

1 Introduction
The tetrathiafulvalene (TTF) moiety is well known as an electroactive organic core with applications in various fields of materials chemistry [1], including electroconducting organic metals (charge-transfer complexes and cation-radical salts), organic ferromagnets, molecular electronics, NLO-phores... Recently, it has also appeared as an important building block in supramolecular chemistry [1].
In this context, much effort has been devoted in the recent years for the preparation of TTF-based hybrid systems exhibiting synergy between electrical conductivity and magnetic interactions [2]. The goal, in this case, is to get crystalline solids for which conducting electrons involved in the π-stacking of TTF units, could interact with localized spins (d electrons of transition metals). In order to optimise the exchange between the mobile electrons and the localized spins in the solid, one may design a system allowing a close proximity of the metal (d electrons) to the electroactive TTF core. One way to achieve such a prerequisite is to covalently associate the TTF framework to a coordinating subunit prone to complex a metal. Among the possible receptors suitable for the complexation of transition metals, polyaza-macrocycles [3], and notably cyclam derivatives (1,4,8,11-tetraazacyclotetradecane) [4], are known to be particularly efficient. Taking advantage on our recent results concerning the preparation of redox-switchable ligands [5, 6], we have extended the synthesis to new redox systems associating the TTF framework (as a tetraalkylsulfanyl-TTF or a bis(pyrrolo)TTF unit) to various polyaza-macrocycles, including a cyclam derivative or a diaza-tetraoxa cyclooctadecane moiety. The X-ray structural determination of one aza-macrocycle is also depicted.
2 Results and discussion
2.1 Synthesis
The target macrocyclic compounds of the tetraalkylsulfanylTTF series (compounds 3–5), were prepared [6] starting from the bis(2-cyanoethylsulfanyl)-3,7-bis(methylthio)tetrathiafulvalene 1 [7] ([Z+E] isomers with respect to the rigid TTF framework) (Fig. 1). Unfortunately, all attempts of N-tosyl or N-triflate cleavages were unsuccessful. On the other hand, reaction of compound 1 under basic medium with an excess of a ω−diodo polyethyleneglycol chain, led to 6a and 6b. The introduction of the polyaza-macrocyclic unit was carried out by using 5,12-dioxocyclam 7, since such macrocycles are known to bind efficiently transition metals [8]. Compound 7, prepared by condensation of ethylene diamine on methyl acrylate [9], allows an original control in the regioselectivity of the bis addition, since two of the four nitrogen atoms are involved in an amide bond, and therefore have lost their nucleophilic character. This protection mode permits to selectively functionalise the tetraazamacrocycle on the two remaining amino groups [10].
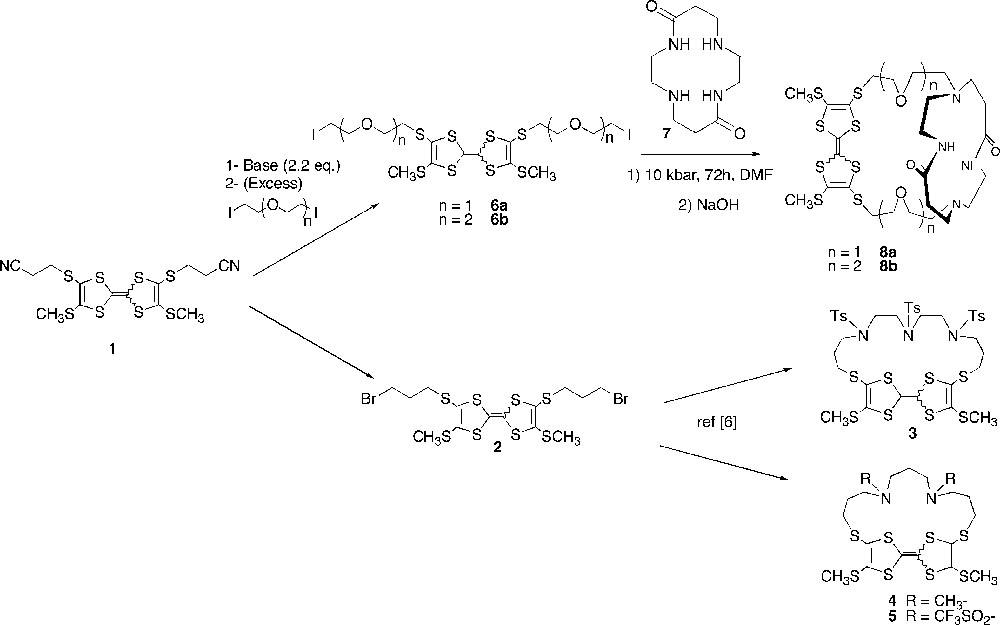
Preparation of the target macrocyclic compounds of the tetraalkylsulfanylTTF series (3–5) starting from the bis(2-cyanoethylsulfanyl)-3,7-bis(methylthio)tetrathiafulvalene 1 ([Z+E] isomers with respect to the rigid TTF framework).
