

1 Introduction
The zirconium-catalysed ethylmagnesation of terminal alkenes attracted great interest 〚1〛. Similarly to alkenes, imines readily give addition products with ethyl Grignard reagents (EtMgX, X = Cl, Br, Et) in the presence of a catalytic amount of Cp2ZrCl2 (Cp = η5-C5H5) 〚2,3〛. Since imines are weakly electrophilic, the direct addition of organomagnesium reagents in mild conditions is somewhat difficult 〚4〛, and consequently the catalysed reaction favourably competes with the non-catalysed one (see Table 1).
Catalytic ethylmagnesation of imines.
| Imine | R1 | R2 | R3 | Time (h) | Yield (%)a,b |
| 1a | Ph | H | Ph | 1.5 | 69 (16) |
| 1b | Ph | H | Bn | 8 | 90 (12) |
| 1c | Ph | H | tBu | 8c | 61 (0) |
| 1d | Ph | Me | Ph | 1.5 | 78 (0) |
| 1e | nC6H13 | Me | Ph | 1.5 | 86 (0) |
| 1f | Ph | Me | Bn | 1.5 | 50 (0) |
In the case of aldimines, the non-catalysed alkylation reaction can take place either slowly (1a–b) or not at all (1c) 〚5〛. On the other hand, when a catalytic amount of Cp2ZrCl2 was used (10 mol%), the formation of addition products became much easier. In the case of ketimines, the title reaction is also effective 〚2〛. This application is particularly interesting since the direct addition of Grignard reagents to ketimines is generally not possible (1d–f) 〚6〛.
2 Results and discussion
Fig. 1 shows a proposed catalytic cycle consistent with what it was postulated for alkenes 〚7〛. The initial step involves the typical formation of zirconocene–ethylene complex B via β-H abstraction of the unstable intermediate A 〚8〛. Ethane is eliminated through this process. In the next step, the insertion of the imine 1d into B gives metallacycle C. The formation of C is supported by the complete deuterium incorporation at the terminal carbon of the amine 3, which was obtained via the stoichiometric reaction of B (prepared from Cp2ZrCl2 (1 equiv) and EtMgBr (2 equiv) from –78 to 0 °C) 〚9〛 with the imine 1d (1 equiv) followed by quenching with D2O/D2SO4 (Fig. 2).
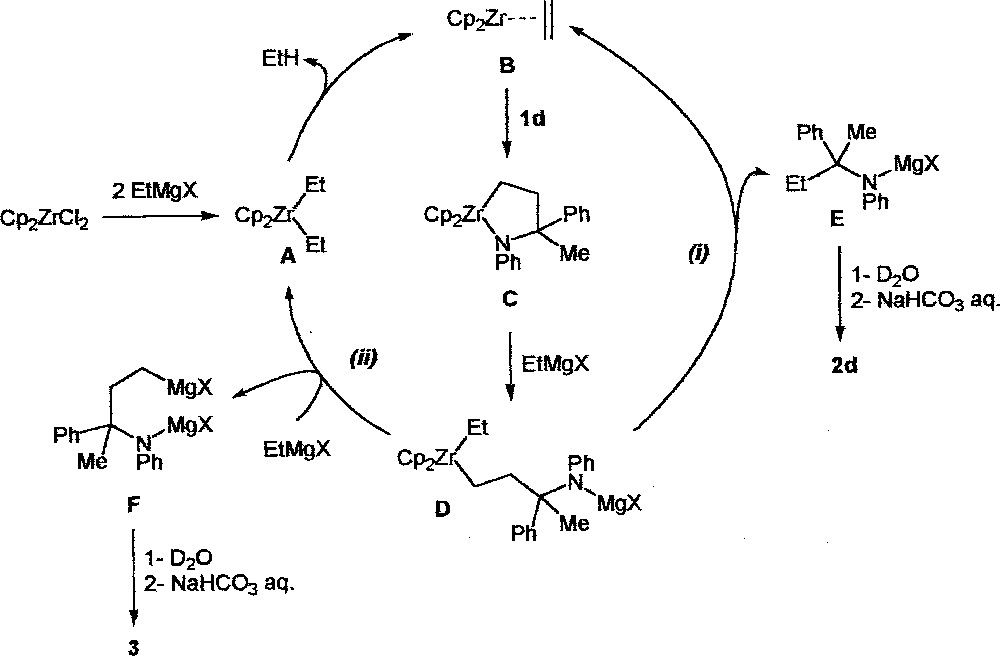
Proposed catalytic cycle for zirconium catalysed ethylmagnesation of imines.
