

1 Introduction
Molecular charge transfer salts combining two different properties, such as conductivity and magnetism, have become an area of increasing interest in the past few years [1–23]. To obtain such materials, organic π-donors of the tetrathiafulvalene (TTF) type are combined with a variety of magnetic metal complex anions with different nuclearities. Up to date, the most significant achievements obtained in this field are sequentially the discovery of coexistence of paramagnetism/metallic conductivity [1, 2], paramagnetism/superconductivity [3], antiferromagnetism/superconductivity [4], ferromagnetism/metallic conductivity [5], and recently field-induced ferromagnetism/superconductivity [6]. Taking into account the crucial role of intermolecular interactions in the conducting and magnetic properties of these molecular organic/inorganic hybrids, our strategy consists in combining organic π donors such as ET (bis(ethylenedithio)tetrathiafulvalene) or DHET–EDTTTF (4,5-bis(2-hydroxyethylthio)-4′,5′-ethylenedithiotetrathiafulvalene) (Fig. 1) with metal complexes containing also external sulphur atoms, with the aim of favouring the intermolecular contacts between the two components [7]. In addition, the DHET–EDTTTF organic π donor contains, out of the central plane, two –S–(CH2)2–OH groups involving oxygen atoms able to give hydrogen bonds with the sulphur atoms of the inorganic part. In this regard, we have recently shown that the mono- and di-nuclear anions [Cr(NCS)6]3– and [MM′(C2O4)(NCS)8]4– (MM′ = CrIIIFeIII and CrIIICrIII) [7–9] are attractive species for such multifunctional molecular solids. We now extend this work describing two new organic/inorganic hybrids based on the combination of the paramagnetic anion [Cr(NCS)6]3– with an organic π donor. We describe here the syntheses, crystal structures and physical properties of (ET)4[Cr(NCS)6].PhCN (1) and (DHET–EDTTTF)2[NEt4][Cr(NCS)6] (2). Two other related salts based on a paramagnetic [M(NCS)6]3– anion, (BMDT–TTF)4[Cr(NCS)6] (BMDT–TTF = bis(methylenedithio)tetrathiafulvalene) and (ET)4[Fe(NCS)6]·CH2Cl2, have been also recently reported [22–23].
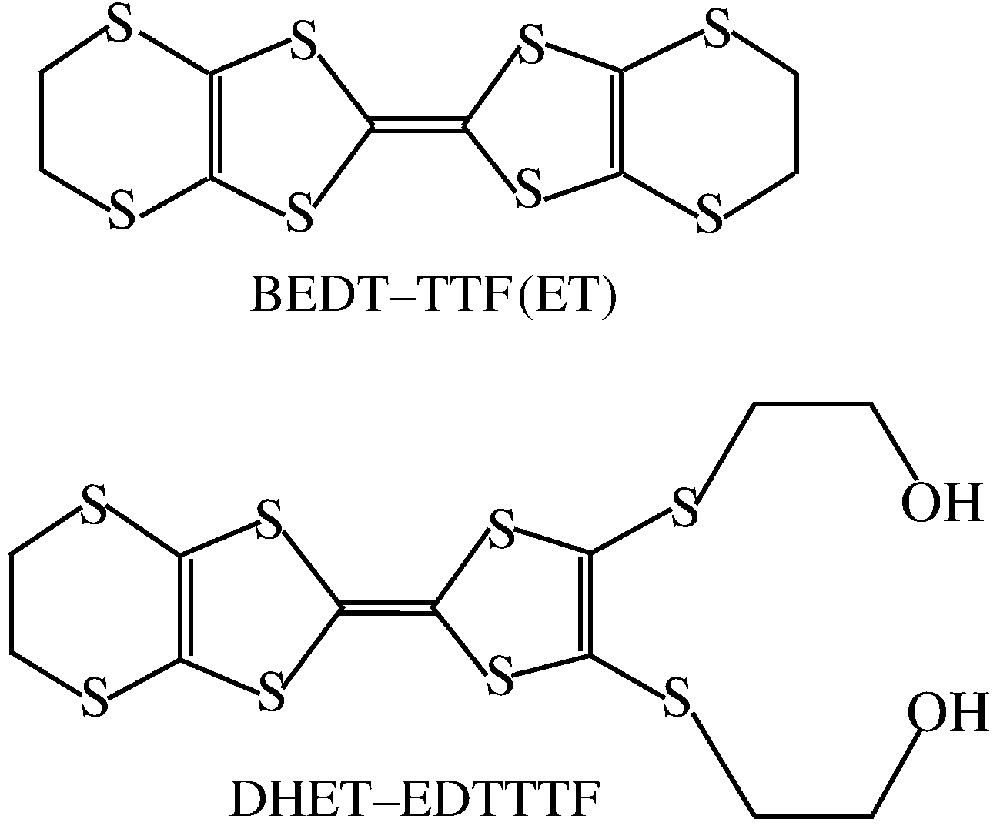
ET (bis(ethylenedithio)tetrathiafulvalene) and DHET–EDTTTF (4,5-bis(2-hydroxyethylthio)-4′,5′-ethylenedithiotetrathiafulvalene).
