

1 Introduction
The catalytic carbonylation of nitroaromatic substrates by various group-VIII metal compounds has great potential in the industrial production of aromatic isocyanates and carbamates (equations (1a)–(1b)) 〚1–4〛:
In particular, the homogeneous catalytic system of a Pd(II) precursor, an aromatic diimine ligand (1,10-phenanthroline or o-phen) and a Brönsted acid promoter is among the most active and selective for such carbonylation reactions 〚5–15〛. Typically, such systems are operative at 130–180 °C under pressures of 40–120 bar of carbon monoxide (equation (2)) 〚16–20〛:
We have previously put forward a mechanism for the catalytic transformation within these specific Pd-based systems 〚7, 15〛. The conversion of the nitroaromatic substrate into carbamate was proposed to occur through a series of stepwise deoxygenation/carbonylation steps involving metallacyclic intermediates (Fig. 1).
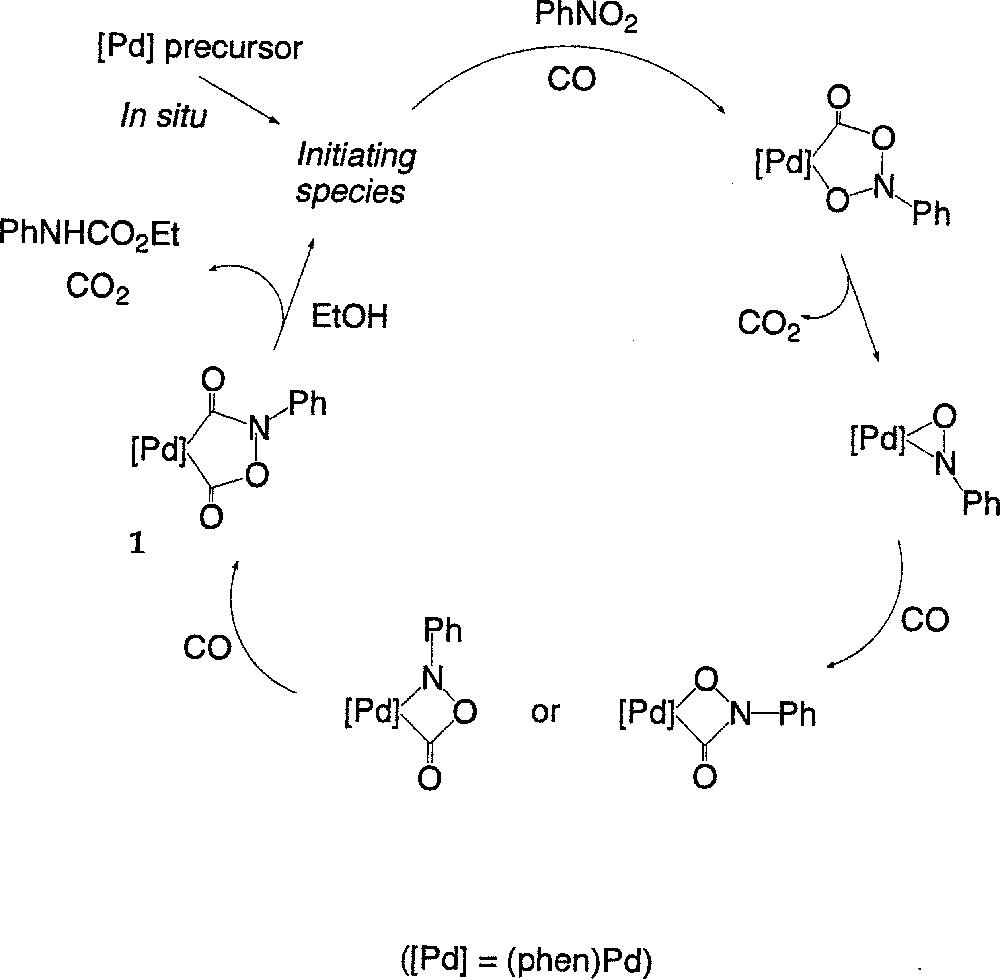
Conversion of the nitroaromatic substrate into carbamate occurring through a series of stepwise deoxygenation/carbonylation steps involving metallacyclic intermediates.
Our proposal was supported by the isolation and characterisation of the palladacycle 1, believed to be an intermediate in the process. This complex constitutes the main Pd-containing species under conditions milder than those used in the catalytic process conditions (80 °C, 30 bar) 〚15〛. During the catalytic cycle, the carbamate would be generated by reaction of 1 with the alcohol, concomitantly with (or following the) decarboxylation of 1. According to this mechanism, phenyl isocyanate should be generated in place of the N-aryl alkyl carbamate in the absence of alcohol 〚7〛.
In order to challenge this proposal, we investigated the course of the reaction in a non-alcoholic solvent. Notably, we wanted to check if complex 1 would still be formed and if phenyl isocyanate would be present in the medium. As briefly mentioned in a short note 〚21〛, these investigations confirmed the formation of 1 in the absence of alcohol and quite unexpectedly, led us also to isolate other palladacycles, like 2, 3 and 4, which apparently were not intermediates of the catalytic cycle (Fig. 2). We now report the complete account of our investigations regarding these new compounds. Notably, their reactivity with small molecules like carbon monoxide, aromatic isocyanates and protons provides useful information about the chemistry occurring under catalytic conditions when no alcohol is present. Hence, based on these experiments, a mechanistic rationale is proposed, which nicely explains the loss of activity of the catalyst, as well as the formation of several pervasive side-products, such as N,N’-diphenylurea (DPU) and N,N’,N”-triphenylbiuret (TPB), typically formed during catalytic runs conducted in non-alcoholic media.
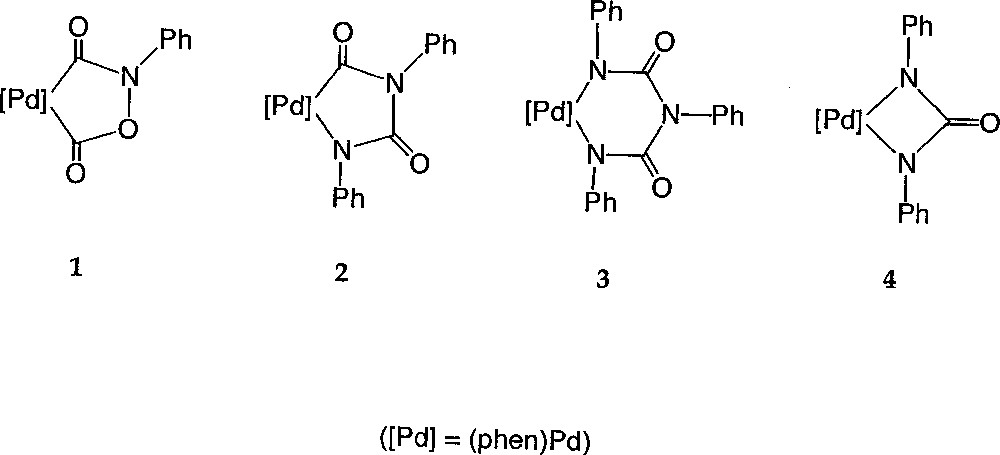
Compounds 1, 2, 3 and 4.
2 Experimental
2.1 General
Unless otherwise specified, all reagents and solvents were purchased from commercial suppliers and used without further purification. Carbon monoxide (N-45 purity grade) was purchased from Air Liquide. Complexes (o-phen)Pd(OAc)2 〚22〛, (o-phen)Pd(OTMB)2 〚23〛, (o-phen)PdCl2 〚23〛, (o-phen)Pd(DBA) 〚24〛 and 1a 〚15〛 were obtained following related syntheses. Reactions under pressure were performed in a 50 ml stainless steel SOTELEM reactor with a 750 W programmable oven. Melting points were measured on an Electrothermal apparatus. 1H–NMR spectra were obtained on a Brucker SY 200 (200 MHz) or SY 400 (400 MHz) Fourier Transform spectrometers. 1H–NMR chemical shifts are reported in units of parts per million relative to residual protiated solvent. Infrared spectra were recorded on Perkin-Elmer 597 or IFS 66 Fourier Transform spectrometers. UV–visible spectra were recorded on a CARY 3 spectrometer, using quartz cells. GC analyses were performed on a Hewlett Packard S II-5890 Gas Chromatograph, equipped with a 10 m methyl silicone semi-capillary column (HP-1). Quantitative analyses were obtained integrating peak areas versus appropriate external or internal standards. Response factors relative to the standard were determined by using authentic samples of the products. FAB–MS mass spectral studies and elemental analyses were submitted to the corresponding services of the University Louis-Pasteur (Strasbourg, France).
2.2 Preparation of five-member palladacycles 2a–g
2.2.1 Typical procedure
120 mg of palladium acetate (0.54 mmol), 260 mg of anhydrous 1,10-phenanthroline (1.4 mmol) and 760 mg (4.26 mmol) of 2,3,5,6-tetramethylbenzoic acid (TMBA) are dissolved in 10 to 12 g of corresponding substituted nitrobenzene (60–100 mmol). ortho-Dichlorobenzene (12 ml) was used as the solvent when the nitroaromatic was not a liquid under standard conditions. The reactor is then flushed with carbon monoxide three times under stirring, pressurised to 40 bar and heated to 80 °C (within 30 min). The stirring is continued for 2 h. After cooling and depressurisation, the resulting pale suspension is filtered and washed several times with ethanol and diethylether. A yellow air-stable precipitate is isolated, which can be recrystallised from hot ortho-dichlorobenzene (120 °C). Crystalline needles of most complexes could be grown by slow diffusion of diethylether or n-pentane in a saturated dichloromethane solution.
(o-phen)Pd〚C(O)N(C6H5)C(O)N(C6H5)〛 (2). Yield: 60%. Colour: Yellow. Melting point: 317 ± 5 °C (dec.). Elemental analysis. Found: C, 59.29; H, 3.47; N, 10.46; C26H18N4O2Pd requires: C, 59.50; H, 3.46; N, 10.67. Infrared (Nujol/KBr, cm–1): 1676, 1623 (s, CO). UV (CH2Cl2, nm, ϵ in M–1 cm–1): 267.5 (51 100), 294.5 (sh, 24 500), 362.0 (sh, 3950). NMR 1H (200 MHz, CD2Cl2, δ, ppm): 10.43 (d, J = 5 Hz, 1 H, H2 or H9/phen), 8.55 (d, J = 8 Hz, 1 H), 8.34 (d, J = 8 Hz, 1 H), 7.96 (m, 3 H), 7.48-7.29 (m, 11 H), 6.82 (d, J = 5 Hz, 1 H, H2 or H9/phen). FABMS (positive mode, m-nitrobenzyl alcohol, m/z): 525 (M+1), 497 (M+1–CO), 406 (M+1–PhNCO), 377 (M-CO–PhNCO), 286 (M–2PhNCO).