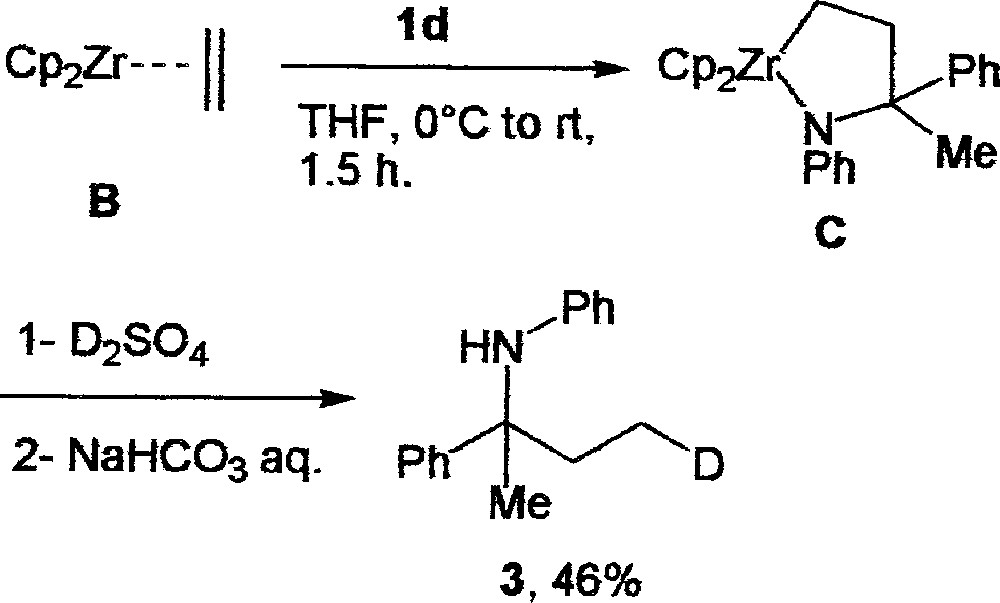
Synthesis of 3 via the stoichiometric reaction of B with the amine 1d (1 equiv), followed by quenching with D2O/D2SO4.
Starting from C, the transmetallation by EtMgX would result in the selective Zr–N bond cleavage to provide the new intermediate D. The complex D undergoes a specific β-H abstraction that regenerates B and affords the monomagnesated product E (path (i)). On the other hand, the transmetallation of D with EtMgX gives the dimagnesated product F and regenerates A (path (ii)).
The coexistence of these two competing pathways is supported by deuteration and gas monitoring experiments. First, the treatment of 1d with 10 mol% Cp2ZrCl2 and 2 equiv EtMgBr and quenching with D2O/D2SO4 led to a mixture of 2d and 3 in a 1:1.5 ratio (Fig. 3).
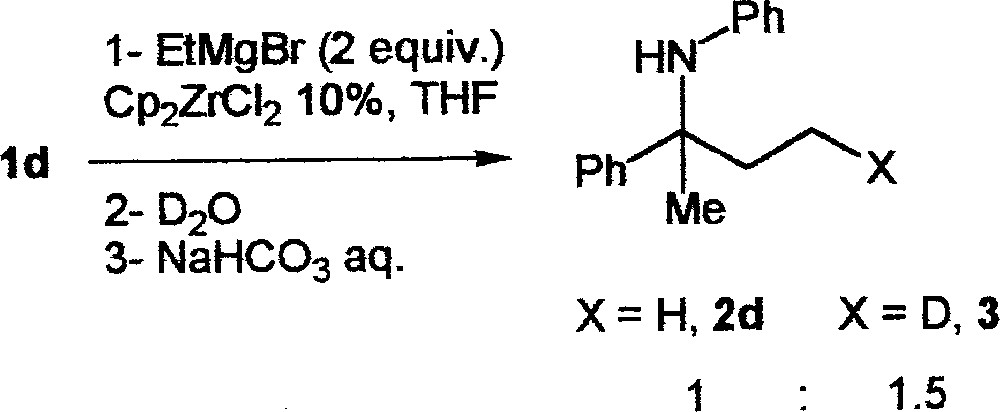
Treatment of 1d with 10 mol% Cp2ZrCl2 and 2 equiv EtMgBr and quenching with D2O/D2SO4, leading to a mixture of 2d and 3 in a 1:1.5 ratio.