Since no good results were obtained by using high dilution conditions, dioxocyclam 7 and TTF derivative 6a (or 6b) were subjected to a [1+1] cyclocondensation under high pressure conditions (10 kbar, 72 h) [11]. The tedious isolation of the different macrocycles in fine necessitated the use of a preparative scale size exclusion chromatography (Sephadex LH20) to afford the target TTF cryptands 8a and 8b in 10% yield. The complete structural characterization of compounds 8a and 8b could be carried out thanks to two-dimensional NMR spectroscopy (DQF COSY, HMQC and HMBC). It is interesting to note that the Z and E isomers (if both isomers are formed), are not distinguishable, contrary to the case of macrocycles 3–5 [6].
In the case of the bis(pyrrolo)TTF series (compounds 12a,b), the coordination unit introduced was a N2O4 macrocycle (Fig. 2); grafting of a N2O4 macrocyclic unit to tetraakylsulfanyl-TTF has already been described [12]. Therefore, targets 12a,b were obtained according to a three steps strategy from naked bis(pyrrolo)TTF 9 [13]. Alkylation of both pyrrolo nitrogen atoms of 9 [5d] produced chlorinated intermediates 10a,b, which were then submitted to a Cl/I exchange. The final step involved a cyclocondensation under high-dilution conditions between iodide intermediates 11a,b, and 1,4,10,13-tetraoxa-7,16-diazacyclooctadecane.

Synthesis of 12a,b from 9.
2.2 X-ray structural determination, electrochemical properties and complexation studies
Slow diffusion of pentane in a dichloromethane solution of macrocycle 5 afforded single crystals suitable for an X-ray diffraction study. The diaza-crown TTF 5 appears in the [Z] configuration (Fig. 3), with a packing mode showing a segregation between the electroactive TTF unit and the diaza fragment along the a axis (Fig. 4). The TTF skeleton in 5 [Z] appears partially distorted, with a bending angle of 21° from the planarity (Fig. 3). This distortion is of course reminiscent of the structural constraints imposed by the lateral diaza-chain. Such a value appears very similar to the one observed for compound 4 [6], which presents the same size for the lateral chain, but far less important than for other aza-crown TTF systems with shorter chains [14], for which a high bending angle value results in poor electrochemical characteristics (high oxidation potential and irreversible redox processes). The weak distortion observed in the case of compound 5 should favour a good π-delocalization. Consequently, we can anticipate that all of these polyaza macrocyclic TTFs present large enough lateral chains to present good π-donating properties, which is confirmed by cyclic voltammetry experiments, where compounds 3–5, 8a, 8b present two reversible redox processes with the usual E1ox and E2ox values for (RS)4TTF derivatives (ca 0.56 V and 0.83 V vs AgCl/Ag respectively). Nevertheless, we could notice in the case of compounds 8a,b a progressive passivation of the Pt working electrode upon scanning to potentials higher than 0.80 V, which could be attributed to an irreversible oxidation of the cyclam moiety. TTF-cryptands 12a,b present as expected a higher π-donating ability as usually observed for bis(pyrrolo)TTF derivatives and Nevertheless, here again we could observe some passivation phenomena upon cycling potentials up to 1.00V.

X-ray structure of (Z)-5.
