

1 Introduction
The synthesis of polydentate hemilabile ligands, which generally combine a hard and soft donor functionality, is a subject of considerable current interest, in particular with respect to the development of new functional materials and homogeneous catalysts. Numerous metal complexes containing P,O or P,N type ligands are catalytically active in a wide range of reactions of both academic and industrial interest 〚1–3〛. We have recently described a series of cationic Pd(II) complexes, in which the heterofunctional phosphine ligand Ph2PNHC(O)Me 1 behaves as rigid and/or hemilabile P,O chelate 〚4〛. These complexes were prepared in high yield in a one-pot procedure by reacting 1 with 〚PdCl(Me)(COD)〛 (COD = 1,5-cyclooctadiene) and a two-electron donor ligand L, in the presence of a halide abstractor (e.g. AgBF4) in CH2Cl2. Interestingly, these complexes could also be obtained by ligand substitution from the acetonitrile complex 2 by reaction with L (Fig. 1). The chelating ability of 1 was compared to that of other potential P,O chelates. Based on competition experiments, 1 was found to be a stronger chelate than the keto- or amidophosphine ligands Ph2PCH2C(O)Ph and Ph2PCH2C(O)NPh2, respectively. Thus, in complexes 3 and 4, these phosphines act as monodentate P ligands, whereas 1 chelates the Pd centre through P and O coordination (Fig. 1).
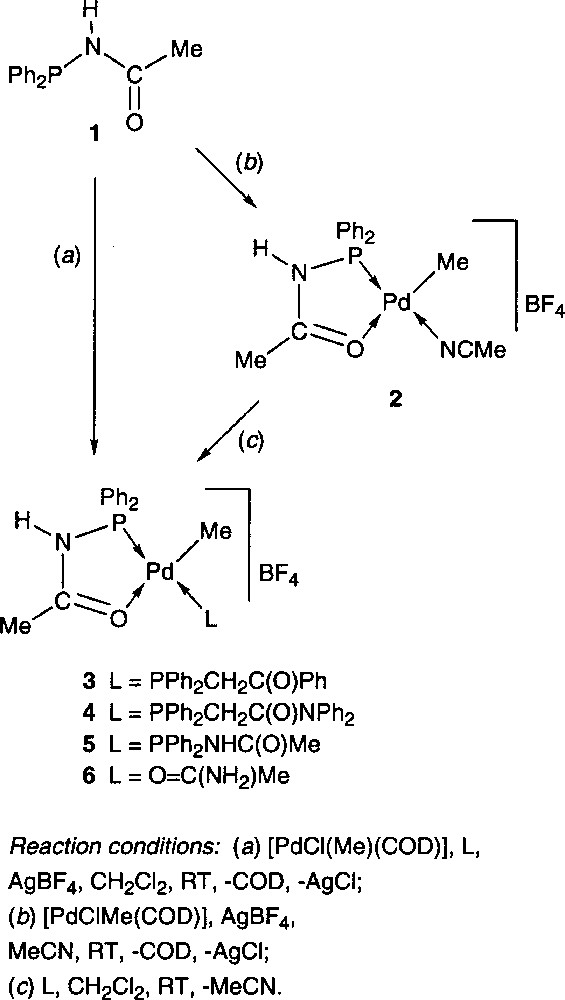
Furthermore, complex 2 showed a higher stability, both in solution and in the solid state, than its Ph2PCH2C(O)Ph analogue, which reflects a greater stabilizing effect of 1. The dynamic behaviour of the latter in complex 5 evidences the hemilabile properties of this ligand. This led in solution to a fast equilibrium resulting from Ph2PNHC(O)Me alternatively acting as P,O chelate or P-monodentate ligands 〚4〛. Taking advantage of the stability of this family of complexes, the reactivity of 2 was investigated towards the insertion of CO, ethylene and methylacrylate into the Pd-Me bond, reactions which are of considerable current interest 〚5–8〛. This allowed us to isolate and completely characterise the first intermediates resulting from successive ethylene/CO/methylacrylate coupling, a model system for a terpolymerisation process 〚9〛. During this work, a complex containing acetamide in its coordination sphere was serendipitously obtained, which we describe here. This complex was subsequently prepared in a more rational way.
