure réductibilité des oxydes CuO et Fe2O3.
3.1.2 Effet de la proximité oxyde–zéolithe
Il a été rapporté dans une étude préliminaire que la proximité oxyde–zéolithe joue un rôle très important sur les performances catalytiques. Un rapport massique oxyde/zéolithe = 1/3 est utilisé pour vérifier cette hypothèse dans le cas des catalyseurs hybrides préparés avec un l’oxyde métallique (CFA). Ces derniers (CFA + HZSM-5) ont été préparés par mélange mécanique, soit les catalyseurs hybrides lit simple et lit intime (Fig. 1).
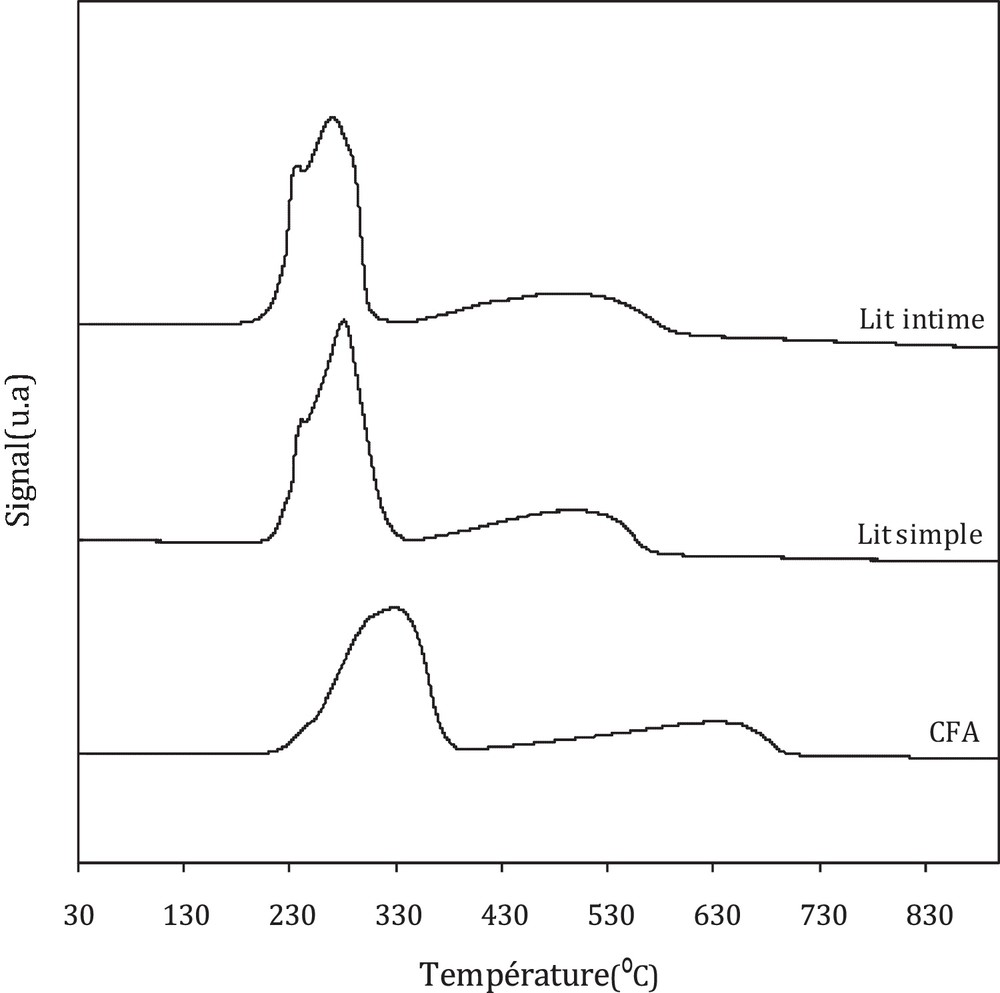
L’effet de proximité oxyde–zéolithe sur la réductibilité des catalyseurs pour une gamme de températures comprise entre 30 et 900 °C est présenté sur la Fig. 6. Les profils obtenus sont normalisés à la masse de l’oxyde CFA.