(o-phen)Pd{C(O)N〚p-O2CCH3–(C6H4)〛C(O)N〚(p-O2CCH3–(C6H4)〛}(2a). Yield: 50%. Colour: yellow. Melting point: > 300 °C.. Elemental analysis, found: C, 56.37; H, 3.48; N, 8.43; C30H22N4O6Pd requires: C, 56.22; H, 3.46; N, 8.74. Infrared (Nujol/KBr, cm–1): 1670, 1615 (s, CO). NMR 1H (200 MHz, CD2Cl2, δ, ppm): 10.40 (d, J = 5 Hz, 1 H, H2 or H9/phen), 8.55 (d, J = 8 Hz, 1 H), 8.39 (d, J = 8 Hz, 1 H), 7.96 (m, 3 H), 7.60 (dd, J1 = 8 Hz, J2 = 5 Hz, 1 H), 7.47 (d, J = 7 Hz, 2 H), 7.37 (d, J = 7 Hz, 2 H), 7.11 (s, 4 H), 6.75 (d, J = 5 Hz, 1 H, H2 or H9/phen), 2.35 (s, 3 H, –O2CCH3), 2.31 (s, 3 H, –O2CCH3).
(o-phen)Pd{C(O)N〚m-CF3–(C6H4)〛C(O)N〚m-CF3–(C6H4)〛} (2b). Yield: 10%. Colour: yellow. Melting point: > 300 °C. Elemental analysis, found: C, 51.57; H, 3.60; N, 8.19; C28H16N4O2F6Pd requires: C, 50.89; H, 2.44; N, 8.48. Infrared (Nujol/KBr, cm–1): 1675, 1625 (s, CO). NMR 1H (200 MHz, CD2Cl2, δ, ppm): 10.39 (d, J = 5 Hz, 1 H, H2 or H9/phen), 8.58 (d, J = 8 Hz, 1 H), 8.43 (d, J = 8 Hz, 1 H), 7.99 (m, 3 H), 7.78–7.73 (m, 8 H), 7.42 (dd, J1 = 8 Hz, J2 = 5 Hz, 1 H), 6.97 (d, J = 5 Hz, 1 H, H2 or H9/phen).
(o-phen)Pd〚C(O)N(p-F–C6H4)C(O)N(p-F–C6H4)〛 (2c). Yield: 48%. Colour: yellow. Melting point: > 320 °C. Elemental analysis, found: C, 55.69; H, 3.08; N, 9.90; C26H16N4O2F2Pd requires: C, 55.71; H, 2.88; N, 10.05. Infrared (Nujol/KBr, cm–1): 1670, 1610 (s, CO). NMR 1H (200 MHz, CD2Cl2, δ, ppm): 10.40 (d, J = 5 Hz, 1 H, H2 or H9/phen), 8.56 (d, J = 8 Hz, 1 H), 8.42 (d, J = 8 Hz, 1 H), 7.98 (m, 3 H), 7.45 (m, 3 H), 7.31 (dd, JHH = 9 Hz, JHF = 6 Hz, 2 H), 7.13 (t, JHH = JHF = 9 Hz, 2 H), 7.09 (t, JHH = JHF = 9 Hz, 2 H), 7.02 (d, J = 5 Hz, 1 H, H2 or H9/phen).
(o-phen)Pd{C(O)N〚p-CH3–(C6H4)〛C(O)N〚p-CH3–(C6H4)〛} (2d). Yield: 55%. Colour: yellow. Melting point: > 329 °C. Elemental analysis, found: C, 60.98; H, 4.23; N, 9.99; C28H22N4O2Pd requires: C, 56.86; H, 4.02; N, 10.13. Infrared (Nujol/KBr, cm–1): 1675, 1625 (s, CO). NMR 1H (200 MHz, CD2Cl2, δ, ppm): 10.43 (d, J = 5 Hz, 1 H, H2 or H9/phen), 8.55 (d, J = 8 Hz, 1 H), 8.39 (d, J = 8 Hz, 1 H), 8.01 (m, 3 H), 7.38 (dd, J1 = 8 Hz, J2 = 5 Hz, 1 H), 7.33 (d, J = 7 Hz, 2 H), 7.23 (d, J = 7 Hz, 2 H), 7.21 (s, 4 H), 6.87 (d, J = 5 Hz, 1 H, H2 or H9/phen), 2.43 (s, 3 H, –CH3), 2.38 (s, 3 H, –CH3).
(o-phen)Pd{C(O)N〚p-C(CH3)3–(C6H4)〛C(O)N〚p-C(CH3)3–(C6H4)} (2e). Yield: 33%. Colour: yellow. Melting point: > 361 °C. Elemental analysis, found: C, 64.15; H, 5.40; N, 8.99; C34H34N4O2Pd requires: C, 64.10; H, 5.38; N, 8.79. Infrared (Nujol/KBr, cm–1): 1675, 1625 (s, CO). NMR 1H (200 MHz, CD2Cl2, δ, ppm): 10.41 (d, J = 5 Hz, 1 H, H2 or H9/phen), 8.54 (d, J = 8 Hz, 1 H), 8.36 (d, J = 8 Hz, 1 H), 7.95 (m, 3 H), 7.46 (m, 4 H), 7.37 (d, J = 8 Hz, 2 H), 7.22 (m, 3 H), 6.62 (d, J = 5 Hz, 1 H, H2 or H9/phen), 1.43 (s, 9 H, –C(CH3)3), 1.37 (s, 9 H, –C(CH3)3). FABMS (positive mode, m-nitrobenzyl alcohol, m/z): 637 (M+1), 609 (M+1–CO), 462 (M+1–tBu(C6H4)NCO), 433 (M–CO–tBu(C6H4)NCO), 286 (M–2tBu(C6H4)NCO).
2.2.2 Evolution of 1 and 2 in the medium during the reaction without the acid promoter
The reactor is charged with 120 mg of palladium acetate (0.54 mmol), 260 mg of 1,10-phenanthroline (1.4 mmol) in 12 ml of nitrobenzene (118 mmol), heated at 80 °C, as previously described (§ 2.2.1) and nine aliquots (1 ml) are sampled at various reaction times over 17 h (t0, t0 + 25 min, t0 + 55 min, t0 + 95 min, t0 + 140 min, t0 + 225 min, t0 + 530 min, t0 + 1185 min, t0 + 1640 min). For sampling, chronometer and heating are stopped and the autoclave is purged. The aliquot is then immediately quenched by 5 ml of ethanol and cooled to 0 °C. Once the aliquot taken, the autoclave is again pressurised and heating is continued, the chronometer being re-started only when the inner temperature reaches 80 °C. For each aliquot, the precipitate that forms after 2 h is filtered, dried in vacuum and analysed by 1H–NMR, while the filtrate is being subjected to GC–MS (the PhNCO formed is present as N-phenyl ethyl carbamate). The quantitative estimation of the total yield in 1 and 2 at different reaction times is determined by four parallel experiments, in which the totality of the reaction medium was precipitated each time following the same procedure and weighted.
2.2.3 Complex 2f isolated during the synthesis of 3a
The reaction of para–(trifluoromethyl)phenyl isocyanate with 1 (described in § 2.3.1.) leads to the formation of the five-member metallacycle 2f, which could be purified by fractional precipitation.
(o-phen)Pd{C(O)N〚p-CF3–(C6H4)〛C(O)N〚p-CF3–(C6H4)〛} (2f). Yield: 15%. Colour: yellow. Melting point: > 338 °C. Elemental analysis, found: C, 50.80; H, 2.53; N, 8.27; C28H16N4O2F6Pd requires: C, 50.89; H, 2.44; N, 8.48. Infrared (Nujol/KBr, cm–1): 1675, 1625 (s, CO). NMR 1H (200 MHz, CD2Cl2, δ, ppm): 10.36 (d, J = 5 Hz, 1 H, H2 or H9/phen), 8.59 (d, J = 8 Hz, 1 H), 8.43 (d, J = 8 Hz, 1 H), 7.99 (m, 3 H), 7.69 (d, J = 5 Hz, 2 H), 7.62 (s, 4 H, phenyl), 7.53 (d, J = 9 Hz, 2 H), 7.41 (dd, J1 = 8 Hz, J2 = 5 Hz, 1 H), 7.01 (d, J = 5 Hz, 1 H, H2 or H9/phen).
2.3 Preparation of six-member palladacycles 3a–c
2.3.1 Typical procedure
To 50 mg of complex 1a (0.11 mmol) suspended in 5 ml of freshly distilled nitrobenzene, 4.40 mmol of corresponding aromatic isocyanate are added and the reaction medium is stirred at 142 °C. The yellow heterogeneous medium becomes homogeneous and red at 120 °C. After 6 h, the medium is cooled down and 150 ml of diethylether, containing 2 ml of ethanol are added. The pale orange precipitate is filtered and washed with diethylether. In several cases, the precipitate (58 mg) is a mixture of corresponding six- and five-member metallacycles (3/3a–c and 2c/2g; see § 2.2.3.). In each case, the dominant six-member palladacycle could be obtained in a pure form after recrystallisation of the crude precipitate from a dichloromethane/diethylether mixture.
(o-phen)Pd〚N(C6H5)C(O)N(C6H5)C(O)N(C6H5)〛 (3). Yield: 90%. Colour: yellow–orange. Melting point: 255 ± 5 °C (dec.). Elemental analysis, found: C, 62.07; H, 3.98; N, 11.21; C32H23N5O2Pd requires: 62.40; H, 3.76; N, 11.37. Infrared (Nujol/KBr, cm–1): 1644, 1626 (s, CO). UV (CH2Cl2, nm, ϵ in M–1 cm–1): 268.5 (49 400), 348.5 (2300), 374.0 (sh, 2000). NMR 1H (200 MHz, CD2Cl2, δ, ppm): 8.48 (m, 6 H), 8.22 (d, J = 5 Hz, 2 H), 7.98 (s, 2 H), 7.57 (dd, J1 = 5 Hz, J2 = 8 Hz, 2 H), 7.45 (d, J = 8 Hz, 2 H), 7.35 (t, J = 6 Hz, 2 H), 7.24 (m, 5 H), 6.97 (t, 2 H). FABMS (positive mode, m-nitrobenzyl alcohol, m/z): 661 (M+1), 497 (M+1–PhNCO), 377 (M–2PhNCO), 286 (M–2PhNCO–NPh).