2 Results and discussion
The reaction of 1, 〚PdCl(Me)(COD)〛 and methylacrylate in CH2Cl2, followed by the addition of AgBF4, afforded after work-up a yellow powder. Its 31P{1H} NMR spectrum showed two signals at δ 80.3 and 60.4 ppm (ratio: 2:1), indicating the presence of two products. This was confirmed in the 1H NMR spectrum, which showed two sets of signals, the methylacrylate remaining apparently unreacted. The structure of the complex to which the 31P resonance at δ 80.3 ppm was ascribed has been determined by X-ray diffraction (Fig. 2). It showed the unexpected formation of the cationic Pd(II) complex 6, in which the fourth coordination site is occupied by an acetamide ligand, coordinated to the metal centre by the oxygen atom (Fig. 1). This is, to the best of our knowledge (SciFinder search), only the second complex of this type with coordinated acetamide to a Pd centre 〚10〛 and the first one to be structurally characterised. This surprising result prompted us to determine the origin of this ligand. Since a similar reaction carried out in the absence of methylacrylate led to the same mixture of products, we could rule out a possible involvement of methylacrylate in the formation of 6. We therefore came to the conclusion that acetamide arose from partial decomposition (or hydrolysis?) of the phosphine ligand Ph2PNHC(O)Me, after storage for several months in a Schlenk flask. In retrospect, this is not too surprising if one considers that P–N bonds are known to be reactive, as for example in diphosphazanes where the P–N bond is easily cleaved in protic solvents 〚11〛.
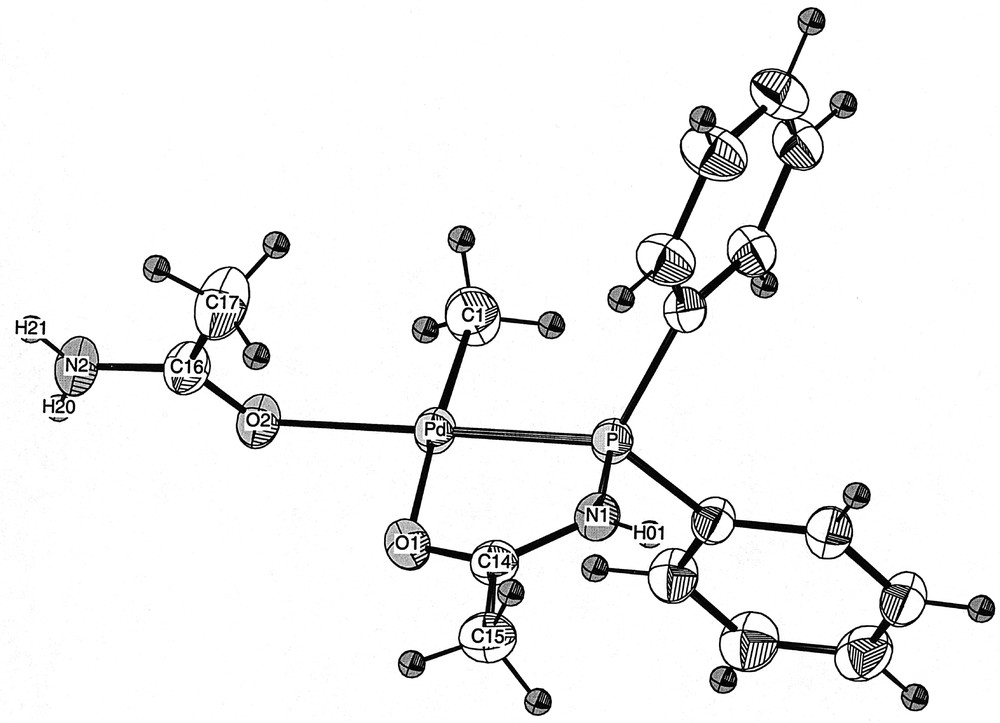
View of the molecular structure of the cation in 6·0.5 CH2Cl2.