Effet de la proximité oxyde–zéolithe sur la réductibilité des catalyseurs.
Comme on l’a vu précédemment, le profil RTP de l’oxyde CFA comporte essentiellement deux pics de consommation d’hydrogène. Dans le cas des catalyseurs hybrides, l’ajout de la zéolithe induit un changement dans la réductibilité et, par conséquent, un décalage des pics de réduction vers les basses températures est observé ; cette tendance est accentuée avec la proximité oxyde–zéolithe. Les deux pics observés sont attribués à la réduction de CuO en Cu et de Fe2O3 en Fe3O4 vers les basses températures et à la réduction de Fe3O4 en Fe pour des températures plus élevées. Ce décalage des pics est attribué à un effet de synergie entre les deux sites actifs, induit par les interactions solide–solide entre l’oxyde et la zéolithe [36].
Par ailleurs, cette amélioration de réductibilité peut être assignée à un hydrogène épandu. Afin de confirmer ce postulat, des tests d’adsorption d’hydrogène à température ambiante sur des catalyseurs pré-réduits ont été réalisés.
Le Tableau 3 présente l’effet de la proximité oxyde-zéolithe sur la quantité d’hydrogène adsorbée et sur l’hydrogène épandu. Les valeurs de Hspillover sont calculées par la différence entre la quantité d’hydrogène adsorbée sur les catalyseurs hybrides et celle adsorbée sur le catalyseur oxyde de référence.
Effet de la proximité oxyde–zéolithe sur la quantité et sur l’hydrogène épandu.
| Catalyseurs | Hads (μmol/gcat) | Hads (μmol/gCFA) | Hspillovera (μmol/gCFA) |
| HZSM-5 | 0 | – | – |
| CFA | 0,6 | 0,6 | 0 |
| Lit simple | 0,4 | 1,3 | 0,9 |
| Lit intime | 0,7 | 2,9 | 2,3 |
a Hspillover = Hads (μmol/gcat) − Hads (μmol/gCFA).
Une quantité de 0,6 μmol/gCFA est adsorbée sur l’oxyde CFA alors que, dans le cas des catalyseurs hybrides, la quantité d’hydrogène adsorbée est multipliée par un facteur compris entre 2 et 5 avec l’augmentation de la proximité oxyde–zéolithe, soit de 1,3 et 2,9 μmol/gCFA pour le lit simple et le lit intime, respectivement.
L’augmentation de la quantité d’hydrogène adsorbée avec la proximité oxyde–zéolithe peut être expliquée par l’hydrogène épandu ou par une diminution de la taille des particules du cuivre métallique. Cependant, d’après les résultats obtenus par diffraction X (Tableau 1), tous les catalyseurs après réduction possèdent la même taille de particules. Par conséquent, la surconsommation d’hydrogène provient de l’épandage de l’hydrogène et sa valeur (Tableau 3) augmente avec la proximité entre les sites actifs favorisant ainsi une meilleure réductibilité.
3.2 Réactivité catalytique
Les tests catalytiques ont été réalisés en utilisant un catalyseur tamisé dilué avec du carborundum (masse du lit catalytique est fixée à 5 g) dans les conditions de travail suivantes : 350 °C, 30 bars, H2/CO2 = 3 et VVHCFA = 5460–2730 h−1. Avant chaque test, le catalyseur est réduit pendant 4 h sous hydrogène pur à 350 °C et à pression atmosphérique.
3.2.1 Effet de l’ajout de la zéolithe
L’effet de l’augmentation de la masse de la zéolithe sur les performances catalytiques est montré dans le Tableau 4.
