

1 Introduction
The design and synthesis of small molecule libraries is steadily growing in importance [1]. Many of these libraries are directed towards a particular biological target, while others are constructed in an effort to explore uncharted areas of molecular diversity. In designing libraries of the latter type, the following requirement should be taken into account:
- • the scaffold should be readily available and derivatisable;
- • the scaffold should be, when possible, suitable for the generation of a variety of distinct, complementary, and well-defined substitution patterns;
- • the scaffold should preferably bear groups having some resemblance with portions of biologically active compounds.
We have recently synthesised a new type of scaffold [2] named BTAa (Fig. 1), obtained by the combination of tartaric acid and α-amino acid derivatives (Bicycles from Tartaric acid and Amino acid) [2−4], which seems suitable for that purpose.
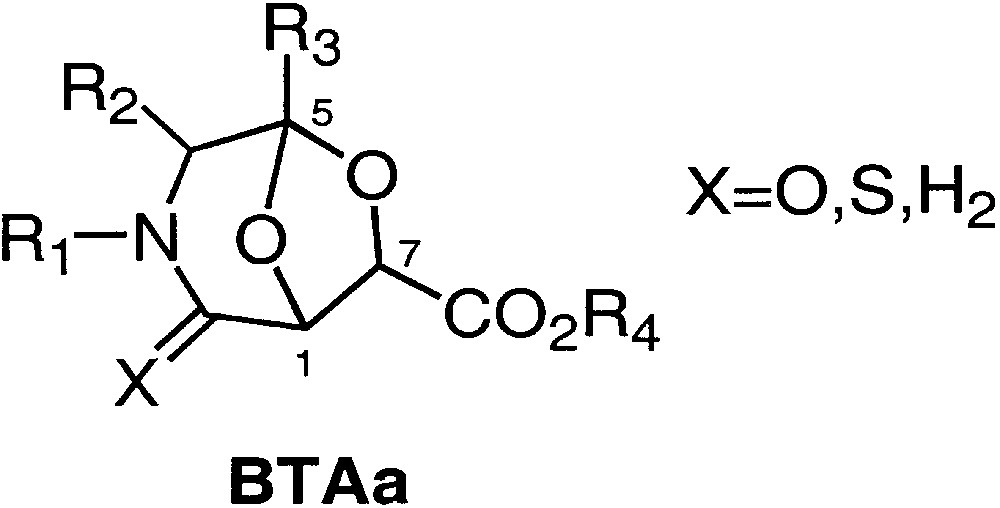
Structure of BTAa.
These scaffolds present a high degree of molecular diversity that can be varied through modifications on the configuration of the stereocentres and the functionalities introduced on the scaffold, by choosing suitable starting reagents. Here we report the preliminary results concerning the generation of a library of amides obtained by neat reaction of two scaffolds with fifteen different amines (Fig. 2).

Generation of amides by neat reaction of two scaffolds with different amines.
2 Results and discussion
The ester 1 was prepared in four steps according to the procedure previously reported by us [2]. The ester 2 was prepared in good yield starting from 1 by treatment with Lawesson’s reagent [5]. Having these scaffolds in our hands, the derivatisation of ester functionality was investigated. During the course of our studies on the reactivity of BTAas, we observed that the ester groups of these scaffolds were highly reactive. Thus methyl amide of ester 1 could be immediately obtained by adding dropwise a solution of methylamine in ethanol. In fact, the reaction was completed at the end of the addition. This observation prompted us to perform additional experiments to optimise the conditions of formation of other amides starting from ester 1 and 2. By mixing the scaffold 1 with an excess of piperidine (10 equiv), without solvent or any other reagent, and by maintaining the temperature at 50–60 °C, we obtained a 50% conversion to the corresponding amide after 4 h and a complete conversion after 16 h. The degree of conversion was evaluated by GC-MS and 1H-NMR analysis. This high reactivity is unusual for esters. Direct aminolysis is generally known to be a difficult reaction, as shown by the high number of methods invented to facilitate it [6−15]. However, these methods utilise different reagents that may limit their use for a library synthesis. Thus only unsubstituted amides and few primary amides can by prepared by direct aminolysis of ester [16, 17]. In our case, the amidation occurs either with primary or secondary amines under mild conditions without using activating reagents, catalysts or solvents (Fig. 2).
After having established the above standard conditions, a small library was generated through parallel synthesis by assembling the building blocks shown in Fig. 3. All the amines utilised are commercially available.

Amines and scaffolds employed.
Esters 1 and 2 were completely converted into the corresponding amides when treated with the primary amines P1–P7. All the new compounds were fully characterised by spectroscopic and analytical means. The same result was obtained with the α-branched primary amines P8 and P9. In the case of secondary amines, all led to the corresponding amides, except the hindered amine S4. The aromatic amines A1 and A2 were also tested, but no amides were obtained, even though the reaction was conducted at higher temperature (100–150 °C). Therefore, it appears that the main use of this methodology lies in the parallel synthesis of amides derived from aliphatic primary and secondary amines.
A typical procedure for the synthesis of amides is as follows: ester 1 (4 mg, 0.014 mmol) and piperidine (S2)(14 μl, 0.14 mmol) were heated in a sealed micro-reaction vessel at 60 °C for 16 h. The mixture was then diluted with MeOH and rapidly filtered through a short column of Amberlyst 15 H+ ion-exchange resin. After concentration, pure amide was obtained in quantitative yield. Clear oil. 1H NMR (CDCl3) δ 7.17–7.37 (m, 5 H, Ph), 5.79 (d, 1 H, H-5, J = 2.2 Hz), 5.12 (s, 1 H, H-1), 4.89 (s, 1 H, H-7), 4.67–4.37 (m, 2 H, CH2Ph), 3.66–3.28 (m, 7 H), 3.11–3.05 (m, 3 H), 3.03–2.93 (m, 2 H). MS m/z (%): 330 (M+, 30), 218 (5), 166 (25), 112 (100), 91 (95), 69 (55). Anal. calcd for C18H22N2O4 (330.38): C, 65.44; H, 6.71; N, 8.48. Found: C, 65.64; H, 6.32; N, 8.16.
In conclusion, we have developed a rapid and practical method for parallel synthesis of a small molecule amide library, delivering products of acceptable purity for biological screening. Full details on the synthesis of larger libraries and their biological evaluation will be reported in due course.
Acknowledgments
We thank MURST and the University of Florence (COFIN 2000-2002), and the ‘Consiglio Nazionale delle Ricerche’ (CNR) for financial support. Mr Sandro Papaleo and Mrs Brunella Innocenti are acknowledged for their technical support.