The 31P{1H} NMR resonance at δ 60.4 ppm was ascribed to 〚PdMe{PPh2NHC(O)Me}{PPh2NHC(O)Me}〛〚BF4〛 5, which has been previously synthesised and characterised 〚4〛. We have rationalised the synthesis of 6 by carrying out the reactions described in Fig. 1 in the presence of acetamide. The one-pot procedure also led to a mixture of 5 and 6 (ratio: 1/2). This may be due to a competition between non-coordinated 1 and acetamide for the fourth coordination site at Pd. We also verified that once 5 is formed, acetamide is not capable of displacing the Ph2PNHC(O)Me ligand in this complex. However, the two-step procedure allowed the synthesis of pure 6 by displacement of the acetonitrile ligand of 2 by acetamide. Complex 6 is rather stable, even in solution, which suggests that compounds of this type may be present in palladium-catalysed reactions such as the amidocarbonylation to form amino acid derivatives 〚12〛 or the hydration of nitriles to carboxamides 〚13〛.
3 Crystal structure of 〚PdMe{PPh2NHC(O)Me}{O=C(NH2)Me}〛〚BF4〛·0.5 CH2Cl2
X-ray quality single crystals of 6·0.5 CH2Cl2 were grown by slow diffusion of pentane into a concentrated dichloromethane solution of 6. The crystals contain 0.5 molecule of dichloromethane per molecule of 6. The molecular structure of 6 is represented in Figs. 2 and 3. Selected bond lengths and angles are given in Table 1.
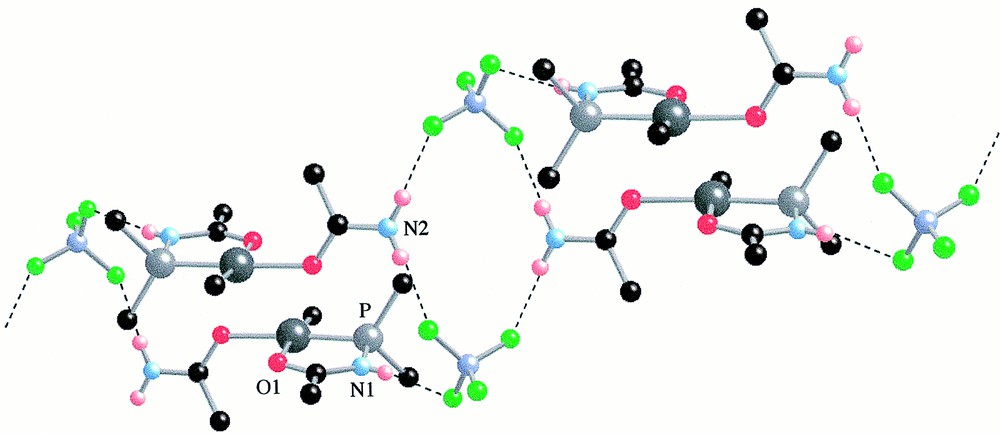
View of the packing of 6 in 6·0.5 CH2Cl2 showing the ‘dimeric’ units connected through H···F bonding (see text). Dichloromethane molecules of solvation and phenyl groups, except the ipso carbon atoms, have been omitted for clarity.