Effet de la masse de la zéolithe sur les performances catalytiques : 700 min, 350 °C, 30 bars et H2/CO2 = 3 et VVHCFA = 2730 h−1.
| Masse de la ZSM5 (g) | Conversion CO2 (%) | Rendements (C-mol %) | Coke (%) | |||||
| CO | HC | C1 | C2 | C3 | C4+ | |||
| 0 | 48,0 | 4,3 | 43,7 | 8,9 | 7,8 | 9,2 | 17,7 | 2,7 |
| 1 | 36,9 | 15,5 | 21,5 | 10,3 | 4,6 | 3,3 | 3,2 | 0,7 |
| 3 | 36,4 | 14,4 | 22,0 | 7,7 | 4,7 | 4,1 | 5,5 | 0,6 |
En absence de la zéolithe, le CO2 est converti à 48 % principalement en hydrocarbures (43,7 %), avec une faible quantité de CO (4,3 %). Les hydrocarbures obtenus présentent une distribution d’ASF avec un facteur de croissance α = 0,6, indiquant que le catalyseur CFA est typique au procédé de Fisher–Tropsh.
La formation majoritaire des hydrocarbures au détriment du CO a été rapportée par différents auteurs [10] et, selon ces derniers, les hydrocarbures sont obtenus via deux réactions [8] et [9].
Réaction inverse de la conversion du gaz à l’eau (RWGS)
| (8) |
Synthèse de produits hydrocarbonés selon le procédé de Fischer–Tropsch (FT)
| (9) |
Par ailleurs, après ajout de la zéolithe, on observe une diminution de la conversion du CO2 et du rendement en hydrocarbures, alors que le rendement en CO augmente. La diminution de la conversion en présence de la zéolithe a été rapportée dans la littérature [29,37,38]. Selon ces auteurs, les interactions solide–solide entre l’oxyde et la zéolithe induit un changement dans les performances catalytiques.
Dans le Tableau 4, on constate une légère augmentation du CH4 et une diminution des hydrocarbures en présence de la zéolithe, attribuée aux réactions de craquage des hydrocarbures lourds sur les sites acides de la zéolithe [39–41]. De plus, en présence de la zéolithe, le taux de carbone formé sur la surface du catalyseur est faible comparé au catalyseur oxyde.
3.2.2 Effet de la proximité oxyde-zéolithe CFA-HZSM-5(1/3)
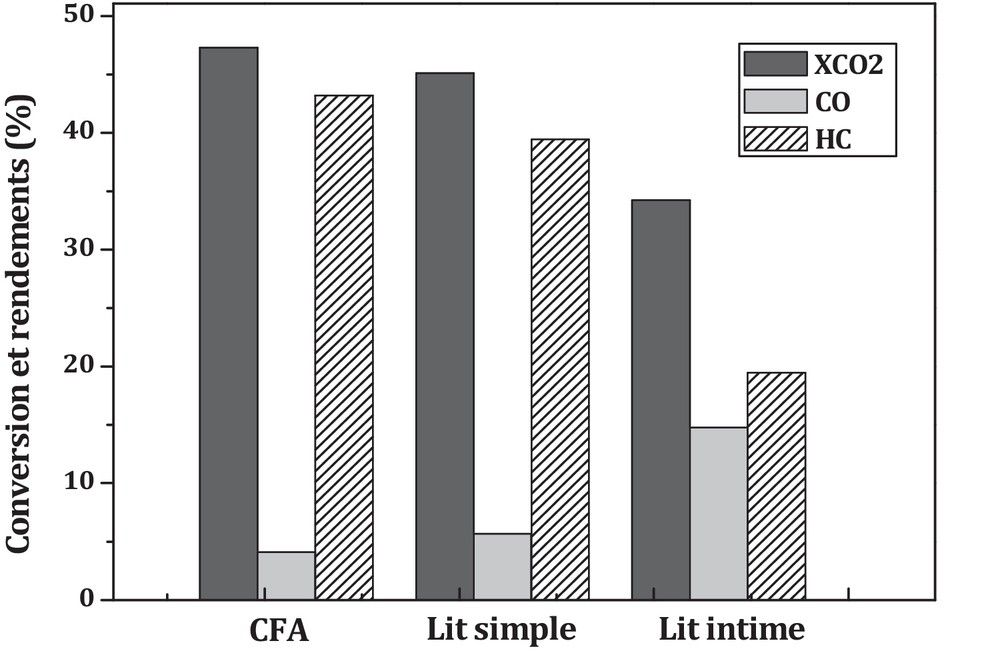
L’effet de la proximité oxyde–zéolithe sur les performances catalytiques est présenté sur la Fig. 7. De cette figure, il ressort que l’augmentation de la proximité CFA-ZSM5 induit une augmentation du rendement en CO au détriment du rendement en hydrocarbures et de la conversion du CO2. Les mêmes résultats ont été relevés dans la littérature pour l’hydrogénation du CO en présence d’un catalyseur hybride [41–43]. Li et al. [42] ont expliqué ce résultat par les interactions entre l’oxyde et la zéolithe.
