

1 Introduction
Perovskite-type ABO3 mixed oxides [1–3] and related A2BO4 structures [4–7] had been investigated as catalysts for the exhaust gas after-treatment of automobiles since around 30 years. Their interest is still under study [8–11] and, recently, their introduction to automotive series application, in combination with precious metals, was announced by a car manufacturer [12]. Perovskites are of high thermal stability in oxidation reactions and allow the incorporation of a large number of different metal ions [13]. In this context, we have shown that the partial substitution of copper in La2CuO4 by small quantities of palladium [4] or iron [7] leads to an activity enhancement in the CO + NO reaction. In the present paper, we investigate the influence of the Ln3+ rare-earth ion in the Ln2CuO4 binary oxide. Therefore we replaced La by its homologues Pr and Nd, leading to Pr2CuO4 and Nd2CuO4. As described in former publications, the catalysts were tested in the CO + NO reaction.
2 Experimental
The catalysts were prepared by the so-called evaporation–decomposition method, as exactly described in a former work [5]. Starting from the corresponding nitrates and passing by an intermediate decomposition at 500 °C, the final calcination temperature was 1000 °C.
For chemical analysis, solids were dissolved in a mixture HF/HCl/HNO3. The quantification of Cu was done by AAS on a Perkin Elmer AAS 1100 using an air/C2H2 flame. For the lanthanides, the apparatus used was a Spectroflame D with an Argon-ICP.
XRD was carried out on a counter tube diffractometer Siemens D500 with a copper anticathode. The spectra were recorded in air at room temperature from 3 to 80°(2 θ;) with a resolution of 0.02°(2 θ;).
BET area was measured on 1 g of sample by adsorption of N2 at –196 °C. Previously, a desorption treatment was carried out under a vacuum of 10–2 Pa for 2 h at 500 °C.
Temperature programmed reduction (TPR) experiments under 1% H2/Ar (1.2 l h–1) were performed up to 900 °C with 50 mg of non pre-treated sample, diluted in 150 mg of SiC. The heating rate was 5 °C min–1 and the H2 uptake was detected by thermal conductivity.
The catalytic test was carried out between 150 and 650 °C on 200 mg of catalyst, with a reaction mixture of 2000 ppm CO and 2000 ppm NO diluted in He. Conditions were previously described in full detail [5]. It is important to note here that the temperatures corresponding to 50 and 80% conversion (T50, T80) and the activity values given in this paper were obtained after preconditioning the catalyst under reaction conditions, i.e. in the so-called stabilised state [5].
3 Results and discussion
3.1 Characterisation of the fresh solids
Chemical analysis of the prepared solids shows that the expected Ln/Cu atomic ratios were obtained within a tolerance below 10% in all cases (Table 1). However, a slight excess of copper with respect to the lanthanide stoichiometry can be noticed. Probably the rare earth salts used suffered from hydration, giving rise to a lack of the Ln-component. The specific surface areas of the Ln2CuO4 mixed solids were found to be very close to that of La2CuO4, about 0.5 m2 g–1. The observed phases are the so-called K2NiF4-type structures, La2CuO4, Pr2CuO4 and Nd2CuO4. Besides very weak diffraction lines characteristic of compounds originating from the corresponding lanthanide precursors, La2O3, PrO2 and Nd(OH)3, were identified.
Characteristics of the Ln2CuO4 catalysts
| Catalyst | BET area (m2 g–1) | Analysis | XRD | |
| La2CuO4 | 0.6 | La 68.1% (68.5)a | La2CuO4 | sb |
| Cu 15.8% (15.7) | La2O3? | vw | ||
| [La2Cu1.02Oz]c | ||||
| Pr2CuO4 | 0.4 | Pr 64.9% (68.8) | Pr2CuO4 | s |
| Cu 15.8% (15.5) | PrO2 | vw | ||
| [Pr2Cu1.08Oz] | ||||
| Nd2CuO4 | 0.5 | Nd 65.7% (69.3) | Nd2CuO4 | s |
| Cu 15.2% (15.3) | Nd(OH)3 | vw | ||
| [Nd2Cu1.05Oz] |
a Theoretical values for ideal stoichiometry.
b Intensity of the diffraction peaks: s: strong, vw: very weak.
c Formula calculated from chemical analysis.