Second, in the course of the reaction of 1d with EtMgX (X = Cl, Br, Et, 2 equiv) and Cp2ZrCl2 (10 mol%), ethane was detected (benzene-d6 was saturated with the contents of the collecting gas burette) supporting the formation of B from A. As shown in Fig. 4, the molar quantity of ethane collected is much higher than that of the catalyst. This suggests that A is regenerated during the catalytic cycle. It is also noteworthy that the molar quantity of ethane is lower than that of the amine 2d; this suggests that 2d is reached through both pathways.
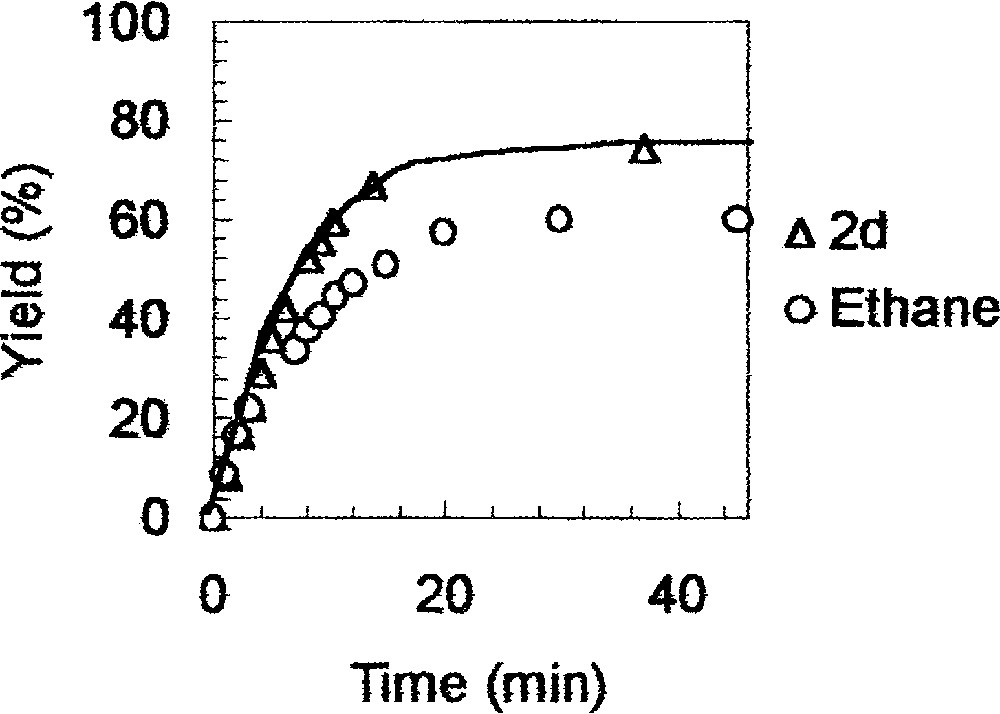
GC and ethane monitoring during the reaction of 1d 〚200 mmol l–1〛 with EtMgCl 〚400 mmol l–1〛 and Cp2ZrCl2 〚20 mmol l–1〛.
Additional experiments were carried out to support the mechanism. First, treatment of 1d with CD3CH2MgBr and Cp2ZrCl2 (10 mol%) followed by quenching with water gave a mixture of two dimagnesated compounds (4 and 5) and two trimagnesated compounds (6 et 7) in a 6:6:1:1 estimated ratio (Fig. 5, equation (1)). On the other hand, quenching with D2O/D2SO4 led to a 1:1 mixture of 5 and 7. This deuterium scrambling is in favour of an intermediate metallacycle and inconsistent with a direct alkylation (7 should be the only product). With the presence of trideutetrated products after hydrolysis, the β-D abstraction comes to light. The comparison of the H2O and D2O/D2SO4, quenching indicates that the formation of 4 and 5 is due to the hydrolysis of the dideuterated regioisomers derived from F, whereas the formation of 6 and 7 is due to the hydrolysis of the trideuterated regioisomers derived from E. The difference between the product ratios in Figs 3 (1:1.5) and 5 (6:1) is likely due to a primary isotope effect, which slows the β-D abstraction down. Consequently, the selective Zr–N bond cleavage of metallacycle C is emphasized. The isotope effect can be estimated to kH/kD = 4.3, which is consistent with other values reported for related reactions 〚6〛. Second, complex 8 was prepared by the reaction of Cp2Zr(Me)Cl with benzylphenylamine lithium salt, followed by insertion of ethylene into the intermediate zirconaaziridine (equation (2)) 〚10〛. Metallacycle 8, whose structure is very similar to the one of C, proved to be catalytically active in the reaction of 1d with ethyl Grignard reagents.