2 Experimental section
2.1 Syntheses of salts 1 and 2
The (NEt4)3[Cr(NCS)6] salt and the organic π donor DHET–EDTTTF were prepared according to the references [24] and [25], respectively. The ET organic π donor was purchased from Fluka and used as received. The two radical salts (1 and 2) were obtained on platinum-wire electrodes by anodic oxidations of 2.6 × 10–3 mol l–1 solutions of the corresponding organic donor in different solvents (1:1 PhCN/CH3CN mixture for 1 and 1:1 CH2Cl2/CH3CN mixture for 2) in U-shaped electrocrystallisation cells under a low current density of 0.5 μA cm–2 in presence of the salt (NEt4)3[Cr(NCS)6] as supporting electrolyte (3.8 × 10–3 mol l–1). After few days (three for 1 and six for 2) of electrochemical oxidation, single crystals were obtained as black needles for 1 and black plates for 2. In both cases, they were washed with acetonitrile and acetone to remove any portion of the tetraethylammonium salt of the supporting electrolyte, and then air-dried.
2.2 Physical techniques
The magnetic susceptibility measurements were performed on polycrystalline samples at 0.1 T in the temperature range 2–300 K with a SQUID magnetometer MPMS-XL-5 from Quantum Design. The molar susceptibility was corrected from the sample holder measured in the same conditions and from the diamagnetic contributions of all atoms (Pascal's tables). Temperature dependence of the dc electrical conductivity over the range 2–300 K was performed on single crystals using the standard four-contact dc method. Contacts to the crystals were made by platinum wires (25 μm diameter) attached with graphite paste.
2.3 X-ray data collection
Data collections of 1 and 2 were made on Enraf-Nonius CAD4 and Nonius Kappa CCD diffractometers, respectively at 293 K for 1 and 173 K for 2. The crystal structures of both compounds were solved by direct methods and successive Fourier difference syntheses, and were refined on F2 by weighted anisotropic full-matrix least-square methods. For both compounds, all non-hydrogen atoms were refined anisotropically, except for the PhCN molecule in 1 and for the disordered NEt4+ ion in 2, which were refined isotropically. The hydrogen atoms were calculated [C–H = 0.95 Å] and thereafter included as a fixed contribution to Fc with thermal parameters taken as Uiso = 1.3 Ueq(C). For 1, the non-centrosymmetric NEt4+ cation is located on the inversion centre (0 0) and therefore disordered; consequently, its hydrogen atoms were not included in the refinements. Drawing made with ORTEP [26] and all calculations performed on Alpha-station 255 4/233 computer using OpenMoleN package [27]. Crystal data for 1 and 2, selected bond lengths and bond angles and pertinent S···S contacts for 1, and selected bond lengths and bond angles for 2 are listed respectively in Tables 1–4. Complete crystallographic details for both structures are included in the Supporting information.
Crystallographic data for 1–2.
| 1 | 2 | |
| Empirical formula | CrC53H37N7S38 | CrC38H48N7O4S22 |
| Molecular weight | 2042.36 | 1424.25 |
| Crystal system | monoclinic | triclinic |
| Space group | P21 | |
| a (Å) | 12.002(1) | 10.790(2) |
| b (Å) | 20.396(2) | 11.661(2) |
| c (Å) | 16.439(2) | 13.204(4) |
| α(°) | 78.54(2) | |
| β (°) | 100.46(2) | 81.35(1) |
| γ (°) | 68.51(2) | |
| V (Å3) | 3957.0(1) | 1509.6(7) |
| Z | 2 | 1 |
| Dcalcd (g cm–3) | 1.71 | 1.57 |
| μ (Mo Kα) (mm–1) | 1.15 | 0.96 |
| Measured reflections | 9529 | 7813 |
| Independent reflections with I ≥ n σ(I) | 5346 (n = 3) | 3801 (n = 4) |
| Number of variables | 851 | 316 |
| a R1 | 0.047 | 0.062 |
| b w R2 | 0.076 | 0.099 |
| c Goodness-of-fit | 1.748 | 1.877 |
| ▵σmax/min (e Å–3) | +0.970/–0.137 | 0.694/–0.435 |
Selected bond lengths (Å) and bond angles (°) in compound 1.