The TPR profiles of the three Ln2CuO4 are given in Fig. 1 . The mixed oxides are reduced within the temperature range 350–750 °C, but with marked differences between each other. La2CuO4 exhibits two main peaks at 420 and 495 °C, which may be attributed to reduction of Cu2+ to Cu0 via the intermediate formation of Cu+ [14]. When Pr or Nd substitutes for La, the reduction occurs at higher temperature, with maxima at 540–625 °C for Pr2CuO4 and 600–650 °C for Nd2CuO4. The calculation of the H2 consumption up to 750 °C shows an almost complete reduction of Ln2CuO4 in Cu0 and Ln2O3, the reduction percentage in metallic copper being 104, 95 and 97%, respectively, for La2CuO4, Pr2CuO4 and Nd2CuO4.

TPR profiles of the three Ln2CuO4 mixed oxides.
3.2 Catalytic activity of Ln2CuO4
The T80 and T50 temperatures for the NO conversion are plotted in Fig. 2 as a function of the ionic radius of the rare earth ion. The dashed lines represent the activation observed between the first temperature rise of the test and after catalytic stabilisation, i.e. when two complete cycles of heating and cooling under reaction conditions had been run. This phenomenon of catalytic activation under reaction conditions had already been described in previous work [5]. The values plotted here are those after stabilisation of the catalysts. The T80 and T50 values for Nd2CuO4 and Pr2CuO4 are by around 100–140 K higher than for La2CuO4, which therefore is the most active of the three catalysts.
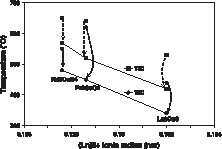
Light-off temperatures for 50 and 80% conversion in the CO + NO reaction as a function of the lanthanide ionic radius. The upper points correspond to the first run, whereas the lower points are those obtained after stabilisation of the activity. The points are connected by dashed arrows in order to show the activation of the catalysts under reaction conditions (decrease of T50 and T80).
In addition to N2, some N2O is formed. The selectivity to N2O is close to 100% at low temperature (200 °C) and then decreases with temperature. It is about 10% at 450 °C. However, the maximum N2O production (≈100 ppm) was observed at around 400 °C for Pr2CuO4 and Nd2CuO4, as it is observed for this type of solids [6].
After stabilisation of the activity, the apparent activation energies given in Table 2 are equal with values around 45 kJ mol–1 for the three Ln2CuO4 oxides, whatever the lanthanide ion is. Before activation under reaction conditions the values differ between the catalysts, 103 kJ mol–1 for Pr2CuO4, 88 kJ mol–1 for Nd2CuO4 and 31 kJ mol–1 for La2CuO4. However, in the latter case, the Arrhenius plot varies strongly during the temperature rise and is not really reliable. In the low temperature domain, it is higher than 150 kJ mol–1.
Apparent activation energies of the Ln2CuO4 oxides before and after activation under CO + NO reaction conditions
| Apparent activation energies Ea a (kJ mol–1) | ||
| catalyst | before | after |
| La2CuO4 | (31)b | 45 |
| Pr2CuO4 | 103 | 46 |
| Nd2CuO4 | 88 | 44 |
a All values with an uncertainty of ±5 kJ mol–1.
b Questionable value given in brackets, energy variable during the temperature rise.
The activity per gram of La2CuO4 shown in Fig. 3 was found to be 4 times higher than for the oxide containing Nd and still 2–3 times higher than for the Pr-based oxide, depending on the temperature at which the activity was measured. Thus, as seen in Fig. 3, the catalytic activity seems to increase almost linearly with the ionic radius of the rare-earth element.
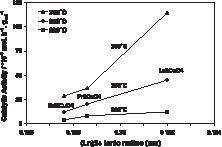
Specific catalytic activity in the CO + NO reaction plotted as a function of the lanthanide ionic radius.
3.3 Modifications of Ln2CuO4 under reaction conditions
We had previously found [5] that La2CuO4 is subject to modifications under CO + NO reaction conditions, leading to the formation of metallic copper and lanthanum hydroxide resulting from hydration of the oxide after exposure to air. In order to check whether this reduction is restricted to lanthanates or is also possible for other rare earth oxides, we recorded X-ray diffraction patterns of the Pr2CuO4 and Nd2CuO4 samples issued from the catalytic test. In Table 3, their phase composition is compared to La2CuO4 in the same state.