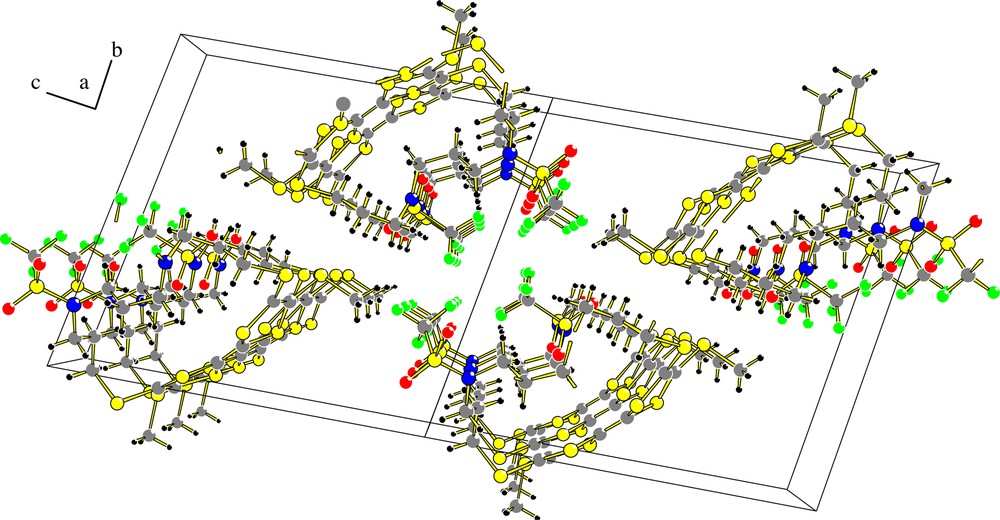
Stacking mode of (Z)-5 along the a axis.
Preliminary complexation studies led on these TTF macrocycles gave best results for TTF receptors 12a,b, for which LSIMS measurements showed a good affinity for Groups-I and II cations, which was notably confirmed by UV–Vis titration experiments. Binding constants of ca log(K) = 3.0 to 4.1 (CH2Cl2/CH3CN, 1:1) (Benesi–Hildebrand method) were obtained for complexation of Ca2+, Sr2+ and Ba2+ by either 12a or 12b, without any evidence of selectivity. The 1:1 stoichiometry of the complexes was shown by occurrence of isobestic points (e.g., 264, 315, 324 nm in the case of 12a/Sr2+) and confirmed by the titration curves, which did not evolve anymore above 1 equiv of added cation.
In conclusion, we have synthesized a series of TTF receptors incorporating different polyaza macrocyclic receptors. The planarity of the π-system is sufficiently preserved in those systems, as shown by an X-ray structural determination, which allows them to exhibit the usual electrochemical behaviour of TTF derivatives. Some of these derivatives show good binding properties for alkaline-earth cations. Attempts to get single crystals of a complex associating one of these polyaza receptors to various transition metals are under investigation.
3 Experimental section
1H-NMR (500.13 MHz) and 13C-NMR (125.75 MHz) spectra were recorded on a BRUKER Advance DRX 500 spectrometer. Chemical shifts (δ) are expressed in ppm related to the tetramethylsilane (TMS) signal and J values in Hz. FAB and ESI mass spectra were achieved on a Jeol JMS 700 B/ES and MALDI–TOF spectra on a Bruker Biflex III. Cyclic voltammetry experiments were carried out on a potentiostat–galvanostat EG&G PARK models 273 or 273A, with solvents and electrolytes of electrochemical grades. For synthesis, anhydrous solvents were obtained by distillation on the appropriate drying agent (sodium/benzophenone for THF, sodium for toluene, P2O5 for dichloromethane, CaH2 for acetonitrile).
3.1 (Z/E)-2,6(7)-Bis(5-iodo-3-oxa-1-pentylsulfanyl)-3,7(6)-bis(methylsulfanyl) tetrathiafulvalene 6a
Sodium methylate (8.5 ml at 2.26 M, 2.2 equiv, 19 mmol) was added slowly under N2, to tetrathiafulvalene derivative 1 (4.0 g, 1 equiv, 8.6 mmol) in dry DMF (100 ml). The solution was stirred during 15 min and afterwards, bis(2-iodoethyl) ether (45 g, 8 equiv, 138 mmol) was added quickly. After stirring overnight, the solvent was removed in vacuo. The red residue was dissolved in dichloromethane, washed with water, dried over magnesium sulphate and concentrated in vacuo. The compound was purified by chromatography on a silica gel column (petroleum ether/dichloromethane 50:50) to furnish a red solid (74%, 4.8 g). 1H NMR (CDCl3): 2.45 (s, 6H, SCH3); 3.01 (t, 4H, 6.6 Hz, SCH2CH2); 3.27 (t, 4H, 6.9 Hz, CH2I); 3.71 (t, 4H, 6.6 Hz, SCH2CH2); 3.76 (t, 4H, 6.9 Hz, CH2CH2I). 13C NMR (CDCl3): 2.4 (CH2I); 19.1 (SCH3); 35.4 and 35.4 (SCH2); 69.5 and 71.7 (OCH2); 110.8 and 110.9 (central C=C); 124.0, 124.2, 131.2 and 131.4 (lateral C=C); MS (PDMS): 756.