| Cr–N1 | 2.017(7) | C19–C20 | 1.31(1) |
| Cr–N2 | 1.999(8) | C21–C22 | 1.40(1) |
| Cr–N3 | 2.012(7) | C23–C26 | 1.37(1) |
| Cr–N4 | 2.015(6) | S24–C30 | 1.745(7) |
| Cr–N5 | 2.021(7) | S24–C31 | 1.725(7) |
| Cr–N6 | 1.961(7) | S25–C29 | 1.740(7) |
| S8–C10 | 1.745(8) | S25–C31 | 1.715(8) |
| S8–C11 | 1.728(7) | S27–C32 | 1.735(7) |
| S9–C9 | 1.755(7) | S27–C33 | 1.770(8) |
| S9–C11 | 1.710(8) | S30–C32 | 1.699(8) |
| S11–C12 | 1.726(7) | S30–C36 | 1.747(7) |
| S11–C13 | 1.743(8) | C29–C30 | 1.37(1) |
| S12–C13 | 1.751(7) | C31–C32 | 1.39(1) |
| S12–C14 | 1.814(9) | C33–C36 | 1.36(1) |
| C9–C10 | 1.36(1) | S32–C40 | 1.767(8) |
| C11–C12 | 1.40(1) | S32–C41 | 1.760(6) |
| C13–C16 | 1.34(1) | S33–C39 | 1.773(6) |
| S16–C20 | 1.715(8) | S33–C41 | 1.733(7) |
| S16–C21 | 1.688(7) | S35–C42 | 1.742(6) |
| S17–C19 | 1.762(7) | S35–C43 | 1.753(7) |
| S17–C21 | 1.738(8) | S38–C42 | 1.776(7) |
| S19–C22 | 1.684(7) | S38–C46 | 1.742(6) |
| S19–C23 | 1.748(8) | C39–C40 | 1.34(1) |
| S22–C22 | 1.738(8) | C41–C42 | 1.325(9) |
| S22–C26 | 1.718(6) | C43–C46 | 1.366(9) |
| N1–Cr–N2 | 178.2(3) | N3–Cr–N6 | 88.7(3) |
| N1–Cr–N3 | 88.8(3) | N4–Cr–N5 | 89.4(3) |
| N1–Cr–N4 | 89.2(3) | N4–Cr–N6 | 179.3(3) |
| N1–Cr–N5 | 91.2(3) | N5–Cr–N6 | 91.2(3) |
| N1–Cr–N6 | 90.4(3) | Cr–N1–C1 | 179.2(7) |
| N2–Cr–N3 | 91.1(3) | Cr–N2–C2 | 176.4(8) |
| N2–Cr–N4 | 89.1(3) | Cr–N3–C3 | 177.0(6) |
| N2–Cr–N5 | 88.9(3) | Cr–N4–C4 | 167.4(8) |
| N2–Cr–N6 | 91.3(3) | Cr–N5–C5 | 173.0(7) |
| N3–Cr–N4 | 90.7(3) | Cr–N6–C6 | 157.8(8) |
| N3–Cr–N5 | 180.0(3) |
Shortest intermolecular S···S contacts in compound 1.
| S···S contacts | ||
| NCS–ET | S1–S36 | 3.644(3) |
| S3–S28(iii) | 3.565(3) | |
| S5–S15(iv) | 3.646(3) | |
| A–B | S9 –S17 | 3.568(4) |
| S11–S19 | 3.557(4) | |
| A–C | S7 –S29(ii) | 3.626(4) |
| S8 –S29(ii) | 3.620(3) | |
| S10–S27(i) | 3.524(4) | |
| S10–S28(i) | 3.587(4) | |
| S12–S26(i) | 3.462(4) | |
| S13–S23(ii) | 3.474(3) | |
| S13–S24(ii) | 3.590(4) | |
| A–D | S7 –S37(ii) | 3.590(4) |
| S8 –S37(ii) | 3.532(3) | |
| S10–S35(i) | 3.476(3) | |
| S10– S36(i) | 3.535(4) | |
| S11–S34(i) | 3.531(3) | |
| S13–S32(ii) | 3.486(4) | |
| B–C | S15–S26 | 3.572(5) |
| S21–S28 | 3.619(5) | |
| B–D | S15–S37(ii) | 3.434(4) |
| S16–S37(ii) | 3.363(3) | |
| S18–S35(i) | 3.572(3) | |
| S18–S36(i) | 3.552(4) | |
| S19–S34(i) | 3.435(3) | |
| S20–S34(i) | 3.456(3) | |
| S21–S31(ii) | 3.386(4) | |
| S21–S32(ii) | 3.477(3) |
Selected bond lengths (Å) and bond angles (°) in compound 2.