(o-phen)Pd{N〚p-CF3–(C6H4)〛C(O)N〚p-CF3–(C6H4)〛C(O)N〚p-CF3–(C6H4)〛} (3a). Yield: 85%. Colour: Yellow. Melting point: 297 ± 5 °C (dec.). Elemental analysis, found: C, 51.43; H, 2.52; N, 8.38; C35H20N5F9O2Pd requires: C, 51.97; H, 2.46; N, 8.54. Infrared (Nujol/KBr, cm–1): 1650, 1630 (s, CO). NMR 1H (200 MHz, CD2Cl2, δ, ppm): 8.57 (m, 6 H), 8.22 (d, J = 5 Hz, 4 H), 8.05 (s, 2 H), 7,66 (dd, J1 = 8 Hz, J2 = 5 Hz, 2 H), 7,64 (s, 4 H, phenyl), 7.48 (d, JHH = 9 Hz, 2 H).
(o-phen)Pd〚N(p-F-C6H4)C(O)N(p-F-C6H4)C(O)N(p-F-C6H4)〛 (3b). Yield: 90%. Colour: yellow. Melting point: 263 ± 5 °C (dec.). Elemental analysis, found: C, 57.50; H, 3.26; N, 10.52; C32H20N5F3O2Pd requires: C, 57.57; H, 3.01; N, 10.45. Infrared (Nujol/KBr, cm–1): 1635, 1615 (s, CO). NMR 1H (200 MHz, CD2Cl2, δ, ppm): 8.51 (d, J = 8 Hz, 2 H), 8.43 (dd, JHH = 9 Hz, JHF = 5 Hz, 4 H), 8.18 (d, J = 5 Hz, 2 H), 7.99 (s, 2 H), 7.60 (dd, J1 = 8 Hz, J2 = 5 Hz, 2 H), 7.40 (dd, JHH = 9 Hz, JHF = 4 Hz, 2 H), 7.08–6.92 (m, 6 H).
(o-phen)Pd{N〚p-CH3–(C6H4)〛C(O)N〚p-CH3–(C6H4)〛C(O)N〚p-CH3–(C6H4)〛} (3c). Yield: 80%. Colour: Yellow. Melting point: 301 ± 5 °C (dec.). Elemental analysis, found: C, 63.72; H, 4.29; N, 10.62; C35H29N5O2Pd requires: C, 63.88; H, 4.44; N, 10.64. Infrared (Nujol/KBr, cm–1): 1640, 1615 (s, CO). NMR 1H (200 MHz, CD2Cl2, δ, ppm): 8.47 (d, J = 8 Hz, 2 H), 8.32 (d, J = 8 Hz, 4 H), 8.22 (d, J = 5 Hz, 2 H), 7.96 (s, 2 H), 7.57 (dd, J1 = 8 Hz, J2 = 5 Hz, 2 H), 7.29 (d, J = 8 Hz, 2 H), 7.14 (d, J = 8 Hz, 2 H), 7.04 (d, J = 8 Hz, 4 H), 2.35 (s, 3 H, –CH3), 2.29 (s, 6 H, –CH3). FABMS (positive mode, m-nitrobenzyl alcohol, m/z): 658 (M+1), 525 (M+1–TolNCO), 391 (M–2TolNCO), 286 (M–2TolNCO–NTol).
2.3.2 Reaction starting from 1a
480 mg (4.40 mmol) of phenyl isocyanate are added to 50 mg of 1a (0.11 mmol) suspended in 10 ml of freshly distilled ortho-dichlorobenzene and the reaction medium is stirred at 142 °C. After 7 h, the medium is cooled down and two fractions can be successively precipitated by pentane (80 ml). The first fraction is mostly composed of six-member metallacycles (infrared, NMR and FAB-MS) and contains 3c (35%), along with three new metallacycles incorporating bis-meta-trifluoromethyl groups (19F NMR), one of which (35%) presenting also symmetry plane perpendicular to the phenanthroline ligand. The second fraction is mostly composed of two five-member metallacycles, containing respectively one and two phenyl groups with bis-meta-trifluoromethyl substituents. Attempts to separate these compounds were unsuccessful.
2.3.3 Reaction of 1 with PhNCO at 80 °C under carbon monoxide pressure
In the reactor, 120 mg of palladacycle 1 (0.26 mmol) and 1300 mg of phenyl isocyanate (11.60 mmol) are dissolved in 8 ml of nitrobenzene. The reactor is then flushed with carbon monoxide (40 bar) and heated at 80 °C following the usual procedure (§ 2.2.1.). After 49 h, the reactor is depressurised and the orange solution is filtrated through a frit. Two fractions of 5 mg and 115 mg yellow product are then precipitated respectively by successive addition of 15 ml ethanol, followed by 15 ml ether, to the reaction medium. The first fraction is exclusively composed of 3 (3%) and the second one contains essentially 2 (82%).
2.3.4 Synthesis of 3 from palladium acetate and N,N’,N”– triphenylbiuret
Method A. 50 mg of (o-phen)Pd(OAc)2 (0.12 mmol) and 150 mg of N,N’,N”-triphenylbiuret (0.45 mmol) are suspended in 10 ml of methanol and refluxed for 3 h. Subsequently, 20 ml of acetone are added to the medium. The yellow–orange suspension of 3 is centrifuged, washed with several fractions of methanol and diethylether and finally dried in vacuo. Yield 68 mg (92%).
Method B. 50 mg of (o-phen)Pd(OAc)2 (0.12 mmol) and 125 mg of N,N’,N”-triphenylbiuret (0.36 mmol) are suspended in 10 ml of methanol and 15 ml of triethylamine (0.14 mmol) are added. The reaction medium is stirred for 4 h and the precipitate (3) is separated and treated as previously described. Yield: 84% (62 mg).
2.4 Reactions of six–member metallacycles 3 and 3c
2.4.1 With carbon monoxide
Under pressure (40 bar). 50 mg of 3 (0.08 mmol) were suspended in 6 ml of nitrobenzene and stirred under CO (40 bar) at 80 °C (reached in 30). After 2 h, the reactor is depressurised and 100 ml ether are added to the brown solution. The resulting suspension is then cooled to 0 °C, and a brown precipitate composed of 2 and metallic palladium is collected, washed with diethylether and dried under vacuum (40 mg; 93% by 1H–NMR). After recrystallisation from ortho-dichlorobenzene and n-pentane, 37 mg of pure 2 are isolated (90%).
Atmospheric CO pressure. 30 mg of compound 3 (0.05 mmol) are suspended in 5 ml of nitrobenzene and heated to 140 °C. After dissolution of the complex, CO is introduced into the medium. The red solution turns darker and eventually becomes a black suspension. After 6 h, the heating is stopped and the flask is cooled. A dark brown solid consisting of 2 and palladium metal is precipitated by addition of 250 ml of diethylether after cooling the flask for several hours to 0 °C, ether washings and drying (15 mg; ≤ 50).
2.4.2 With aromatic isocyanates
3 and p–TolNCO. To 50 mg of compound 3 (0.08 mmol), 435 mg of para-tolyl isocyanate (40 equiv.) solubilised in 10 ml of nitrobenzene are added. Upon heating to 140 °C with vigorous stirring, the initially orange reaction medium becomes homogeneous and red. After 7 h, the medium is allowed to cool and 2 ml of ethanol are added, while 3c starts to deposit as a yellow precipitate. The precipitation is completed by subsequent addition of 250 ml of diethylether and cooling (–20 °C, 5 h), yielding 54 mg ether-washing and vacuum drying (∼ 100%).
3c and PhNCO. Following the above procedure with 25 mg of complex 3c (0,04 mmol) and 180 mg of PhNCO (40 equiv.) in 5 ml nitrobenzene, 17 mg of compound 3 are isolated in a pure form (73%).
2.4.3 With acid
Two solutions of 3 (10 mg) dissolved in CD2Cl2 were placed in two NMR tubes. One was treated with an excess of HCl (37%) and the other with an excess of tetramethyl-benzoic acid. Comparison of the spectra, before and after the addition, reveals a quantitative conversion of the starting complex to (o-phen)PdCl2 (5a) or (o-phen)Pd(O2CC10H13)2 (5b) and free N,N’,N”-triphenylbiuret respectively. The poorly soluble complex 5a that precipitates spontaneously in the medium can be isolated and further characterised by infrared spectroscopy. The identity of 5b that remains in solution is assessed by comparison with the spectra of an authentic sample, independently synthesised.
2.5 Preparation of four–member metallacycles 4a–b
2.5.1 Typical procedure
0.1 mmol of six-member metallacycle (3 or 3b–c) is suspended in 5 ml of ortho-dichlorobenzene (20 μl of water can be added at this point if the solvent does not contain traces of water) and stirred at 140 °C. The yellow heterogeneous medium becomes homogeneous and its colour turns to deep red. After 1 h, the hot solution is filtrated through a coarse frit and cooled. 150 ml of diethylether is then added and the solution is stored at –20 °C for 4 h. The orange–red precipitate that forms is filtered and washed several times with 5 ml of diethylether. This precipitate mainly consists of the corresponding four-member palladacycle (respectively 4 or 4a–b), contaminated with small amounts of metallic palladium. Recrystallisation of all complexes can be easily achieved by dissolution of the crude precipitate in dichloromethane and layering with diethylether.
(o-phen)Pd〚N(C6H5)C(O)N(C6H5)〛 (4). Isolated yield: 50%. Colour: Red. Melting point: 253 ± 1 °C (dec.). Elemental analysis, found: C, 53.69; H, 3.47; N, 9.44; C25H18N4O1Pd.CH2Cl2 requires: C, 53.68; H, 3.47; N, 9.63. Infrared (Nujol/KBr, cm–1): 1631 (s, CO). UV (CH2Cl2, nm, ϵ in M–1 cm–1): 271.5 (37100), 361.0 (3500), 390.5 (3540). NMR 1H (200 MHz, CD2Cl2, δ, ppm): 8.51 (d, J = 8 Hz, 2 H), 8.42 (d, J = 5 Hz, 2 H), 7.99 (s, 2 H), 7.66 (dd, J1 = 5 Hz, J2 = 8 Hz, 2 H), 7.48 (d, J = 7 Hz, 4 H), 7.32 (t, J = 7 Hz, 4 H), 7.11 (t, J = 7 Hz, 2 H). FABMS (positive mode, m-nitrobenzyl alcohol, m/z): 497 (M+1), 378 (M–PhNCO), 286 (M–PhNCO–NPh).
(o-phen)Pd〚N(p-F-C6H4)C(O)N(p-F-C6H4)〛 (4a). In this case, the precipitate contains always a small amount of starting complex 3a and recrystallisation is required. Isolated yield: 30%. Colour: red. Infrared (Nujol/KBr, cm–1): 1634 (s, CO). NMR 1H (200 MHz, C6D5NO2, 45 °C, δ, ppm): 8.50 (m, 4 H), 8.02 (s, 2 H), 7.96 (dd, J1 = 5 Hz, J2 = 8 Hz, 2 H), 7.79 (dd, JHH = 8 Hz, JHF = 6 Hz, 4 H), 7.22 (t, JHH = JHF = 8 Hz, 4 H).