3.2 (Z/E)-2,6(7)-Bis(8-iodo-3,6-dioxa-1-octylsulfanyl)-3,7(6)-bis(methylsulfanyl) tetrathiafulvalene 6b
Caesium hydroxide monohydrate (1.8 g, 2.5 equiv, 10.8 mmol) in dry methanol was added under N2 to tetrathiafulvalene derivative 1 (2.0 g, 1 equiv, 4.3 mmol) in distilled THF (150 ml). The solution was stirred during 15 min and afterwards, 2,2'-(oxydiethylenoxy)-1,1'-diiodoethane (15.9 g, 5 equiv, 21.5 mmol) was added. The caesium iodide immediately precipitated. The red reaction mixture was stirred for 1 h. The solvent was removed in vacuo and the red residue was dissolved in dichloromethane. The organic phase was washed with water, dried over magnesium sulphate and concentrated in vacuo. Purification of 6b was carried out by chromatography on a silica gel column (petroleum ether/dichloromethane: 25/75 and dichloromethane), to produce a red oil (80% yield, 2.90 g). 1H NMR (CDCl3): 2.44 (s, 6H, SCH3); 3.00 (t, 4H, 6.7 Hz, SCH2CH2); 3.27 (t, 4H, 6.6 Hz, CH2I); 3.59–3.79 (t, 16H, OCH2); 13C NMR (CDCl3): 3.6 (CH2I); 36.0 and 36.1 (SCH2); 70.7, 70.8, 71.1 and 72.6 (OCH2); 111.2 and 111.3 (central C=C); 124.9, 125.0, 131.4 and 131.5 (lateral C=C). MS (FAB): 844.
3.3 1,8-(2,7-Diethyloxysulfanyltetrathiafulvaleno)-5,12-dioxocyclam 8a
Tetrathiafulvalene derivative 6a (378 mg, 1 equiv, 0.5 mmol) and 5,12-dioxocyclam 7 [9] (114 mg, 1 equiv, 0.5 mmol) in solution in DMF (18 ml) were reacted for three days under a pressure of 10 kbar. The solvent was removed in vacuo and the orange residue was dissolved in chloroform. A solution of sodium hydroxide in water (3 equiv) was added and the mixture was stirred for one day. The organic phase was washed with water, dried over magnesium sulphate and concentrated in vacuo. Purification was carried out thanks to the successive following treatments: column chromatography (dichloromethane/methanol 100:5) over alumina deactivated with water (5%), silica gel chromatography – acetone/ NH4PF6 (0 to 4%) –, solubilisation in methanol, and finally gel permeation chromatography on Sephadex (LH20) (dichloromethane/methanol 90:10). Compound 8a appears as an orange oil (10% yield, 36 mg). 1H NMR (CDCl3): 2.38 (m, 4H, CH2CO); 2.46 (s, 6H, CH3S); 2.62 (m, 4H, CH2 (9)); 2.72 (m, 8H, CH2 (8) and CH2 (12)); 2.92 (m, 4H, SCH2); 3.38 (m, 4H, CH2 (10)); 3.53–3.74 (m, 8H, CH2O); 8.64 (s, 2H, NH); HRMS (ESI+) C26H40N4O4S8: obs.: 751.0735 (M+Na)+, th.: 751.0713.