(1) Treatment of 1d with CD3CH2MgBr and Cp2ZrCl2 followed by quenching with water, leading to a mixture of 4, 5, 6 and 7 in a 6:6:1:1 estimated ratio. (2) Synthesis of 8 by the reaction of Cp2Zr(Me)Cl with benzylphenylamine lithium salt, followed by insertion of ethylene into the intermediate zirconaaziridine.
Third, the Zr-catalysed ethylmagnesation of 1d was carried out using various concentrations of EtMgCl. As shown above, quenching with D2O/D2SO4 led to a mixture of 3 and 2d. As expected, the selectivity S = 〚3〛/〚2d〛, estimated by NMR, increases linearly as a function of the concentration (Fig. 6). Path (ii) is thus promoted by a high concentration of the ethylmagnesium compound.
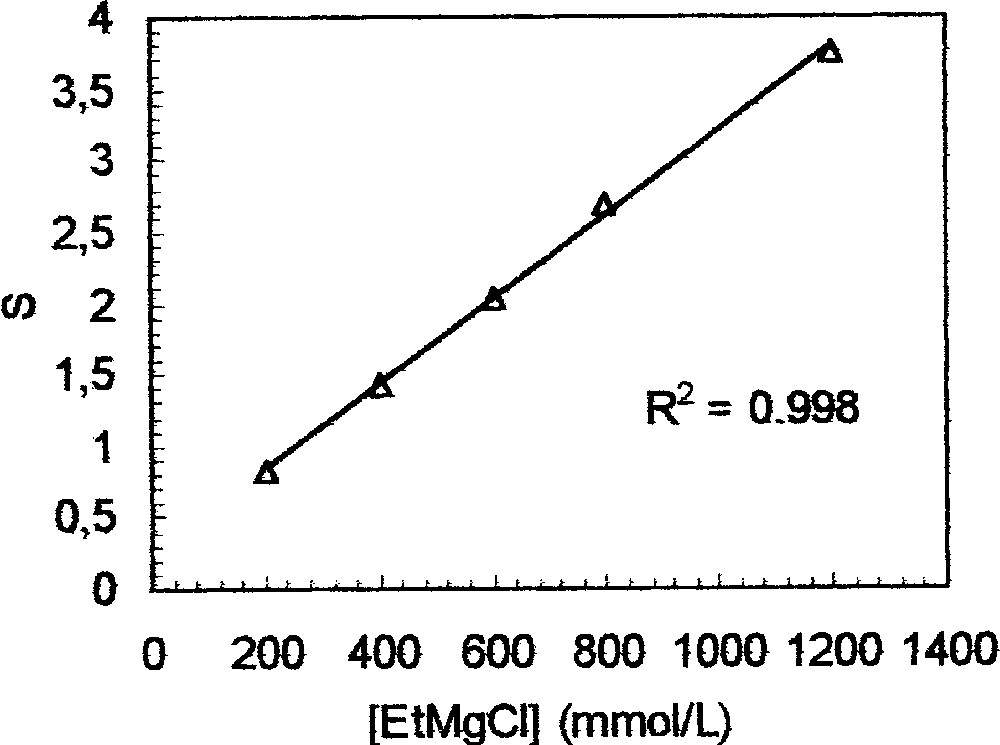
Reaction of 1d 〚100 mmol l–1〛 with Cp2ZrCl2 〚10 mmol l–1〛. S = 〚3〛/〚2d〛.
3 Conclusion
On the basis of deuterium labelling experiments, analysis of by-products, as well as the catalytic activity of an independently prepared intermediate, we have proposed a mechanism for the Zr-catalyzed addition of ethyl Grignard reagents to imines. We have demonstrated the coexistence of two competing ethyl- and (2-magneso)ethylmagnesation pathways that involve a common intermediate resulting from the selective cleavage of an azazirconacyclopentane.
4 Typical experimental procedure of the Zr-catalyzed alkylation of imines
Under an argon atmosphere, a dry Schlenk flask was charged with Cp2ZrCl2 (146 mg, 0.5 mmol) and the imine (5 mmol). THF (20 ml) was added, followed by the ethyl Grignard reagent (1 M in THF, 2 equiv). The reaction mixture was allowed to stir for 1.5 hours. The reaction mixture was carefully quenched with 15% NaOH (2 ml). After dilution with 15% NaOH, the phases were separated and the aqueous phase was extracted three times with ether. The combined organic layers were washed with a saturated solution of Na2CO3, dried over Na2SO4 and concentrated. Amines of greater than 95% purity were obtained by flash chromatography on silica.
Acknowledgements
We thank the Gecom–Concoord organisation board, which invited us to publish this work.