(o-phen)Pd{N〚p-CH3–(C6H4)〛C(O)N〚p-CH3–(C6H4)〛} (4b). Isolated yield: 50%. Colour: red. Melting point: 260 ± 10 °C (dec.). Elemental analysis, found: C, 60.13; H, 3.91; N, 10.23; C27H22N4O1Pd requires: C, 61.78; H, 4.22; N, 10.67. Infrared (Nujol/KBr, cm–1): 1625 (s, CO). NMR 1H (200 MHz, CD2Cl2, δ, ppm): 8.50 (d, J = 8 Hz, 2 H), 8.39 (d, J = 5 Hz, 2 H), 7.97 (s, 2 H), 7.66 (dd, J1 = 5 Hz, J2 = 8 Hz, 2 H), 7.37 (d, J = 8 Hz, 4 H), 7.13 (d, J = 8 Hz, 4 H); 2.31 (s, 6 H, –CH3).
2.5.2 Synthesis of 4c from 1
900 mg of 2,6-di(iso-propyl)phenyl isocyanate (ca. 4.40 mmol) are added to 50 mg of 1 (0.11 mmol) suspended in 5 ml of nitrobenzene and stirred at 142 °C. After 15 h, the medium is allowed to cool and 1 ml ethanol (>> 40 equiv.), followed by 200 ml diethylether are added. The resulting suspension is then kept at –20 °C for several hours. The pure compound 4c that precipitates as an air stable purple solid is filtrated, washed with small fractions of diethylether and dried under vacuum (47 mg, 60%).
(o-phen)Pd{N〚bis-o-CH(CH3)2–(C6H3)〛C(O)N〚bis-o-CH(CH3)2–(C6H3)〛} (4c). Colour: purple. Elemental analysis, found: C, 68.18; H, 6.44; N, 7.96; C37H42N4O1Pd.1/2C6H6 requires: C, 68.22; H, 6.54; N, 7.98. Infrared (Nujol/KBr, cm–1): 1590 (s, CO). NMR 1H (200 MHz, C6D5NO2, 45 °C, δ, ppm): 8.45 (d, J = 8 Hz, 2 H), 7.95 (s, 2 H), 7.71–7.62 (m, 4 H), 7.28–7.16 (m, 6 H), 4.04 (septuplet, J = 7 Hz, –CH(CH3)2, 4 H), 1.29 (d, J = 7 Hz, 12 H, –CH(CH3)2), 1.20 (d, J = 7 Hz, 12 H, –CH(CH3)2).
2.5.3 Preparation of 4 from dilithium salt
Preparation of the dilithium salt ‘〚LiN(C6H5)〛2CO’. Under a nitrogen atmosphere, 2.050 g of a solution of butyl lithium in heptane (commercial solution: 1.6 M, d = 0.680; 2.1 equiv) diluted in 5 ml of diethylether are added dropwise to 500 mg of N,N’-diphenylurea (2.35 mmol) in 80 ml of diethylether at ambient temperature. During the addition, the initial suspension becomes homogeneous and translucid (after 10 min), followed by precipitation of the desired dilithium salt (8) as a white product. After 3 h, the diethylether is evaporated and the dilithium salt 8 is dried in vacuo (ca. 100%).
‘〚LiN(C6H5)〛2CO’ (8). Colour: white. Elemental analysis, found: C, 66.60; H, 6.60; N, 8.73; C13H10Li2N2O1.C4H10O requires: C, 68.47; H, 6.76; N, 9.39. Infrared (KBr windows, cm–1): 3040 (w, C–H), 1585 (s, C=O), 1555 (m, C=N phen). FABMS (negative mode, diglyme, m/z): 226 (M+2), 217 (M-Li), 211 (M+1–2Li), 127 (M+1–Li–PhN).
Synthesis of palladacycle 4. In 30 ml of diethylether, 300 mg of (o-phen)Pd(OAc)2 (5c; 0.74 mmol) and 180 mg of 8 (0.78 mmol) are suspended and stirred at ambient temperature. The initially yellow suspension turns slowly red. After five days, the solvent is evaporated and the precipitate, containing mainly 4 (≥ 75% by 1H–NMR), can be collected. Recrystallisation from dichloromethane/diethylether affords the palladacycle 4 in a crystalline form (red needles). Yield 60%.
2.6 Reactions of 4
2.6.1 With carbon monoxide
20 mg of 4 (0.04 mmol) are suspended in 10 ml of dichloromethane under CO (40 bar). The reactor is depressurised after 15 h and 30 ml of diethylether are added to the yellow suspension. The precipitate (2) is then collected, washed with diethylether and dried under vacuum (18 mg; 85%).
2.6.2 With aromatic isocyanates
4 and phenyl isocyanate. 250 mg of phenyl isocyanate (ca. 2.00 mmol.) solubilised in 15 ml of dichloromethane are added to 20 mg of 4 (0.04 mmol), and the reaction mixture is stirred at ambient temperature. Over 15 h, the red suspension becomes orange. Following addition of 50 ml of diethylether, 3c can be collected, washed by ether and dried in vacuo (20 mg; 81%).
4 and para–tolyl isocyanate. When exactly the same procedure is followed starting from 25 mg of complex 4 (0.05 mmol) and 10 mg of para-tolyl isocyanate (1.5 equiv.) over 3 h, 28 mg of a new compound 3*, analogue of 3, can be isolated (88%).
(o-phen)Pd{N〚p-CH3(C6H4)〛C(O)N(C6H5)C(O)N(C6H5)} (3*). Colour: yellow. Melting point: 233 ± 2 °C (dec.). Elemental analysis, found: C, 63.11; H, 3.97; N, 11.33; C33H25N5O2Pd requires: C, 62.92; H, 4.00; N, 11.12. Infrared (KBr windows, cm–1): 1645 (s, C=O), 1585 (sh, C=O). NMR 1H (200 MHz, CD2Cl2, δ, ppm): 8.47 (m, 4 H), 8.32 (d, J = 8 Hz, 2 H), 8.24 (d, J = 5 Hz, 1 H), 8.20 (d, J = 5 Hz, 1 H), 7.97 (s, 2 H), 7.59 (dd, J1 = 5 Hz, J2 = 8 Hz, 1 H), 7.55 (dd, J1 = 5 Hz, J2 = 8 Hz, 1 H), 7.45 (d, J = 7 Hz, 2 H), 7.34 (t, J = 7 Hz, 2 H), 7.26 (m, 3 H), 7.05 (d, J = 8 Hz, 2 H), 6.96 (t, J = 7 Hz, 1 H), 2.29 (s, 3 H, –CH3). FABMS (positive mode, m-nitrobenzyl alcohol, m/z): 630 (M+1), 511 (M+1–PhNCO), 497 (M+1–TolNCO), 392 (M+1–2PhNCO), 377 (M+1–TolNCO–PhNCO), 286 (M-TolNCO–PhNCO–NPh).
2.6.3 With iso-propyl isocyanate
A solution of 210 mg of iso-propyl isocyanate (2.50 mmol.) in 5 ml of dichloromethane is added to 25 mg of 4 (0.05 mmol). The reaction is stirred as previously described during 3 days and 27 mg (92%) of the corresponding analogue 3# can be isolated following a similar workup. However, this palladacycle is not stable in CD2Cl2 solution at ambient temperature and transforms into a mixture of unidentified compounds, containing presumably other isomeric palladacycles.
(o-phen)Pd{N〚CH(CH3)2〛C(O)N(C6H5)C(O)N(C6H5)} (3#). Colour: yellow. Melting point: 180 ± 10 °C (dec.). Elemental analysis, found: C, 58.28; H, 3.85; N, 10.21; C29H25N5O2Pd requires: C, 59.85; H, 4.33; N, 12.03. Infrared (KBr windows, cm–1): 3045 (s, C–H phen), 1630 (vs, C=O), 1600 (vs, C=O). NMR 1H (200 MHz, CD2Cl2, δ, ppm): 9.23 (d, J = 5 Hz, 1 H), 8.84 (d, J = 7 Hz, 1 H), 8.47 (d, J = 7 Hz, 1 H), 8.34 (d, J = 8 Hz, 2 H), 8.07 (d, J = 5 Hz, 1 H), 8.03 (s, 1 H), 8.01 (s, 1 H), 7.97 (dd, J1 = 5 Hz, J2 = 8 Hz, 1 H), 7,53 (dd, J1 = 5 Hz, J2 = 8 Hz, 1 H), 7.47 (d, J = 7 Hz, 2 H), 7.30 (t, J = 7 Hz, 2 H), 7.19 (m, 3 H), 6.91 (t, J = 7 Hz, 2 H), 3.91 (septuplet, J = 6 Hz, 1 H, -CH(CH3)2), 1.53 (d, J = 6 Hz, 6 H, –CH(CH3)2). FABMS (positive mode, m-nitrobenzyl alcohol, m/z): 582 (M+1), 496 (M–1–iPrNCO), 463 (M+1–PhNCO), 377 (M–1–iPrNCO-PhNCO), 344 (M+1–2PhNCO), 286 (M–iPrNCO–PhNCO–NPh).
2.6.4 With acid
HCl in excess. An excess of hydrochloric acid (37%) was added to a solution of 3 (10 mg) in CD2Cl2 in an NMR tube. The comparison of the 1H–NMR spectra before and after addition reveals a quantitative conversion of the starting complex into free N,N’-diphenylurea and (o-phen)PdCl2 (5a), which precipitates in the medium.
Carboxylic acid in excess. Under similar conditions, 4 is reacted with an excess of acetic or 2,3,5,6-tetramethylbenzoïc acid (>20 equiv.). After addition of the acid, 1H–NMR reveals that a new fluxional species (6), presenting a symmetric o-phen ligand, has formed in the homogeneous medium. This reaction is quantitative (100% in 4) and takes place in less than 1 h. A tentative ‘〚(o-phen)Pd(DPU)〛(O2CC10H13)2〛’ composition can be proposed for 6 (§ 3.2.4.).
1 equiv. TMBA; isolation of 7. When 2.4 mg of 2,3,5,6-tetramethylbenzoïc acid (1 equiv.) are added to a solution of 4 (6.6 mg) in 3 ml deuterated dichloromethane at ambient temperature, a yellow precipitate deposits and the red solution turns orange. 1H–NMR spectrum of the filtrate indicates the presence of a mixture (ca. 3-1) of the symmetric species 〚(o-phen)Pd(DPU)〛(O2CC10H13)2〛 (6) previously observed along with a new non-symmetric compound 7, which also constitutes the yellow precipitate (8 mg; 77%), once collected, washed with diethylether and dried in vacuo.