| Cr–N1 | 2.014(4) | S10–C8 | 1.730(5) |
| Cr–N2 | 2.010(4) | S9–C11 | 1.817(8) |
| Cr–N3 | 1.999(5) | S10–C12 | 1.777(7) |
| S4–C4 | 1.719(5) | S1–C9 | 1.741(6) |
| S4–C6 | 1.735(5) | S11–C14 | 1.803(8) |
| S5–C4 | 1.717(4) | O1–C13 | 1.432(9) |
| S5–C7 | 1.739(6) | O2–C15 | 1.45(1) |
| S6–C5 | 1.724(4) | C4–C5 | 1.388(7) |
| S6–C8 | 1.729(6) | C6–C7 | 1.350(7) |
| S7–C5 | 1.718(5) | C8–C9 | 1.366(7) |
| S7–C9 | 1.735(5) | C10–C11 | 1.460(9) |
| S8–C6 | 1.747(6) | C12–C13 | 1.443(8) |
| S8–C10 | 1.810(6) | C14–C15 | 1.18(1) |
| S9–C7 | 1.738(5) | ||
| N1–Cr–N2 | 90.0(2) | N2–Cr–N3 | 91.0(2) |
| N1–Cr–N3 | 91.1(2) | Cr–N1–C1 | 173.5(5) |
| N1–Cr–N3 | 88.9(2) | Cr–N2–C2 | 171.1(5) |
| N2–Cr–N3 | 89.0(2) | Cr–N3–C3 | 178.1(4) |
3 Results and discussion
3.1 Crystal structures of (ET)4 [Cr(NCS)6]·PhCN (1) and (DHET–EDTTTF)2(NEt4)[Cr(NCS)6] (2)
For 1, the crystal structure determination shows that the asymmetric unit is built up of one [Cr(NCS)6]3– anion, four ET units (A, B, C and D types) and one solvent PhCN molecule, all located on general positions. ORTEP drawings of the asymmetric unit with the atom-labelling scheme are depicted in Fig. 2. The structure consists of alternating organic layers (in which the PhCN molecules are inserted) and inorganic layers along the c direction (Fig. 3). Careful analysis of the organic layers leads to the shortest S···S intermolecular distances summarized in Table 3. These contacts are schematically shown in the projection of the organic layers depicted in Fig. 4. The most important aspects of these interactions are: (i) they are observed between all pairs of ET units, except between the C and D types, and (ii) most of them (and the shortest ones) are along the a direction (connecting A–C-, A–D- and B–D-type ET molecules), whereas very few (and longer) contacts are observed in the b direction (connecting A–B- and B–C-type ET molecules, see Fig. 4 and Table 3). These two structural aspects reflect the structure of the organic layer shown in Fig. 4, which can be seen as a succession along the b axis of a monodimensional chain with weak inter-chain contacts. It is interesting to note that there are three S···S cation-anion contacts shorter than the sum of the van der Waals radii (3.65 Å).
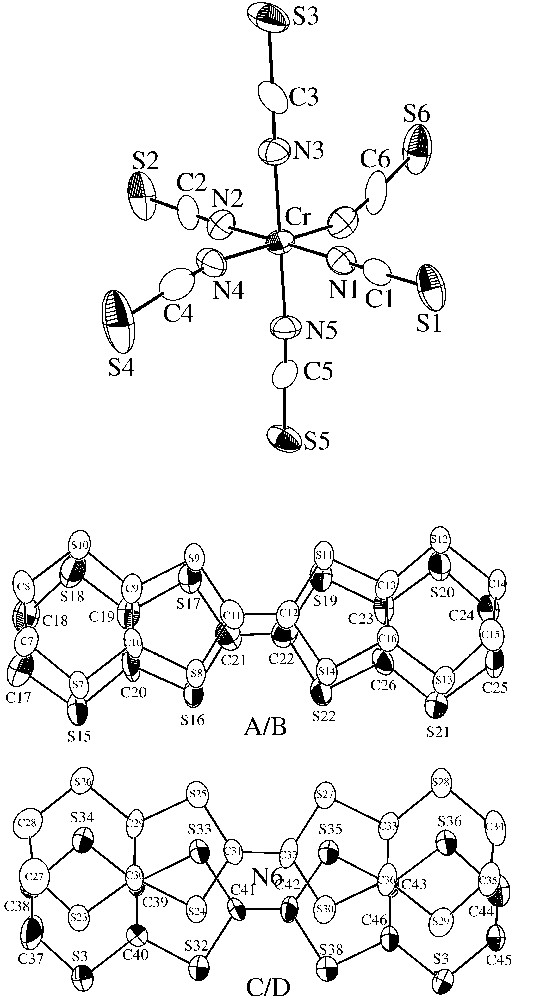
Asymmetric unit showing the atomic numbering and overlaps of the ET units in 1 (A unit: from S7 to S14; B unit: from S15 to S22; C unit: from S23 to S30 and D unit: from S31 to S38).
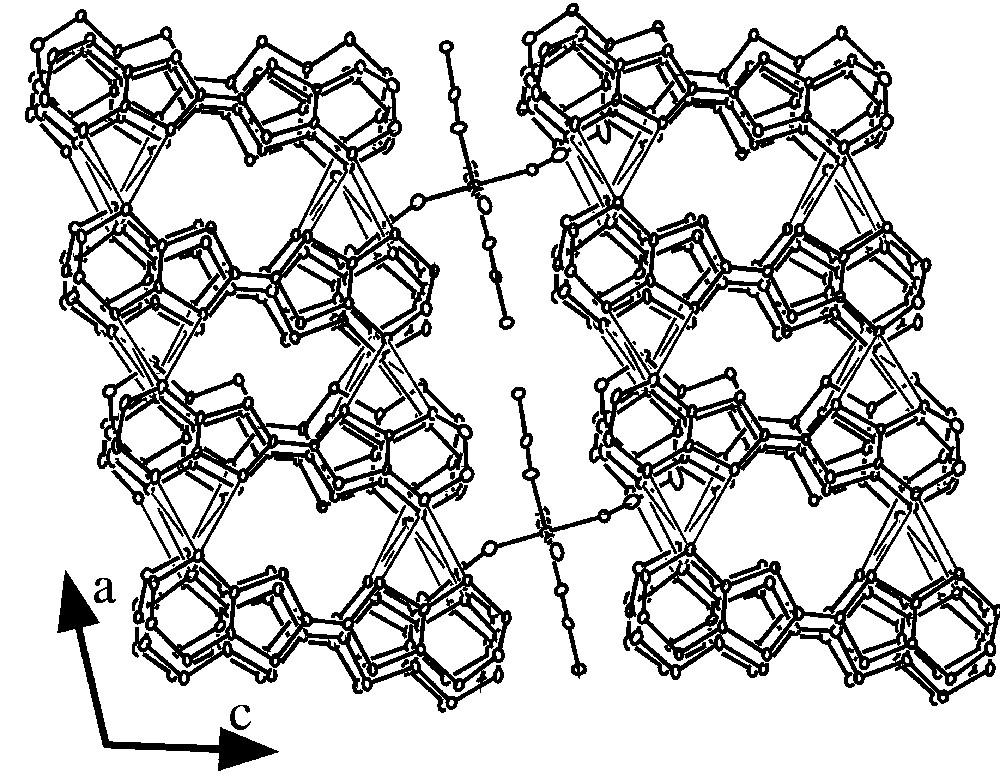
ORTEP structure of 1 in the ac plane, showing the alternating organic/inorganic layers along the [001] direction and the shortest longitudinal intermolecular S···S distances (dS···S < 3.65 Å); the PhCN solvent molecules were omitted for clarity.