Effet de la proximité oxyde–zéolithe sur les performances catalytiques : 700 min, 350 °C, 30 bars et H2/CO2 = 3 et VVHCFA = 5460 h−1.
Si on regarde maintenant l’effet de la proximité oxyde–zéolithe sur la sélectivité des hydrocarbures, on observe que la sélectivité en produits change considérablement avec la proximité oxyde–zéolithe (Fig. 8). En effet, dans le cas du lit intime, on obtient essentiellement CH4 avec une sélectivité de 44 %. Dans le cas du lit simple, on obtient principalement des C4+ avec une sélectivité de 54 %, alors que la sélectivité en CH4 est diminuée de moitié en comparaison du lit intime (22 % contre 44 %). La formation majoritaire des C4+ en présence d’un catalyseur hybride (lit séparé) a été obtenue dans les travaux réalisés par Song et al. [44].
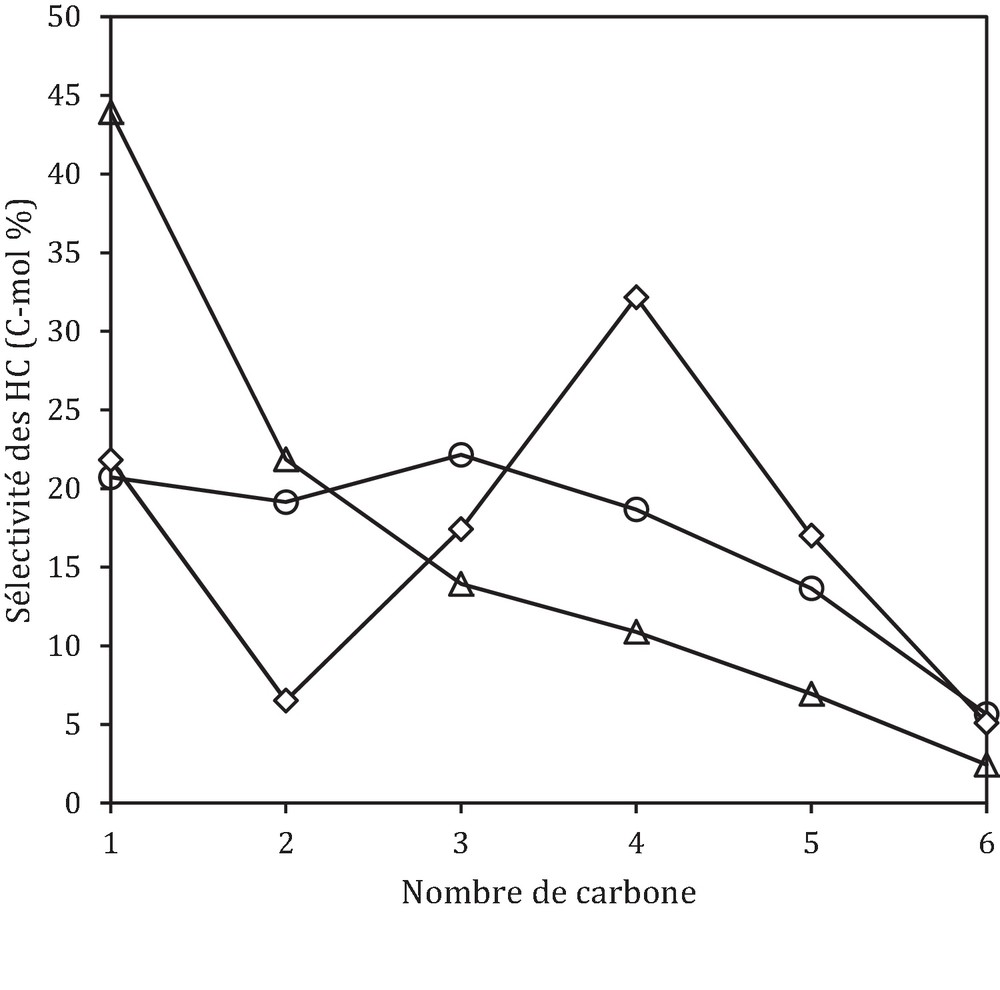
Effet de la proximité oxyde–zéolithe sur la sélectivité des hydrocarbures : 700 min, 350 °C, 30 bars et VVHCFA = 5460 h−1, () CFA, () lit simple et (Δ) lit intime.
En effet, les hydrocarbures (C4+) sont obtenus via des réactions d’oligomérisation du propène (intermédiaire réactionnel) suivies de réactions de craquage et de transfert d’hydrogène.
La formation des hydrocarbures lourds avec la diminution de la proximité oxyde–zéolithe (lit intime vers lit simple) est rapportée dans les travaux réalisés par différents auteurs [40,42]. Selon eux, la croissance de la chaîne carbonée est défavorisée par le contact intime entre l’oxyde et la zéolithe. D’autres auteurs [39,45] ont relié la formation d’hydrocarbures légers à l’hydrogène épandu. En effet, la croissance de la chaîne carbonée est inhibée par l’hydrogène épandu, les oléfines formées sont directement hydrogénées en paraffines correspondantes, d’où la formation des hydrocarbures légers.