Phase identification of the Ln2CuO4 catalysts before and after CO + NO testing
| Catalyst | XRD | |||
| before CO + NO test | after CO + NO test | |||
| La2CuO4 | La2CuO4 | sa | La(OH)3 | s |
| metallic Cu | m | |||
| La2O3 ? | vw | La2CuO4 | vw | |
| Pr2CuO4 | Pr2CuO4 | s | Pr(OH)3 | s |
| PrO2 | vw | metallic Cu | m | |
| Nd2CuO4 | Nd2CuO4 | s | Nd(OH)3 | s |
| Nd(OH)3 | vw | metallic Cu | m | |
| NdCuO2 ? | w |
a Intensity of the diffraction peaks: s: strong, m: middle, w: weak, vw: very weak, (?) phase attribution questionable.
We can state that the initial Ln2CuO4 binary oxide has been completely transformed during the activation under the CO + NO mixture. As previously observed for La2CuO4 [5], the principal phases after reaction and exposition to air are the corresponding rare earth hydroxides Ln(OH)3 and metallic copper. The observed activation under reaction conditions thus corresponds to a reduction of the initial cuprate.
3.4 Discussion
The active sites for the CO + NO reaction should be the same for La2CuO4, Pr2CuO4 and Nd2CuO4. In fact, we found the same apparent activation energy for the three solids after the catalysts had reached their activated state under reaction conditions. We attribute this coincidence to the presence of metallic copper, the common component of the three samples. Indeed for La2CuO4 [7], the surface concentration of metallic copper was found to be related to the catalytic activity, and it was concluded that copper could be considered as the main active species in the reaction. It can be remarked that the presence of metallic copper does not exclude redox mechanisms involving copper in different valence states and occurring at the surface or at the interface with the lanthanide oxide. In the present work, the catalytic activities of the three solids after stabilisation are different and related to the ionic radius of the lanthanide. It suggests that the number of active copper sites should be different for the three solids, which implies some differences in the reduction steps of the original cuprates and in the formation of the copper particles. If we consider the ionic radii of the rare earth element with a coordination number of 12, they are of 0.1286 nm for Pr3+ and 0.1276 nm for Nd3+ compared to 0.1320 nm for La3+ [15]. These differences in ionic radii are small, but they correspond to different energetics of the cuprates crystal lattice. The smaller ionic radii for the Pr- and Nd-based catalysts result in a more compact structure, which makes the oxygen less labile and consequently the reduction of the oxide more difficult. This is evidenced by the TPR study under hydrogen, which clearly shows that the smaller the ionic radius, the higher the reduction temperature. Cu0 is formed less easily when La is substituted by Pr or Nd. However, the link between this difference in reducibility observed by TPR and the changes in catalytic activity requires further discussion. In TPR processes, both thermodynamic and kinetic factors are important to control the initial formation of copper nuclei and then the interface propagation inside the bulk [16]. However, in the present catalytic study, the differences of activities in the CO + NO reaction cannot be related directly to a variation in the reduction rate constants of the cuprates, because the activities were measured on solids after stabilisation, i.e. after formation of the copper particles. In these conditions, the decrease in activity from La to Nd should be related to an increase in the copper particle sizes, which means a lower number of active sites. Indeed, the easier reducibility of La2CuO4 at low temperature favours the formation of a high number of copper nuclei thus inducing the formation of a dispersed copper phase. On the other hand, for the less reducible oxide (Nd2CuO4), the reduction proceeds at higher temperature. There are a smaller number of nuclei and the reduction therefore results in the growth of large particles, all the bigger as the high temperature favours their sintering. Consequently, the number of active sites is decreased and thus the catalytic activity.
4 Conclusion
The replacement of lanthanum in La2CuO4 mixed oxides by praseodymium or neodymium leads to less active catalysts for the CO + NO reaction. As already found for La2CuO4 [5], a reduction under reaction conditions to metallic copper was observed also for Pr2CuO4 and Nd2CuO4. Since Cu0 sites are the active species in the three cases, the increase in the reduction temperatures of the mixed oxides, related to the change in ionic radii of the rare earth element, seems to be at the origin of this decrease in activity, because it results in copper particles with higher particle sizes.
Acknowledgements
The authors want to express their gratefulness to the automobile manufacturer Renault who enabled L.A.C.E. by its financial support to carry out this study. Mrs Laurence Retailleau is also gratefully acknowledged for her technical assistance in the TPR study.