Selected bond distances (Å) and angles (°) for 6·0.5 CH2Cl2a.
| Pd–C(1) | 2.018(3) | N(1)–C(14) | 1.350(3) |
| Pd–O(2) | 2.120(2) | C(14)–O(1) | 1.246(3) |
| Pd–P | 2.1558(6) | O(2)–C(16) | 1.252(3) |
| Pd–O(1) | 2.170(2) | C(16)–N(2) | 1.321(3) |
| P–N(1) | 1.709(2) | ||
| C(1)–Pd–O(2) | 90.27(11) | C(14)–N(1)–P | 119.6(2) |
| C(1)–Pd–P | 91.88(9) | O(1)–C(14)–N(1) | 121.6(2) |
| O(2)–Pd–P | 177.37(5) | C(14)–O(1)–Pd | 114.7(2) |
| C(1)–Pd–O(1) | 174.2(1) | C(16)–O(2)–Pd | 122.5(2) |
| O(2)–Pd–O(1) | 94.28(8) | O(2)–C(16)–N(2) | 120.2(2) |
| P–Pd–O(1) | 83.49(6) |
The geometry at Pd is square planar, as reflected in the P–Pd–O(2) and C(1)–Pd–O(1) angle values of 177.37(5) and 174.2(1)°, respectively. The four atoms coordinated to Pd, as well as the N(1), C(14) and C(15) atoms of the phosphine ligand, lie in the same plane, which also contains the Pd centre. The bond lengths and angles involving the P,O chelate are within the same range as those found in 〚PdMe{PPh2NHC(O)Me}{PPh2NHC(O)Me}〛〚CF3SO3〛 〚4〛 and in 〚PdMe{CH2CH2C(O)Me}{PPh2NHC(O)Me}〛〚PF6〛 〚9〛. The acetamide ligand is coordinated to the Pd centre via its oxygen atom 〚Pd–O(2): 2.120(2) Å〛 in a trans position to the P atom of the P,O ligand. This results in a cis arrangement of the two oxygen atoms around the Pd centre 〚O(1)–Pd–O(2): 94.28(8)°〛. The bond distances and angles involving the acetamide ligand are in the expected range and are similar to those found in other complexes containing an acetamide or acetamidate ligand that have been structurally characterized 〚14–17〛.
In the lattice, molecules of 6·0.5 CH2Cl2 form ‘dimeric’ units, resulting from a head-to-tail packing with the centre of symmetry on the position , 0, 0 of the P21/n space group (Fig. 3). In this arrangement, the oxygen atom of the P,O ligand of one molecule lies above the Pd centre of the other molecule at a Pd···O(1) distance of only 3.087 Å. Furthermore, these ‘dimeric’ units are connected to other ‘dimers’ via hydrogen bonding involving the N–H protons and the BF4 anions (Fig. 3) with average H···F separations of 2.1 Å. The centre of symmetry is on the position , , 0 of the P21/n space group.
4 Experimental section
All reactions were performed using Schlenk-tube techniques under dry nitrogen. Solvents were dried and distilled prior to use under nitrogen. The 1H and 31P{1H} spectra were recorded at 300.1 and 121.5 MHz, respectively, on a FT Bruker AC 300 instrument. The 13C{1H} NMR spectra were recorded at 50.32 MHz on a FT Bruker AC 200 instrument. IR spectra were recorded in the 400–4000 cm–1 range on a Bruker IFS66 FT spectrometer. Elemental C, H and N analyses were performed by the ‘Service de microanalyse du CNRS’ (ULP, Strasbourg).
4.1 Preparation of 〚PdMe{PPh2NHC(O)Me}{O=C(NH2)Me}〛〚BF4〛 6
4.1.1 Method (a)
Solid 〚PdCl(Me)(COD)〛 (0.248 g, 0.936 mmol) was added to a solution of Ph2PNHC(O)Me (0.227 g, 0.936 mmol) in CH2Cl2 (75 ml) at ambient temperature. Then acetamide (0.055 g, 0.936 mmol) was added and the mixture was stirred for 30 min before solid AgBF4 (0.182 g, 0.936 mmol) was added. The solution was then stirred for 30 min before it was filtered. The solvent was evaporated under vacuum to leave a yellow solid, which was washed with pentane (10 ml) and diethyl ether (10 ml) and dried under vacuum. The solid that was isolated (0.350 g) consisted in a mixture of 5 and 6 in a 1:2 ratio.