(o-phen)Pd〚O2C(C10H13)〛〚N(C6H5)C(O)NH(C6H5)〛 (7). Colour: yellow. Elemental analysis, found: C, 63.86; H, 4.65; C36H32N4O3Pd requires: C, 64.05; H, 4.78. Infrared (KBr windows, cm–1): 3210 (w, N–H), 1635 (vs, C=O), 1585 (vs, C=O). NMR 1H (200 MHz, CD2Cl2, δ, ppm): 10.56 (s, 1 H, NH), 8.89 (m, 2 H), 8.61 (m, 2 H), 8.12 (d, J = 7 Hz, 2 H), 8.04 (s, 2 H), 7.91 (dd, J1 = 7 Hz, J2 = 5 Hz, 2 H), 7.54 (d, J = 7 Hz, 2 H), 7.23-7.06 (m, 4 H), 6.88–6.79 (m, 3 H), 1.56 (broad s, 12 H, all-CH3).
Excess of TMBA. When the above reaction is repeated with an excess (4–5 equiv.) of 2,3,5,6-tetramethylbenzoic acid, 1H–NMR monitoring of the reaction reveals that 7 transiently forms before being converted into 6. In this case, the medium stays homogeneous throughout the reaction.
2.7 X–ray structural analysis of 3c·Et2O
Single crystals of 3c·Et2O suitable for X-ray studies were obtained by slow diffusion of diethylether in a dichloromethane solution of the complex at room temperature. Data were collected on a Philips PW1100/16 diffractometer at –100°C using Cu Kα graphite monochromatic radiation (λ = 1.5418 Å), θ/2 θ flying step-scan mode. Table 1 reports all pertinent crystallographic data for the complex. Absorption corrections were applied using the method of Stuart 〚25〛, since face indexation was not possible under the cold gas stream. The structure was solved using the heavy atom method and refined against . All non-hydrogen atoms were refined anisotropically. The hydrogen atoms were introduced as fixed contributors (dC–H = 0.95 Å, BH = 1.3 Beqv(C) Å2). Final results are listed in Table 1. For all computations, the OpenMoleN package was used 〚26〛. Data for this structure have been deposited within the Cambridge Crystallographic Data Centre as Supplementary publication n° CCDC-182799. Copies of the data can be obtained free of charge on application to CCDC, 12 Union Road, Cambridge CB2 1EZ, UK (fax: (+ 44) 1223-336-033; e-mail: deposit@ccdc.cam. ac.uk).
Crystallographic data for complex 3c·C4H10O.
| Crystal data | 3c |
| Formula | C35H29N5O2Pd2·C4H10O |
| fw | 732.18 |
| Crystal system | monoclinic |
| Space group | P21/n |
| a 〚Å〛 | 20.172(6) |
| b 〚Å〛 | 12.922(3) |
| c 〚Å〛 | 12.797(3) |
| β 〚deg〛 | 100.67(2) |
| V〚Å–3〛 | 3278 |
| Z | 4 |
| Colour | yellow |
| Crystal dimensions (mm) | 0.20 × 0.15 × 0.10 |
| Dcalcd (g cm–3) | 1.48 |
| Data collection | |
| F(000) | 1512 |
| μ(Mo Kα) 〚cm–1〛 | 49.43 |
| Trans. min and max | 0.683/1.000 |
| T 〚K〛 | 173 |
| Wavelength 〚Å〛 | 1.54184 |
| Radiation | Cu Kα graphite monochromated |
| Diffractometer | Philips PW1100 |
| Scan mode | ϴ/2ϴ flying step scan |
| hkl limits | –21,20/0,13/0,13 |
| θ limits (°) | 3.0/54.07 |
| Number of data measured | 4407 |
| Number of data with I > 3 σ(I) | 3542 |
| Structure refinement | |
| Number of variables | 428 |
| Final R | 0.045 |
| Rw | 0.075 |
| Goodness of fit/F2 | 1.798 |
| Largest diffraction peak and hole (e Å–3) | 1.160 |
3 Results
3.1 Pd (II) Metallacycles isolated from nitroaromatics
When the nitrobenzene substrate is used as solvent in place of ethyl alcohol, the repetition of the experimental protocol that allowed the isolation of 1 leads to a mixture of complexes 〚7, 15〛. Compound 1 is still formed along with a new palladacycle 2 (equation (3a)) 〚21〛. The latter could be structurally characterised and presents a five-member metallacyclic core resembling that of 1, but where the carboxyl fragment has been replaced by a phenylisocyanato fragment (Fig. 2). Monitoring the concentrations of 1 and 2 reveals that 1 is the first organometallic species formed in the medium. However, at the very beginning of the catalysis, its concentration decreases with the appearance of 2, and reaches a 40–60% ratio after 16 h (Fig. 3).
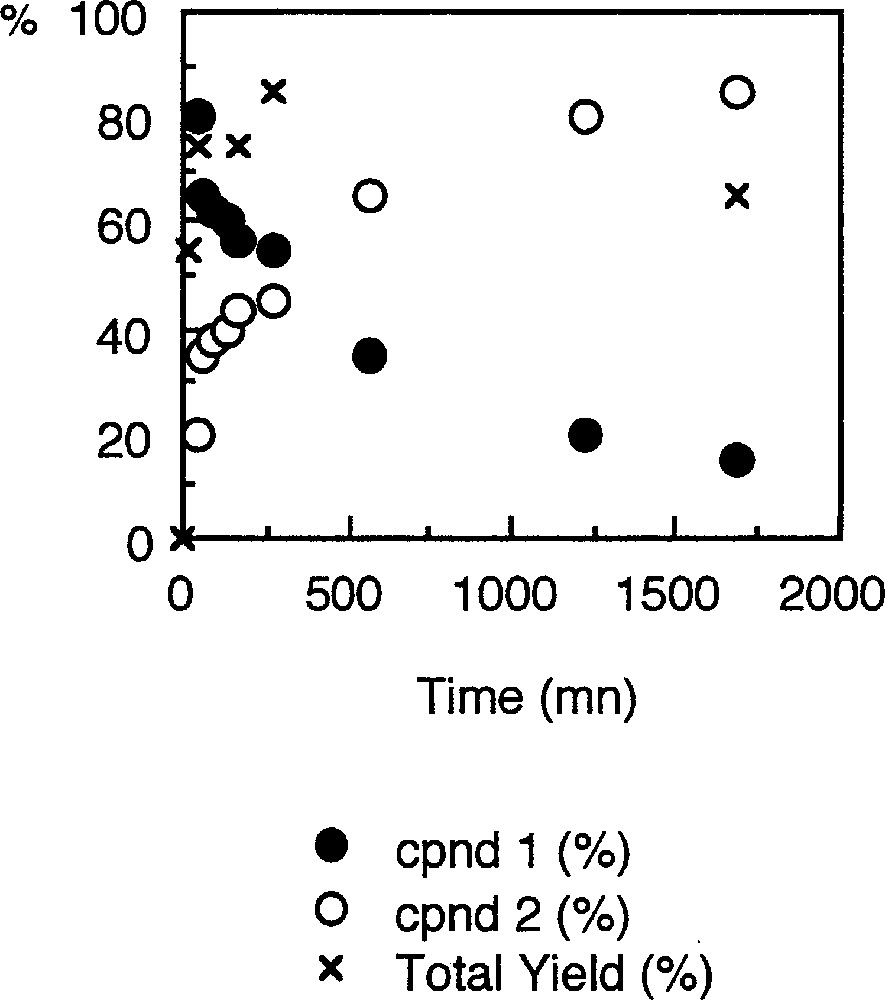
Concentrations of palladacycles 1 and 2, and total yield isolated expressed in percent (%) of the initial Pd concentration introduced in the reactor.
In contrast to 1, complex 2 appears stable in presence of carboxylic acids like 2,3,5,6,-tetramethyl benzoic (TMBA) 〚21〛 – notably, 2 did not react with alcohol or TMBA at temperatures as high as 160 °C, even in presence of carbon monoxide. Using this property, we could devise a synthetic route for the selective isolation of complex 2. Thus, repetition of the reaction in the presence of an excess of carboxylic acid like TMBA allows the isolation of pure 2 in 2 h (equation (3b)). This route was also used to isolate substituted analogues, such as 2a–2e, in 10–60% yield starting from the corresponding nitroaromatic substrate. The palladacycles 2a–2e have been characterised by elemental analysis and also by FAB–MS in some cases. The presence of only one isomer of structure similar to 2 is clearly indicated by the spectroscopic data. As discussed previously 〚15〛, the 1H–NMR downfield shifts of either the 2- or 9-protons of the coordinated o-phen ligand constitute a reliable signature of carbonyl groups located in α-position to the metal centre within five-member metallacycles. For 2/2a–e, the large difference between these shifts can be attributed to the existence of two complementary phenomena; (i) an intramolecular H···O interaction with the oxygen atom of the α-Pd carbonyl group of the metallacycle, deshielding one ortho-phenanthroline proton of the o-phen ligand and (ii) the magnetic ring current of the phenyl ring perpendicular to the metallacycle plane, shielding the symmetric ortho-phenanthroline proton on the other side. These two interactions are also apparent in the solid-state structure of 2 (see supplementary material) 〚21〛.
3.2 Pd(II) Metallacycles isolated from aromatics isocyanates
3.2.1 Six–member metallacycles 3 and 3a–c
In order to check if 2 could be formed by direct substitution of the carboxyl fragment from 1 with isocyanate, we investigated the direct reaction of 1 with PhNCO. A colour change from yellow to red indicated that a reaction was occurring with excess phenyl isocyanate at 140 °C. The palladacycle 1 reacts quantitatively to give a new orange complex 3, for which a six-member ring metallacyclic structure was suggested by the elemental analysis, FAB–MS, infrared and 1H–NMR data (equation (4)) 〚21〛. The temperature control for this reaction is particularly critical, as heating above 145 °C leads to extensive decomposition of 3.
In several instances, however, there was evidence for the formation of the five-member palladacycle 2 in reactions between 1 and PhNCO. Thus, when only 2 equiv. of phenyl isocyanate are reacted, instead of 40 (equation (5a)), 2 is formed (ca. 20%) along with 3 (ca. 40%). This reaction is not clean, and significant amounts of metallic palladium are deposited. Alternatively, when the reaction of 1 with excess PhNCO is conducted under 40 bar of carbon monoxide, 2 (82%) is the main palladacycle formed, along with palladium metal (equation (5b)). Interestingly, the course of the reaction (4) is not markedly affected by an atmospheric carbon monoxide pressure in comparison to carbon monoxide-free conditions. Under such conditions, 3 remains the only complex isolated (75%), apart from metallic palladium. Thus, either a low isocyanate concentration or a high pressure of carbon monoxide appears to favour the formation of 2 over 3, in the reaction between 1 and PhNCO. Finally, we have also checked whether 2 would be converted to 3 by PhNCO. However, 2 does not react with excess phenylisocyanate, not even under forcing conditions (equation (5c)). Obviously, 2 is not an intermediate ‘en route’ to the formation of 3 in the reaction between 1 and PhNCO.