ORTEP projection of 1 in the ab plane showing the shortest intermolecular S···S distances (dS···S < 3.65 Å).
The 1:4 stoichiometry of compound 1 is indicative of a mixed valence state in the organic part: [ET]43+. An estimation of the charge distribution of the ET units [28] leads to a charge of +0.94, +1.06, +1.02 and +0.13 for the A-, B-, C- and D-type ET molecules, respectively, giving a total charge of +3.15, very close to the expected value of +3. These values suggest a charge distribution of +1 for A, B and C units, and a neutral charge for the D unit. This is in good agreement with the molecular structure described above which clearly shows that the D-type ET molecules are more ‘isolated’ than the fully oxidized A, B and C ones.
For 2, the asymmetric unit contains one [Cr(NCS)6]3– and one tetraethylammonium ions, both located on the special positions (0 0 0) and (0 0), respectively, together with an organic donor DHET–EDTTTF, located on a general position. Fig. 5 displays an ORTEP drawing showing the inorganic anion and the organic donor of this asymmetric unit, with the atom-labelling scheme. The organic sublattice contains a centrosymmetric (DHET–EDTTTF)2 dimer centred on the inversion centre ( ). Within the dimer, the overlap mode is of the eclipsed type (Fig. 6) and only two S···S distances are shorter than the sum of van der Waals radii (Fig. 7). Careful examination of the inter-dimer S···S distances reveals that adjacent dimers are essentially isolated. This absence of significant S···S inter-dimer contacts seems essentially due to the steric effects induced by the presence of four tetraethylammonium ions around the organic dimer and also by the two –S–(CH2)2–OH groups that are located out of the central plane of the tetrathiafulvalene group (see Fig. 6). In contrast, short S···S and S···O contacts between the organic units and the inorganic anions are observed (S···S in the range 3.556–3.622 Å and S···O in the range 3.169–3.221 Å). The stoichiometry of 2 is indicative of a fully oxidized state for the organic donors ([DHET– EDTTTF]•+). By using the formula derived for the ET [28], the charge on the DHET–EDTTTF unit is calculated to be +1.06, in good agreement with the expected value of +1.
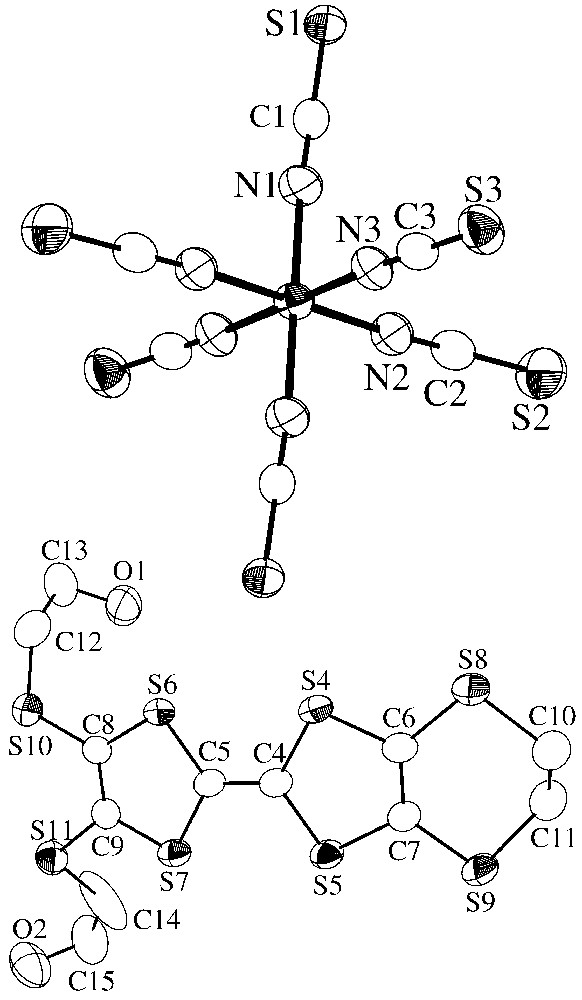
ORTEP view showing the atomic-labelling scheme in the asymmetric unit of 2.