4.1.2 Method (b)
To a solution of 2 (0.100 g, 0.203 mmol) in CH2Cl2 (15 ml) was added acetamide (0.012 g, 0.203 mmol). The solution was stirred for 30 min before it was filtered. The solvent was evaporated under reduced pressure to leave a beige powder, which was washed with pentane (10 ml) and diethyl ether (10 ml) and dried under vacuum (0.090 g, 87% yield). Yellow crystals suitable for X-ray diffraction were obtained by recrystallisation from CH2Cl2/pentane.
4.1.3 Spectroscopic data for 6
IR (CH2Cl2) ν (cm–1): 1654, 1612 (C=O); 1H NMR (CD2Cl2): δ = 0.86 (d, 1JPH = 2.1 Hz, 3H, Pd–Me), 2.32 and 2.34 (two s, 6H, C(O)CH3), 6.15 and 6.35 (br s, 2H, NH2), 9.12 (br s, 1H, NH); 13C{1H} NMR (CD2Cl2): δ = –0.1 (s, Pd–Me), 23.9 (s, NHC(O)CH3), 27.2 (s, NH2C(O)CH3), 183.4 (d, 2+3JPC = 4.3 Hz, NHC(O)CH3), 184.1 (d, 4JPC = 1.9 Hz, NH2C(O)CH3); 31P{1H} NMR (CD2Cl2): δ = 80.3. C17H22BF4N2O2PPd: calculated C, 39.99; H, 4.34; N, 5.49; found: C, 40.02; H, 4.23; N, 5.23%.
4.2 X-ray crystallographic analysis
The relevant data for 6·0.5 CH2Cl2 are summarised in Table 2 〚18,19〛. Data were recorded on a KappaCCD diffractometer, at 173 K, using a Mo-Kα graphite monochromated radiation of 0.71073 Å. All non-hydrogen atoms were refined anisotropically and hydrogen atoms were included in calculated positions with isotropic thermal parameters. Fig. 3 was generated using ATOMS 5.0.7 and Fig. 3 was created with CrystalMaker 4.0.
X-ray experimental data for 6·0.5 CH2Cl2.
| Formula | C35H46B2Cl2F8N4O4P2Pd2 |
| Molecular weight | 1106.01 |
| Color | colorless |
| Crystal system | monoclinic |
| Space group | P21/n |
| a (Å) | 13.046(2) |
| b (Å) | 12.817(2) |
| c (Å) | 13.805(2) |
| β (°) | 95.002(3) |
| V (Å3) | 2299.4(7) |
| Z | 2 |
| Crystal dim (mm) | 0.20 × 0.20 × 0.20 |
| ρ (calcd) (g cm–3) | 1.597 |
| F(000) | 1108 |
| μ (mm–1) | 1.040 |
| Temperature (K) | 173 |
| Reflections collected | 6855 |
| Reflections with I > 2 σ(I) | 5843 |
| R(gt)a | 0.042 |
| Rwa | 0.111 |
| Goodness of fit | 1.047 |
Supplementary material
The crystallographic material has been sent to the Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB2 1EZ, UK, as supplementary material CCDC 166432 and can be obtained by contacting the CCDC (quoting the article details and the corresponding SUP number). See http://www.rsc.org/suppdata/166432 for crystallographic files in .cif format.
Acknowledgements
We are grateful to Drs A. DeCian, N. Gruber and Prof. R. Welter (Strasbourg) for the crystal structure determination. This work was supported by the ‘Centre national de la recherche scientifique’ (France) and the French ‘Ministère de la Recherche’ (PhD grant to CF).