Six-member palladacycles like 3a–c can be isolated in fair yields, similarly to 3, from the corresponding isocyanates (equation (6a)). A similar symmetric six-member ring structure is proposed for these compounds, based on the spectroscopic data and on the solid-state structure established for 3c (Fig. 2). While the corresponding analogue 3c is obtained as the sole product with the para-tolyl isocyanate, five-centre palladacycles similar to the previously characterised complexes 2a–e were also isolated as side-products with aryl isocyanates bearing electron-withdrawing substituents.
With a sterically crowded isocyanate like the bis-2,5–(iso-propyl)phenyl, the reaction with 1 takes again a different course (equation (6b)). This time, in spite of the large excess of isocyanate present, a four-member metallacycle 4c is isolated (see § 3.2.3.). In that case, steric congestion presumably prevents the formation of the expected six-member palladacycle.
Finally, when the reaction with para-tolyl isocyanate is repeated starting from the bis-3,5-CF3-labelled analogue 1a instead of 1 (equation (7)), mixtures of several five- (2d*) and six-member (3c*, 3c**) palladacycles are isolated by fractional precipitation, as shown by 19F-NMR, 1H–NMR and FAB-MS spectroscopies. This shows that, under favourable circumstances, the isocyanate fragment initially present in the five-member metallacycle of the reactant can be incorporated into the metallacyclic core of the complexes formed during the reaction. Notably, in all the reactions conducted with functional isocyanates, the absence of any metallacycle featuring a N-phenyl substituent indicates that the nitrobenzene used as solvent does not actually act as a source of phenyl isocyanate. This makes sense, considering that carbon monoxide was also absent in all of these reactions. The latter usually constitutes the reducing agent needed for such reactions to take place with nitroaromatics 〚1〛.
3.2.2 Reactions of 3 with CO, ArNCO and H+
The reactivity of the palladacycle 3 with carbon monoxide, aryl isocyanate and protons was then investigated. When heated under 1 bar carbon monoxide, 3 is converted to 2 and palladium metal within 6 h (equation (8a)). The reaction is even faster and cleaner with a higher CO pressure (see experimental part, § 2.4.1.). When heated with para-tolyl isocyanate, 3 undergoes an exchange reaction of the phenyl groups of the metallacycle for para-tolyl groups (equation (8b)). The converse transformation of 3c into 3 can also be achieved under similar conditions. Finally, 3 reacts readily with an excess of Brönsted acid like TMBA or HCl, to form N,N’,N”-triphenylbiuret (TPB) and the corresponding palladium(II) salt 5a or 5b (equation (8c)). Compound 3 appears much more reactive than 2 with these three families of reactants. However, heating was nevertheless required for the reactions with carbon monoxide or isocyanate, since no reactions could be observed at room temperature. This observation led us to investigate the thermal stability of this metallacycle.
3.2.3 Four-member metallacycles 4 and 4b–c
When heated in ortho-dichlorobenzene at 140 °C under an inert atmosphere, 3 gradually solubilises to give a deep red solution. Upon prolonged heating, the solution turns yellow and deposits metallic palladium after a few hours, showing that 3 is not stable at this temperature. If a large excess of diethyl ether is added to the red solution immediately after heating, a brick red complex 4 precipitates and can be isolated (Fig. 2). This complex, which can be purified by recrystallisation, was structurally characterised and identified as a four-member metallacycle 〚21〛. Two analogues of the complex could similarly be isolated starting from the corresponding precursor complexes 3b–c (equation (9)). Based on the spectroscopic data, their structure appears to be identical to that of 4. A symmetric four-member palladacycle structure featuring aryl groups in α-positions to the metal is suggested by the unique and quite low carbonyl stretch around 1640 cm–1 along with the same 1H–NMR signal for the 2- and 9-protons of the phenanthroline ligand.
The existence of an equilibrium between the four- and six-member metallacycles (equation (9)) can be demonstrated by 1H–NMR variable temperature studies (see supplementary material). Heating produces a change in the equilibrium toward the right with concomitant extrusion of one equivalent isocyanate from the six-member metallacycle. Traces of water in the solvents possibly contribute to the clean isolation of 4 and 4a–b previously described, since water will react with the liberated PhNCO to form aniline and CO2 and thereby avoiding a quantitative reversal of the equilibrium upon cooling.
3.2.4 Reactions of 4 with CO, ArNCO or iPrNCO and H+
Again, the reactivity of 4 with carbon monoxide, isocyanate or Brönsted acids was tested. Upon exposure to one atmosphere carbon monoxide, 4 is quantitatively converted into 2 at ambient temperature (equation (10a)). Application of a larger CO-pressure leads to the same result. In accordance with the equilibrium situation previously presented (equation (9)), 4 reacts also at 25 °C with excess phenyl isocyanate to regenerate the starting complex 3 (equation (10b)). A non-symmetrical analogue of 3 can also be formed when 4 is reacted with a stoichiometric amount of para-tolyl isocyanate (equation (10c)). A palladacycle of similar structure is also formed when 4 is reacted with excess iso-propyl isocyanate (equation (10d)). For both reactions, the spectroscopic data indicate the presence of only one isomer (respectively 3* and 3#). The loss of planar symmetry in 1H–NMR reveals a palladacycle with a different isocyanate fragment incorporated at the α-position to the metal centre. Complexes 3* and 3# were also characterised by FAB–MS spectrometry. In comparison, complex 4 appears more reactive than 3 with carbon monoxide or isocyanate, since these reactions require no heating.
Finally, 4 reacts readily with excess hydrochloric acid to form N,N’-diphenylurea (DPU) and the corresponding palladium(II) salt 5a, which precipitates spontaneously from the reaction mixture (equation (11a)). However, a slightly different behaviour is observed with acetic acid. The initially red medium also turns orange, but stays homogeneous. 1H–NMR reveals a unique symmetric species (6) present in solution, distinct from 5c, but resembling a DPU-adduct of 5c. DPU must be quite labile in this adduct, as attempts to precipitate 6 only afforded mixtures of free DPU and 5c (equation (11b); § 3.4.). In that respect, the clean formation of 5a observed with hydrochloric acid might be attributed to the weak solubility of this complex in the reaction medium. With TMBA, we have also investigated the reaction of 4 with 1 equiv. of carboxylic acid. In that case, a non-symmetric complex 7 is formed and was identified by 1H–NMR (equation (11c)). Complex 7 precipitates spontaneously from the solution and can be isolated as a yellow powder. It was further characterised by infrared and elemental analysis. Upon further addition of TMBA, this compound re-dissolves, giving a labile species analogous to 6. Obviously, 7 can be envisioned as an intermediate in the protonation process of 4 with TMBA.
3.3 Crystal structure of 3c·Et2O
Suitable crystals of 3c could be obtained by slow diffusion of diethylether into a dichloromethane solution of the complex. Complex 3c·Et2O crystallised in the P–1 triclinic system with one molecule of diethylether solvate in the asymmetric unit (Table 1). The geometry around each metal centre is square planar, as expected for a Pd(II) centre (Fig. 4). The metallacycle adopts a symmetric boat conformation, with the two-phenyl rings located above the mean plane of the phenanthroline ligand. In contrast to a related Cr(IV) metallacycle 〚27〛, no interaction takes place between N5 and the metal centre (Pd–N5 = 3.027 Å). This suggests that the boat conformation for 3c·Et2O is essentially dictated by the crystal packing, possibly in order to release the strain with the 2- and 9- protons of the phenanthroline ligand. Within the six-member ring, all C–N bond lengths are non-equivalent. The C13–N3 (1.341 Å) and C14–N4 (1.394 Å) distances are significantly shorter than C13–N5 (1.410 Å) and C14–N5 (1.434 Å), indicating a stronger bond order in the former. Similar features have been reported for biuret hydrates 〚28, 29〛. Very similar conformations, bond lengths and angles have been observed for a related nickel(II) metallacycle reported by Hoberg and co-workers 〚30〛. As suggested by these researchers, such a metallacycle can be envisioned as corresponding to a transoid bis-N-chelated Pd(II) complex of the N,N’,N”-triphenyl biuret dianion, for which a very similar arrangement has independently been reported for a Cu(II) centre 〚31〛 (Fig. 4).
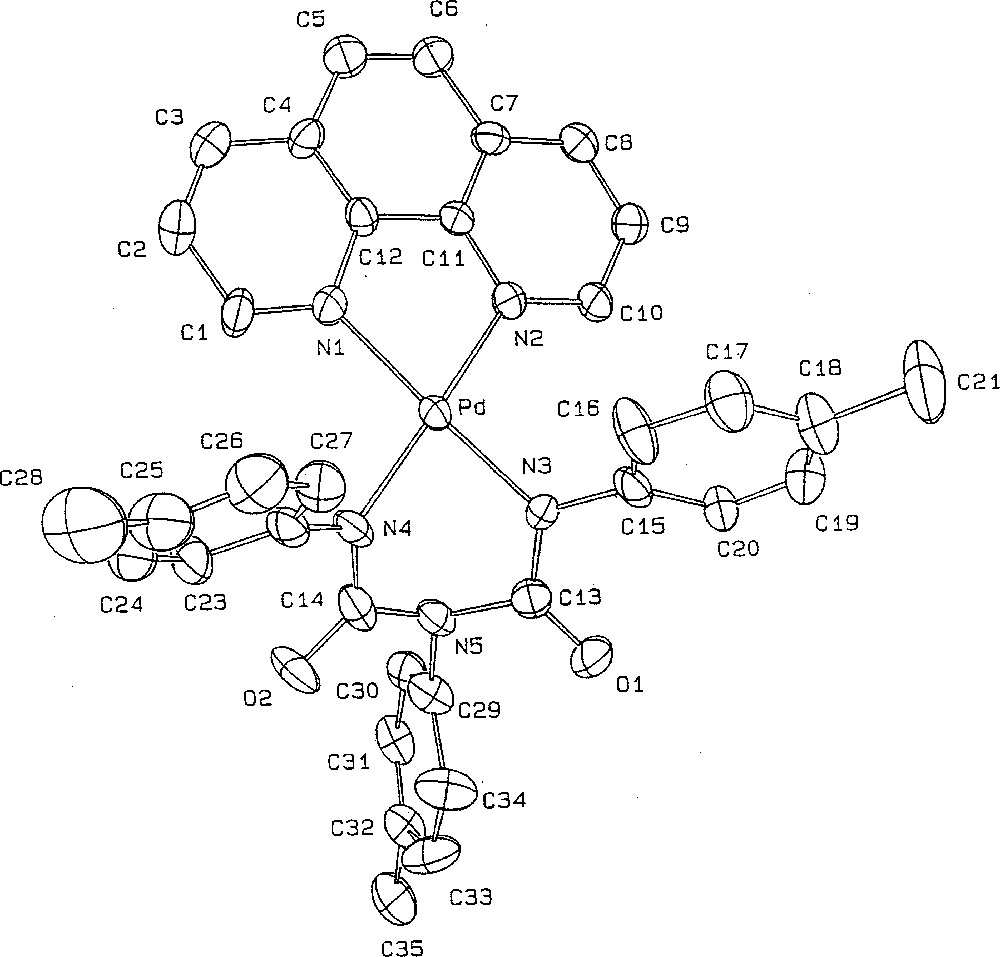
ORTEP drawing of the palladacycle 3c·Et2O. Selected bond distances (Å) and angles (°): Pd–N1, 2.053(4); Pd–N2, 2.046(4); Pd–N3, 1.993(4); Pd–N4, 1.988(4); O1–C13, 1.225(6); O2–C14, 1.227(7); N4–C14, 1.349(7); N5–C14, 1.410(7); N5–C13, 1.434(7); C13–N3, 1.341(6); N3–C15, 1.413(7); N5–C29, 1.440(7); N4–C22, 1.420(7). Bond: N1–Pd–N2, 80.4(2); N1–Pd–N4, 98.0(2); N3–Pd–N4, 85.1(2); N2–Pd–N4, 85.0(2); Pd–N4–C14, 118.8(4); N4–C14–N5, 116.5(5), C13–N5–C14, 122.6(2); N3–C13–N5, 115.3(4); Pd–N3–C13, 121.4(3).