3.4 1,8-(2,7-Triethyloxysulfanyltetrathiafulvaleno)-5,12-dioxocyclam 8b
Compound 8b was prepared according to the same methodology as for 8a. Orange oil, 10% yield. 1H NMR (CDCl3): 2.38 (m, 4H, CH2CO); 2.43 (s, 6H, CH3S); 2.63 (m, 4H, CH2 (11)); 2.72 (m, 4H, CH2 (10)); 2.74 (m, 4H, CH2 (14)); 2.97 (m, 4H, SCH2); 3.38 (m, 4H, CH2 (12)); 3.54–3.68 (m, 16H, CH2O). 8.63 (s, 2H, NH); 13C NMR (CDCl3):19.1 and 19.2 (SCH3); 32.6 (CH2CO); 34.8 (SCH2); 38.2 (CH2NHCO); 50.4 (NCH2 (14)); 51.9 (NCH2 (10)); 53.0 (NCH2 (11)); 68.4, 70.0, 70.2, 70.2, 70.4, 70.5, 70.6 and 70.7 (CH2O); 110.5 (central C=C); 124.0, 124.1, 131.0 and 131.1 (lateral C=C); 172.5 (CO); MS (MALDI-TOF) C30H48N4O6S8: 816 (M+•).
3.5 Synthesis of bis(pyrrolo)TTF derivatives 10a,b
A suitable ω-chlorinated derivative (3 mmol) was added to bis(pyrrolo)tetrathiafulvalene 9 (282 mg, 1 mmol) in dry degassed DMF (50 ml). The mixture was cooled to 0 °C. Sodium hydride (washed with petroleum ether) (3 mmol) was added in one portion. The reaction mixture was stirred 1 h at 0°C and then 2 h at room temperature. The suspension was filtered over Celite and the solvent was removed in vacuo. The residue dissolved in dichloromethane was washed with brine, dried over magnesium sulphate and concentrated in vacuo. The yellow oil was purified by chromatography (SiO2+ 4% triethylamine, petroleum ether/dichloromethane 1:1).
3.6 Bis-(N-(5-chloro-3-oxa-1-pentyl)pyrrolo[3, 4, d]tetrathiafulvalene 10a
Yellow powder, 61% yield. m.p.: 120°C. 1H NMR (CDCl3): 3.56–3.73 (m, 12H, CH2O and CH2Cl); 3.99 (t, 5.4 Hz , 4H, CH2N); 6.51 (s, 4H, CHpyrrole). 13C NMR (CDCl3): 43.1 (CH2Cl); 50.9 (CH2N); 71.5, 71.7 (CH2O); 113.1 (CHpyrrole); 119.5 (C=C); 120.6 (C=Cfulvene). MS (EI) C18H20Cl2N2O2S4: 493.98 (M+•).
3.7 Bis-(N-(9-chloro-3,6-dioxa-1-octyl)pyrrolo[3, 4, d]tetrathiafulvalene 10b
Yellow oil, 57% yield. 1H NMR (CDCl3): 3.56–3.73 (m, 20H, CH2O and CH2Cl); 3.98 (t, 4H, CH2N); 6.51 (s, 4H, CHpyrrole). 13C NMR (CDCl3): 43.8 (CH2Cl); 50.8 (CH2N); 70.7, 70.9, 71.4 and 71.7 (CH2O); 113.9 (CHpyrrole); 118.5 (C=C); 119.8 (C=Cfulvene).
MS (PDMS) C22H28Cl2N2O4S4: 582.6.
3.8 Synthesis of bis(pyrrolo)TTF derivatives 11a,b
Compounds 10a or 10b were refluxed for two days in acetone with a large excess of sodium iodide. The solvent was removed in vacuo and the residue was dissolved in dichloromethane. The organic phase was washed with an aqueous solution of sodium thiosulphate, with water, dried over magnesium sulphate and concentrated in vacuo. Compounds 11a,b were used without further purification.
3.9 Bis-(N-(5-iodo-3-oxa-1-pentyl)pyrrolo[3, 4, d]tetrathiafulvalene 11a
Yellow powder, 89% yield, m.p.: 93°C. 1H NMR (CDCl3): 3.21 (t, 6.6 Hz, 4H, CH2I); 3.64–3.71 (m, 8H, CH2O); 3.99 (large s, 4H, CH2N); 6.52 (s, 4H, CHpyrrole). 13C NMR (CDCl3): 2.9 (CH2I); 51.0 (CH2N); 71.1, 72.2 (CH2O); 113.1 (CHpyrrole); 119.5 (C=C); 120.6 (C=Cfulvene).