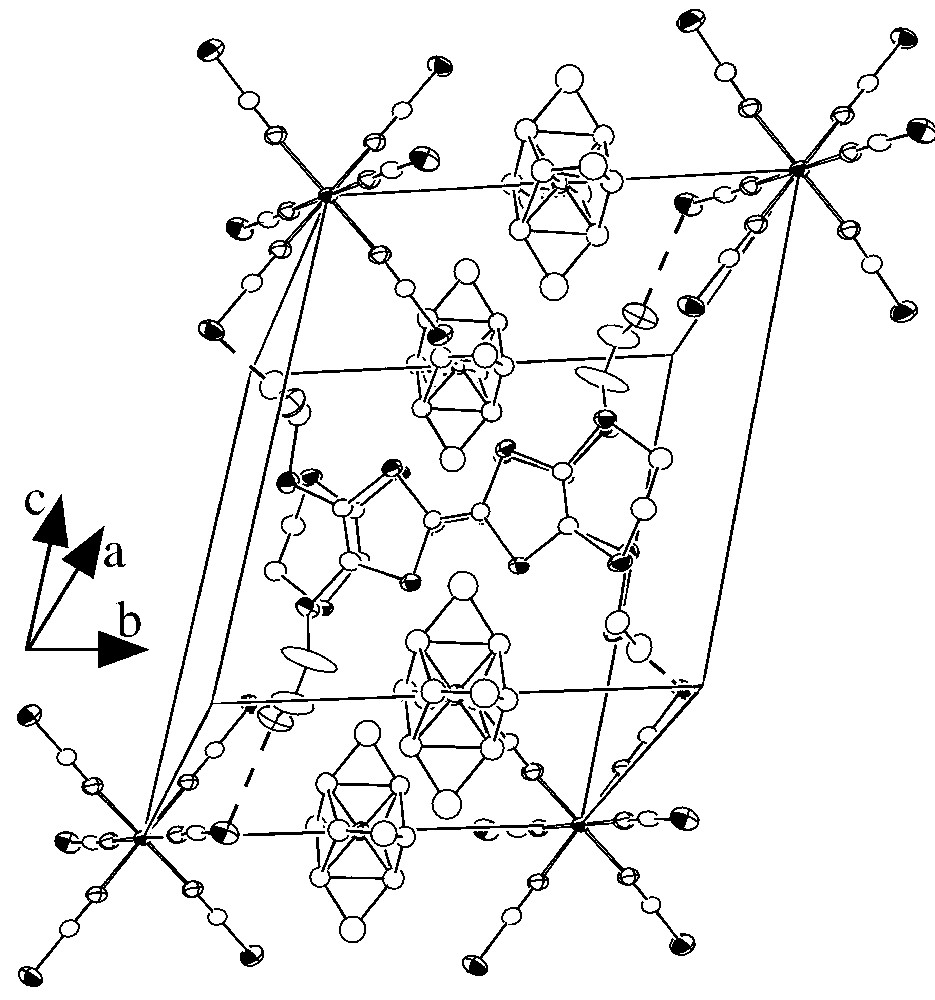
Structure of 2 showing the eclipsed DHET–EDTTTF radical cations (the NEt4+ ions are disordered).

Structure of the organic dimer in 2 showing the shortest S···S distance, d = 3.584(2) Å ((i): 1 – x, 1 – y, 1 – z).
Finally, in the [Cr(NCS)6]3– inorganic anion, the Cr(III) cation has a pseudo-octahedral environment in both cases, but the coordination octahedron is much more distorted in 1, since the Cr(III) ion is located on a general position, while in 2 it lies on the special position (0 0 0). Thus, in 1, the Cr–N-bond lengths range from 1.961(7) to 2.021(7) Å, whereas in 2, they range from 1.999(5) to 2.014(4) Å (Tables 2 and 4). Furthermore, the N–Cr–N bond angles in 1 are more deviated from the expected values of octahedral environment (see Tables 2 and 4). These bond lengths are in good agreement with previously reported values [7–8, 29–31].
3.2 Conductivity measurements
The conductivity measurements were performed on single crystals of both compounds 1 and 2. As expected from the estimated charge distributions (in 1 there are three fully oxidized and one neutral ET molecules and in 2 all the DHET–EDTTTF units are fully oxidized), both salts are semiconductors with room-temperature conductivities of 1.4 × 10–2 and 6.1 × 10–5 S cm–1 and activation energies of 0.24 and 0.23 eV, in 1 and 2, respectively (Fig. 8). The higher room temperature conductivity of salt 1 compared with salt 2 should be attributed to the presence in 1 (and absence in 2) of intermolecular interactions in the organic sub-lattices.