The square planar geometry around the metal and the present Pd–N bond distances for 3c·Et2O compare also quite well to these previously reported for 4·CH2Cl2, however the N3–Pd–N4 angle of the metallacycle was more acute in the latter (65.6° vs 85.1°) 〚21〛. In 4·CH2Cl2, the intra-cyclic C–N (ca. 1.37 Å) bonds were intermediate between the short and long C–N distances in the present metallacycle 3c·Et2O, while the C–O bond length was quite similar (ca. 1.24 Å). Notably, these bond lengths are in the range observed for related W(II), Ru(II) or Os(II) metallacycles 〚32, 33〛. Likewise to 3c·Et2O, 4·CH2Cl2 can also be envisioned as an urylene dianion chelating the metal centre 〚34〛. The metal centre actually seems to only slightly disturb the bonding within the coordinated dianion, as suggested by comparison with dinuclear complexes in which the urylene dianion adopts a bridging conformation between two metal centres 〚35–37〛. Oppositely to 4·CH2Cl2, the aryl rings in 3c·Et2O can adopt a conformation not strictly perpendicular to the metallacyclic rings. These different conformations observed in the solid state are apparently not maintained in solution, as suggested by the observation of very close 1H–NMR shifts for the 2- and 9- protons of the phenanthroline ligand in 3 and 4.
3.4 Pd(II) metallacycles isolated from triphenylbiuret or urylene dianion
Since both palladacycles 3 and 4 can be envisioned as Pd(II) complexes of TPB and DPU N,N’-dianions respectively, we have tried to synthesise these complexes starting from a Pd(II) precursor and the corresponding dianions.
Accordingly, when triethylamine is used to generate the desired dianion in situ, 3 can be quantitatively isolated starting from (o-phen)Pd(OAc)2 (5c) and TBP (equation (12a)). Alternatively, 3 can also be isolated without triethylamine, when (o-phen)Pd(OAc)2 is heated in the presence of TPB (equation (12b)). In that case, the acetate ligand probably plays the role of the base in the process.
In contrast, 4 cannot be isolated using similar conditions starting from DPU. Despite the colour change of the solution from yellow to orange, only mixtures of the starting complex 5c and DPU were recovered by precipitation, even after addition of excess triethylamine. However, another reaction does take place, as the NMR spectra of the medium reveal the presence of a fluxional species (6) in solution (see § 3.2.4.). We have also tried to isolate 4 from the corresponding urylene dilithium salt 8. The latter can simply be generated by reaction of DPU with two equivalents of butyl lithium in diethylether (equation (13a)). Reaction of this dilithium salt and (o-phen)Pd(OAc)2 (5c) leads to the isolation of the expected palladacycle 4 in good yields (75%, equation (13b)).
The air-sensitive dilithium salt 8 was also isolated and characterised by negative FAB–MS. The shift of the carbonyl vibration to lower wavenumbers (1590 cm–1) suggests a strong delocalisation of the charge in this dianion 〚38, 39〛.
4 Discussion
4.1 Interconnections between the metallacycles 1, 2, 3 and 4
The present work has demonstrated a rich chemistry of 1 and of the related palladacycles 2–4 with CO, isocyanate or protons, and also revealed several chemical interconnections between these families of compounds. In contrast to 1, which was a completely new metallacycle, hetero-metallacycles like 2, 3 and 4 are not unprecedented regarding their structures and reactivity 〚32, 33〛, especially with metals belonging to the Ni triad 〚30, 34, 40, 41〛. Five- and six-member nickellacycles, analogous to 2 and 3 respectively, have been isolated after reaction between Ni(0) precursors and phenyl isocyanate by Hoberg and co-workers 〚30, 40〛, and four-member metallacycles analogous to 4 have been isolated after reaction of Pd(0) or Pt(0) complexes by Beck. Related four-member platinacycles were isolated from aryl-phosphaketenes and Pt(0) or Pd(0) precursors 〚42, 43〛.
Lately, such palladacycles were also isolated from urea and Pt(II) precursors 〚34〛. Each time, the formation of hetero-metallacycles like 2–4 from isocyanate was proposed to be initiated by low-valent metal complexes and to involve intermediates like (A)–(D) 〚30, 41〛 (Fig. 5).
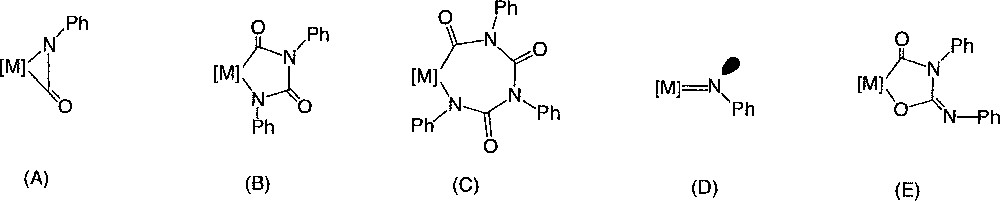
Intermediates involved in the formation, initiated by low-valent metal complexes, of hetero-metallacycles like 2–4 from isocyanate.
It appears likely that in all reactions involving 1 and aryl isocyanate where metallacycles like 2–4 were isolated as products, 1 acted as a source of Pd(0) and PhNCO upon heating, in accordance with its postulated role as intermediate in the catalytic carbonylation mechanism (Fig. 1) 〚7, 15〛. Thus, in the absence of trapping agents like ethanol, we propose that 1 does generate a π-isocyanato complex, like 9, by thermal decarboxylation (Fig. 6). Such isocyanato complexes similar to (A) have been isolated with Ni(0) complexes 〚44, 45〛. The existence of an intermediate like 9 would be in line with the formation of mixed metallacycles observed when the reaction is conducted from 1a (equation (7)). In the presence of an excess of phenyl isocyanate, 9 would further react to generate the intermediates 10 and 11, featuring two and three isocyanate molecules in their structure. The latter (11) would then generate the six-member palladacycle 3 upon decarbonylation, in equilibrium with the four-member palladacycle 4 (equation (9)). The exact structure of the two intermediates 10 and 11 is presently not known.
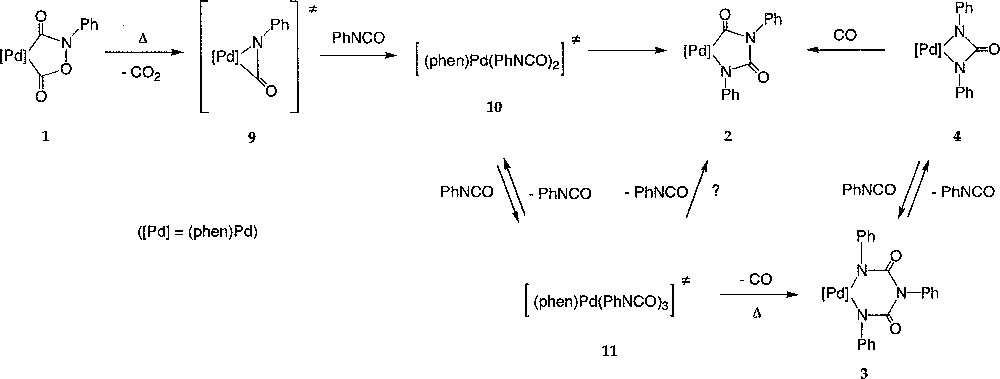
Generation of a π-isocyanato complex, like 9, by thermal decarboxylation.
A seven-member ring structure analogous to (C) appears likely for 11 considering the mechanistic proposal of Hoberg et al. 〚30〛. Decarbonylation of such an intermediate to generate 3 would be favoured by the large size of the cycle. Such a palladacycle must be of lower stability than 2, 3 and 4, since it was never isolated nor observed in carbonylation reactions of 3 (equation (8a)), or in the reaction of 2 with phenylisocyanate (equation (5c)). Regarding the structure of 10, our results clearly indicate that this intermediate is different from 2 (equation (5c)). The latter apparently consists of a stable palladacycle that does not react anymore with isocyanate. An isomeric structure like (E) might be put forward for 10 〚46〛. The latter (E) would then result from coupling of η2-CO and η2-NC coordinated isocyanates. Interconversion between the η2-CO and the η2-NC coordination mode is expected to be easy, considering calculations on the isolobal Ni(0) ketene model complexes 〚47〛. In that case, 2 would be simply formed by isomerisation of 10. At the present time, such a hypothesis is not fully satisfying, since isomerisation would also be required after further reaction with isocyanate to give the postulated seven-member intermediate 11.
The temperature-dependent equilibrium evidenced between 3 and 4 (equation (9)) nicely explains the reactivity observed for 3 with carbon monoxide (equation (14a)) or aryl isocyanates (equation (14b)). The isolation of 2, 3* and 3# from 4 suggests also that the insertion of any incoming unsaturated molecule in the four-centre metallacycle will essentially take place in α-position to the metal centre. Thus, in order to achieve the complete replacement of the original isocyanate, two successive insertions have to occur on the ‘same side’. This helps understanding the need of an excess of isocyanate to observe a clean exchange of all original isocyanate fragments in 3.