4 Conclusion
Un catalyseur oxyde à base de cuivre et de fer et une série de catalyseurs hybrides ont été préparés. Ces derniers ont été caractérisés par différentes techniques afin d’évaluer leurs propriétés physicochimiques. En effet, la diffraction des rayons X (DRX) a révélé la présence des phases cristallines CuO et Fe2O3 dans les catalyseurs calcinés et des phases Cu et Fe3O4 dans les catalyseurs réduits. La réduction en température programmée (RTP) a montré que l’ajout de la zéolithe au catalyseur oxyde améliore la réductibilité et que ce phénomène est accentué avec la masse de la zéolithe et la proximité oxyde–zéolithe. Cette amélioration de réductibilité est attribuée à l’hydrogène épandu (hydrogen spillover).
Les résultats des tests catalytiques ont montré que le catalyseur oxyde CFA est très actif en hydrogénation du CO2 et sélectif en hydrocarbures. En effet, le CO2 est converti principalement en hydrocarbures (C4+) avec une distribution d’Anderson–Schulz–Flory (ASF). Dans le cas des catalyseurs hybrides préparés par mélange intime entre l’oxyde et la zéolithe, une augmentation du rendement en CO au détriment du rendement en hydrocarbures et de la conversion du CO2 sont obtenus. Sur la base de ces résultats, on peut conclure que le catalyseur oxyde CFA est le plus actif pour la synthèse des hydrocarbures en comparaison des catalyseurs hybrides.