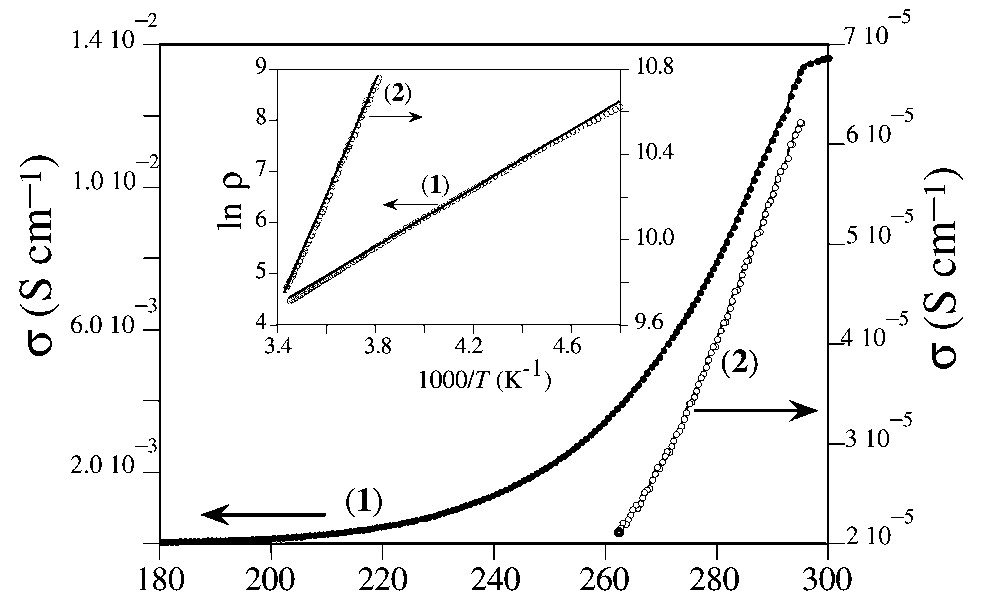
Temperature dependence of the dc electrical conductivity for compounds 1 and 2.
3.3 Magnetic properties
The magnetic properties of both compounds (1–2) are displayed in Fig. 9 as the thermal variation of the product of the molar susceptibility times the temperature (χm T). In order to estimate the contribution of the organic part, they are compared with that of the corresponding NEt4+ salt, (NEt4)3[Cr(NCS)6] (3).
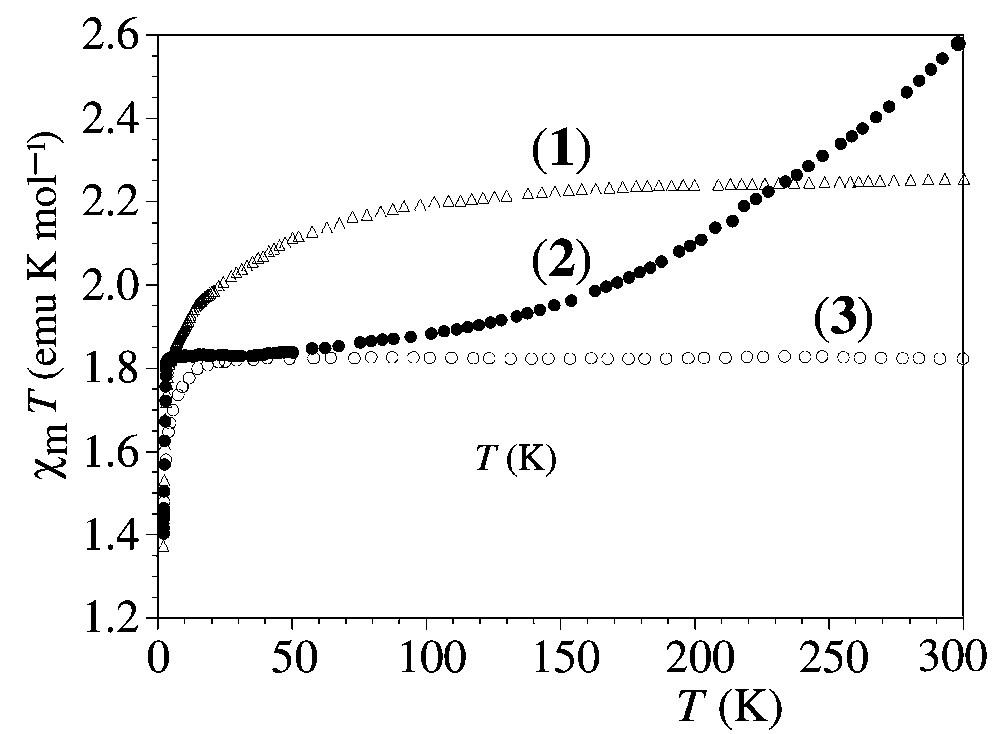
Thermal variations of the magnetic susceptibility in the forms χm T vs. T for compounds 1–2 and for (NEt4)3[Cr(NCS)6] (3).
In salt 3, the room temperature χm T product (1.83 emu K mol–1, very close to the expected value 1.875 for S = 3/2 with g = 2) remains constant down to ca 20 K, where a decrease is observed, reaching a value of 1.45 emu K mol–1 at 2 K. This decrease is very typical of the paramagnetic salts of CrIII (S = 3/2) and can be attributed to weak antiferromagnetic coupling of the CrIII ions or/and to the presence of a zero-field splitting (ZFS) in this ion.