4.2 Protonation reactions of palladacycles 3 and 4
While 2 appears insensitive to the presence of acid, 3 and 4 react readily with protons. In contrast to 1 〚7〛, the reaction does not apparently lead to a multiple fragmentation of the metallacycle, but TPB or DPU are quantitatively formed from 3 and 4, respectively. Similar protonolysis reactions leading to the formation of TPB or DPU have already been reported for metallacycles analogous to 3 or 4 with Ni and Pd/Pt, respectively 〚30, 41〛. Interestingly, DPU was also obtained from the five-member nickellacycle analogue of 2, which appears far less reactive among the palladium complexes. The isolation of the non-symmetric complex 7 (11c) from TMBA and 4 suggests that these deprotonation reactions occur in a stepwise fashion.
The syntheses of 3 and 4 starting from (o-phen)Pd(OAc)2 (5c) and the corresponding dianions correspond to the reverse reactions. Taken together, these also provide information on the protonolysis mechanism. The failure to isolate 4 from DPU and 5c, while this reaction worked readily from the N,N’-diphenyl-urylene dianion 8 (equation (13b)), indicates that the acetate ligand or the triethylamine are not basic enough to deprotonate the coordinated urea. Their successful use as bases with TPB and 5c (equation (12a–b)) can be explained by the lower basicity of the amide hydrogens in TPB complexes. This makes sense considering the characterised modes of coordination of biurets 〚48–50〛 or ureas 〚51–53〛 on metal centres. A chelating conformation should be adopted by TPB 〚50〛, while an end-on coordination should be favoured for DPU in the Pd(II) complex initially formed. Typically with platinum metals, the O-coordination (V) of urea is kinetically favoured over the N-coordination (VI), the latter being thermodynamically more stable 〚54–56〛. In the biuret complexes, N,N”-chelation is usually the coordination mode adopted after deprotonation, even when N,N”-complexation was not initially observed 〚31〛. Our results can then easily be rationalised considering a equilibrium between 5a–c, DPU or TPB and the corresponding palladacycles 3 or 4 (Fig. 7). In the absence of external base, the deprotonation of the coordinated amide could also proceed via an inter or intramolecular process involving the counter ions of the positively charged metal centre 〚57〛. Thus, the Lewis-acidic Pd(II) centre would assist the deprotonation of both amides groups for TPB, but not for DPU. As a consequence, the equilibrium is not displaced toward the formation of 4. Instead, a fluxional process sets in, allowing the observation of ‘6’. According to this scheme, this labile intermediate would actually correspond to the kinetic average of signals belonging to 5c and to complexes like (F) and (G). On the other hand, the non-symmetric Pd(II) complex 7, which corresponds to (H), can be isolated after protonating 4 with one equivalent of TMBA (equation (11c)). The poor solubility of 7 undoubtedly favours its clean isolation.
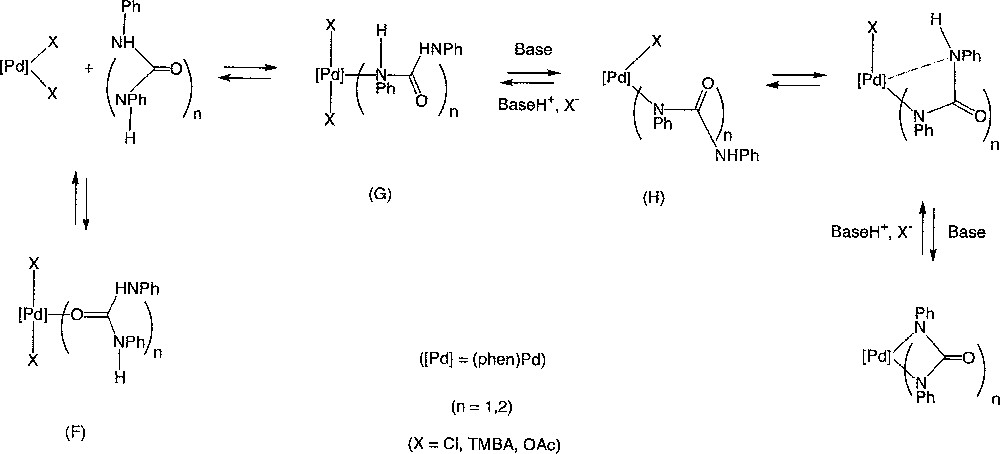
Equilibrium between 5a–c, DPU or TPB and the corresponding palladacycles 3 or 4.
The reactions of DPU or TPB with 5c to give reactive metallacycles like 4 and 3 respectively constitute interesting examples of activation of these organic substrates on a Pd(II) centre. Similar reactions have been reported 〚41〛, and recently a platinacycle similar to 4 could be isolated from a bis-chloro Pt(II) precursor, DPU and silver oxide (Ag2O) in refluxing dichloromethane 〚34〛.
4.3 The influence of CO pressure; connection with the catalytic carbonylation of nitroaromatics in non-alcoholic solvents
Coming back to the reactions leading to the isolation of 2 (equations (3a–b)), it seems now highly probable that 2 is formed competitively to 1 and results from the trapping of an active species of the cycle like (A) by the catalytically generated aryl isocyanate, as indicated by the monitoring of their respective concentrations (Fig. 3). Such an hypothesis is also supported by the changes in the PhNCO concentration (derivatised as (N-ethyl)phenyl carbamate), which amounts to the concentration of the palladium(II) precursor at the very beginning of the reaction (0.045 M) immediately after the start of the reaction and then drops to a steady state level around 0.007 M (see supplementary material). In accordance with the reactivity studies, presented above, of the palladacycles 2, 3 and 4, only 2 is expected to be formed under these conditions (PCO = 40 bar, 80 °C), because of the carbon monoxide pressure. Moreover, 2 appears to be thermally stable and does not react with CO, PhNCO or protons under these conditions. Thus, 2 can be envisioned as a thermodynamic sink for the active species at 80 °C under 40 bar CO and would correspond to a deactivated state of the catalyst, regardless of the presence of a carboxylic acid like TMBA (Fig. 8). Indeed, we know that TMBA, which promotes the catalytic transformation, does not react with 2 at 80 °C. Its presence should therefore only speed up the conversion of 1 into 2. In addition, TMBA will also protonate 3 or 4, if such species are actually formed under these conditions, and generate DPU or TPB as organic side-products. DPU and TPB were detected by GC–MS in many experiments 〚1, 7〛. Notably, such an interpretation remains valid, regardless if 1 is actually part of the catalytic cycle converting the nitroaryl in aryl isocyanate or just a resting state of the active species.
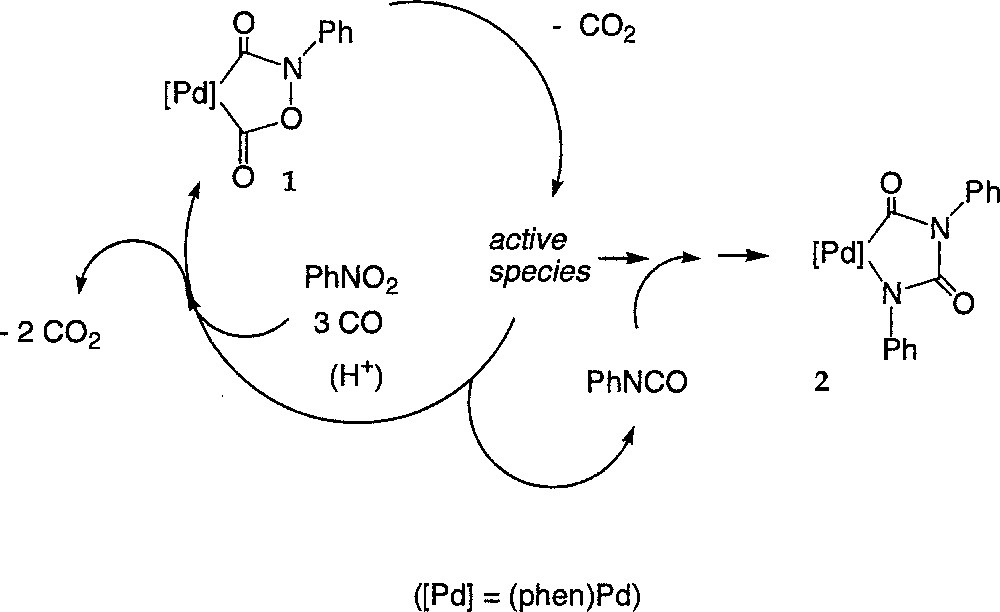
Conversion of 1 into 2.
Whether such a picture is valid under harsher conditions like those traditionally used for catalytic nitroaromatic carbonylations in non-alcoholic media (140–200 °C, 40–60 bar) is still an open question 〚1〛. The poor yields in isocyanate under such conditions are usually attributed to the high chemical reactivity of the formed isocyanate, leading to the formation of polymers or tars 〚1, 58, 59〛. Self-poisoning of the catalyst, as demonstrated by the process under milder conditions, also constitutes a plausible explanation for the lower activity (in non-alcoholic solvents) of otherwise very active catalysts, like the (Pd+/o-phen/H+) system. Accordingly, Cenini and co-workers have reported that when this system is used repetitively in dry benzene with a large ratio of carboxylic acid, the catalyst deactivates progressively 〚8〛. Concomitantly with its deactivation, DPU constituted the most abundant side-product, along with the expected phenyl isocyanate and ‘non-identified’ organic compounds, believed to be ‘polymers of isocyanate’.
5 Conclusion
Several families of four-, five- and six-member new palladacycles were characterised. Their reactivity with small molecules like CO and PhNCO reveals a rich chemistry and provides an almost complete picture of the possible interconversions existing between them. Reversible reactions of 3 and 4 with acids have also been observed. While reactions with H+ generate TPB and DPU respectively in the reaction medium, the converse reactions are also interesting, since they actually correspond to an activation of such organic substrates. More specifically, in the context of the catalytic carbonylation of nitroaromatic effected by the (Pd/o-phen/H+) system, this contribution shows that when the carbonylation is conducted in the absence of an agent able to trap the formed isocyanate at 80 °C, the latter leads to self-poisoning of the catalyst, concomitantly with the formation of palladacycle 2. Such a process might constitute a clue of the unexpected deactivation reported for this catalytic system under harsher conditions, when the carbonylation reaction is conducted in non-alcoholic solvents. Additional studies are now needed to test this hypothesis.
Supplementary material
(i) Evolution of the N-ethyl phenylcarbamate concentration vs reaction time (equation (3a)), (ii) PLUTO drawing of the palladacycle 2, (iii) variable temperature 1H–NMR spectra of 3c in nitrobenzene-d6, (iv) X-ray structural information for 3c·Et2O including tables of atomic positional parameters, bond distances and angles, anisotropic and isotropic thermal displacement parameters (PDF file, 14 p).
Acknowledgements
F.P. would like to thank Dr G. Argouarch (UMR 6509) and S. Woodhouse for their help while writing the manuscript. This research has been supported by the CNRS and Rhône-Poulenc Recherches.