MS (EI) C18H20I2N2O2S4: 678.
3.10 Bis-(N-(9-iodo-3,6-dioxa-1-octyl)pyrrolo[3, 4, d]tetrathiafulvalene 11b
Yellow oil, 86% yield. 1H NMR (CDCl3): 3.25 (t, 6.9 Hz, 4H, CH2I); 3.55–3.75 (m, 16 H, CH2O); 3.99 (large s, 4H, CH2N); 6.52 (s, 4H, CHpyrrole). 13C NMR (CDCl3): 3.7 (CH2I); 54.5 (CH2N); 70.2, 70.8, 71.4 and 72.6 (CH2O); 113.9 (CHpyrrole); 118.5 (C=C); 119.8 (C=C fulvene). MS (EI) C22H28I2N2O4S4: 766.
3.11 Synthesis of N,N′-Bis(pyrrolo) tetrathiafulvalene cryptands 12a,b
A suspension of 4,13-diaza-18-crown-6 (76 mg, 0.29 mmol) and potassium carbonate (400 mg, 10 equiv, 2.9 mmol) in dry degassed acetonitrile (25 ml) was refluxed during 30 min. The mixture was diluted with acetonitrile (425 ml) and tetrathiafulvalene derivative 11a or 11b (1 equiv) was added. The reaction mixture was refluxed during five days. Then, the solvent was removed in vacuo. The residue was dissolved in dichloromethane, washed with water, dried over magnesium sulphate and concentrated in vacuo. The oil was purified by chromatography on a silica gel column (SiO2 + 4% triethylamine, petroleum ether/dichloromethane 50:50 and dichloromethane/methanol 100:1).
3.12 Compound 12a
Yellow oil, 32% yield. 1H NMR (CDCl3): 2.58 (t, 4.8 Hz, 4H, CH2N); 2.64 (t, 5.6 Hz, 8H, CH2N); 3.33 (t, 5.6 Hz, 4H, CH2O); 3.40 (t, 4.8 Hz, 8H, CH2O); 3.48 (s, 8H, CH2O); 3.57 (t, 4.3 Hz, 4H, CH2O); 3.95 (t, 4.3 Hz, 4H, CH2N); 6.49 (s, 4H, CH). 13C NMR (CDCl3): 51.1 (NCH2); 54.5 (NCH2); 55.6 (NCH2); 69.7, 70.3, 70.3 and 70.7 (OCH2); 112.6 (CHpyrrole); 118.7 (CAr); 122.0 (C=C). HRMS (ESI+) C30H44N4O6S4: obs.: 685.2192 (M+H)+ , th: 685.2222.
3.13 Compound 12b
Yellow oil, 41% yield. 1H NMR (CDCl3): 2.80 (large s, 4H, CH2N); 2.88 (large s, 8H, CH2N); 3.51–3.61 (m, 24H, CH2O); 3.70 (s, 4H, CH2O); 3.97 (s, 4H, CH2N); 6.57 (s, 4H, CH). MS (ESI+) C34H52N4O8S4: obs.: 773.2753 (M+H)+, th 773.2746.
3.14 X-ray structure analysis of compound 5
Data were collected at 293 K on a Nonius MACH3 four-circle diffractometer, using MoKα graphite-monochromated radiation. The structural determination was achieved using the MolEN package.
Crystal Data for (Z)-5: orange plates, triclinic, a = 6.873(5) Å, b = 14.786(5) Å, c = 15.728(0), α = 99.89°, β = 94.89°, γ = 95.24°, V = 1559(8) Å3, Z = 2, 2774 data with I > 3 σ(I), R = 0.057, Rw = 0.066, GOF = 1.807 (CCDC 198842).
4 Supplementary material
The supplementary material has been sent in electronic format to the Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB2 1EZ, UK, as Cif files No. CCDC 198842, and can be obtained by contacting the CCDC.
Acknowledgements
The ‘Service commun d’analyses spectroscopiques’ (University of Angers) is greatly acknowledged for spectroscopic analyses, as well as the MENRT, CNRS, SMARTON (GT) and CMEP 99MDU431 (MM) for their financial support.