The magnetic behaviour of salt 1 shows a χm T value of 2.25 emu K mol–1 at room temperature that decreases with decreasing temperature, as shown in Fig. 9. The room χm T value of 2.25 emu K mol–1 corresponds to the sum of a paramagnetic CrIII ion plus an extra contribution that must be attributed to the organic sub-lattice. The organic contribution (2.25–1.83 = 0.42 emu K mol–1) at room temperature suggests the presence of one organic radical (S = 1/2) which is magnetically isolated at room temperature and one (ET2)2+ dimer, in which the electrons are strongly antiferromagnetically coupled (S = 0) in the whole temperature range studied. These observations indicate that both organic and inorganic parts are magnetically independent and the organic units are antiferromagnetically coupled. For 1, we can assume, from the structural data and charge distribution, that the magnetic contribution of the organic sub-lattice may correspond to that of a regular chain of spins 1/2, which are antiferromagnetically coupled. Accordingly, we have fitted the magnetic behaviour of salt 1 to the sum of isolated S = 3/2 ions with a ZFS [32] plus a regular antiferromagnetically coupled S = 1/2 chain [33]. This model reproduces very satisfactorily the magnetic behaviour of salt 1 in the whole temperature (J = –19.0 cm–1 and D = 2.5 cm–1), but the D value is higher than those previously reported for isolated Cr(III) ions, suggesting that weak antiferromagnetic exchange interactions between the Cr(III) ions must be present. The magnetic behaviour of salt 2 shows a χm T value of 2.60 emu K mol–1 at room temperature, which decreases with decreasing temperature to reach, at 50 K, a constant value of 1.83 emu K mol–1, observed for the NEt4+ salt. As in salt 1, the χm T product of salt 2 exceeds that expected for a CrIII ion, indicating that the organic sub-lattice contributes to the magnetic properties in this salt. This organic contribution of 0.77 emu K mol–1 at room temperature can be attributed to the presence of two isolated organic radicals that are antiferromagnetically coupled at low temperature, since the χm T product decreases when decreasing the temperature to reach, at 50 K, the value observed for 3. These observations, which are in agreement with structural data (see above), suggest, as in compound 1, that the organic and inorganic parts are magnetically independent. For 2, we can assume, from the structural data and charge distribution, that the magnetic contribution of the organic sub-lattice can be reproduced with a model of antiferromagnetically coupled S = 1/2 dimer. Accordingly, we have fitted the magnetic behaviour of salt 2 with a model that takes into account the ZFS in the CrIII ions and an antiferromagnetically coupled S = 1/2 dimer [32]. Although this model reproduces the general trends observed in the magnetic properties of 2, the fit is not very satisfactory.
The χm T decreases observed in the low temperature region (T < 10 K) for the three compounds (1–3) suggest the presence of a ZFS in the Cr3+ ion and/or weak antiferromagnetic interactions between the Cr3+ ions. A close inspection of this χm T parameter shows that the magnetic behaviours of compounds 1–3 are slightly different; this may be explained by slight differences in the magnitudes of the ZFS effect of the Cr(III) ions, as expected from the structural data found for the Cr(III) anions in 1 and 2.
4 Concluding remarks
The two radical salts described here are obtained by the combination of the organic π donors ET and DHET–EDTTTF with the paramagnetic anion [Cr(NCS)6]3–. In both cases, the magnetic measurements indicate that the organic units are antiferromagnetically coupled and the organic and inorganic parts are magnetically independent, in agreement with structural data.
With the aim of favouring the magnetic interactions between the organic and the inorganic parts, we are currently using associating inorganic ions with new functionalised π-electron donors [25, 34–36]. With such donors, intra- or intermolecular interactions between the organic and the inorganic parts in the solid state might be favoured via a possible covalent bonding when starting from inorganic magnetic fragments involving one or more free coordination sites; furthermore, these donors should also lead to interesting intermolecular interactions via hydrogen bonding contacts.
Supplementary materials
Crystallographic data (excluding structure factors) for the structures of compounds 1 and 2 have been deposited with the Cambridge Crystallographic Data Centre as supplementary publication Nos. CCDC-195708 (1) and CCDC-195709 (2). Copies of the data can be obtained free of charge on application to CCDC, 12 Union Road, Cambridge CB2 1EZ, UK [Fax: (internat.) + 44-1223/336-033; E-mail: deposit@ccdc.cam.ac.uk].
Acknowledgements
This work was supported by CNRS (‘Centre national de la recherche scientifique’, UMR 6521, France), the University of Brest, France (SUCRI 2E) and by the Spanish ‘Ministerio de Ciencia y Tecnología’ (Project MAT2001-3507-C02-01). F.T. and F.B. thank the French ‘Ministère de l’Éducation nationale, de la Recherche et de la Technologie’ for postgraduate grants. S.B. and J.-M.F. thank the CMEP, the Algerian ‘Ministère de l’Éducation supérieure’ and the French ‘Ministère des Affaires étrangères’ for their support.