

1 Introduction
Over the last few years, various solid-state magic angle spinning NMR experiments have been proposed to determine the atomic connectivities in solids [1–12]. In rigid solids, the dipolar interactions are usually several orders of magnitude larger than the scalar J couplings. A large number of methods have been thus developed to reintroduce the through-space dipolar interaction under magic angle spinning (MAS), leading to the observation of nearest-neighbour spatial proximities in two-dimensional (2D) correlation spectra [1–7]. However, it has been recognised for several years that weak J-couplings can be used to obtain through-bond correlation spectra in rigid solids, providing a direct identification of atomic connectivities [8–12]. Recently, we have shown that the refocused INADEQUATE experiment allows a straightforward determination of the through-bond P–O–P connectivities in crystalline and disordered phosphates [13]. In this work, we illustrate the applications of the refocused INADEQUATE experiment to the structural characterisation of MP2O7 crystalline compounds (M = Zr, Sn) and lead phosphate glasses.
ZrP2O7 belongs to the M4+P2O7 family of compounds that are of interest because several members of the family show low or negative coefficients of thermal expansion [14,15]. At high temperature, these compounds usually have a simple cubic structure, with Z = 4 and space group symmetry , which can be described as a network of corner-sharing MO6 octahedra and P2O7 pyrophosphate groups [15]. In this ideal cubic structure, the P2O7 groups are on 3-fold axes with the P–O–P bridging oxygen atoms at inversion centres, constraining by symmetry the P–O–P bond angles to 180°. At low temperature, these energetically unfavourable linear bond angles are partially or completely avoided by distortion of the ideal cubic structure, resulting in a room temperature structure with a tripled (3 × 3 × 3) unit cell (Z = 108) [15–17]. Assuming phase transitions with group sub-group relationship, symmetry considerations indicate that the room temperature structure can adopt one of 12 possible space groups derived from the symmetry of the ideal high temperature phase [15]. These various space group symmetries give rise to very subtle differences in the reflections observed by powder X-ray diffraction, but, as the symmetry of the possible space group is lowered, the number of crystallographically distinct P sites and the number of different types of P2O7 groups in the asymmetric unit dramatically increases (see Table 1). As previously demonstrated in the case of SiP2O7 [18] and TiP2O7 [19], through-bond correlation NMR methods, which allow the determination of the number of independent P sites in the asymmetric unit and the phosphorus connectivity scheme, can be used to discuss the possible space groups adopted at room temperature by these materials.
Number of different types of P2O74– groups and number of unique crystallographic P sites expected for the possible space group symmetries adopted at room temperature by MP2O7 materials [15,35]
| Space group | No. of P2O7 groups with two equivalent P sites | No. of P2O7 groups with two inequivalent P sites | No. unique P sites |
| (1 × 1 × 1) | 1 | 1 | |
| 1 | 5 | 11 | |
| P213 | 11 | 22 | |
| 2 | 18 | 38 | |
| R3 | 38 | 76 | |
| Pbca | 1 | 13 | 27 |
| Pca21 | 27 | 54 | |
| P212121 | 27 | 54 | |
| P21/c | 2 | 26 | 54 |
| Pc | 54 | 108 | |
| P21 | 54 | 108 | |
| 4 | 52 | 108 | |
| P1 | 108 | 216 |
In the case of phosphate glasses, magic-angle-spinning NMR provides evidence of the basic building units of the disordered network [20]. The various PO4 tetrahedral units (Qn with n: number of bridging oxygen atoms per tetrahedron) are usually partly resolved in 1D MAS spectra, depending on glass composition. Dipolar correlation techniques have been used to probe the spatial proximities of these Qn units, from which the Qn pair connectivities were deduced [21–23]. The refocused INADEQUATE experiment, which proves to be very useful for crystalline materials, can also be applied to obtain through-bond correlation spectra in phosphate glasses, allowing characterization of the different Qn units and their connectivities in the disordered network.
2 Experimental
2.1 Synthesis
High-purity crystalline ZrP2O7 was prepared from ZrOCl2 and H3PO4 (85%), which were mixed in a 2:1 molar ratio in a platinum crucible and heating to 350 °C for 1 h. The resultant powder was washed with distilled water, and heated to 750 °C then 1000 °C for a 12-h period. The high purity of the ZrP2O7 sample was confirmed by X-ray and neutron powder diffraction.
The PbOx(P2O5)1–x glasses (x = 0.52, 0.59, 0.66) were prepared from reagent-grade PbO and (NH4)2HPO4. The glass samples were melted in Pt crucibles for 1 h under air at temperatures varying from 700 to 850 °C depending on composition and quenched by partly immersing the crucible in water. Glass compositions were checked by microprobe (1 mol% uncertainties).
2.2 Solid-state NMR
The solid-state NMR experiments were performed at room temperature on a Bruker DSX 300 spectrometer (7.0 T) with a 4-mm MAS probehead operating at a Larmor frequency of 121.4 MHz for 31P. The one-dimensional (1D) 31P MAS spectra of the crystalline and glassy samples were recorded at various spinning frequencies (from 3 to 15 kHz) using single-pulse (π/8) acquisition. The recycle delay varied from 80 to 20 s for the crystalline sample and the glasses, respectively, to ensure no saturation.
The two-dimensional (2D) 31P through-bond single-quantum-double-quantum (SQ–DQ) MAS NMR correlation spectra were acquired using the refocused INADEQUATE [24] sequence [12] (Fig. 1a). The π/2 pulse duration was 2.3 μs. The delay τ in the excitation and reconversion periods was synchronized with the rotor period and was set to 10 ms for ZrP2O7, and 5 ms for the glassy samples. For ZrP2O7, the spinning frequency was 12 kHz and 70 t1 increments, with 32 transients each, were collected using a recycle delay of 20 s. For the PbOx(P2O5)1–x glasses, 64 t1 increments, with 32 transients each, were recorded at 14 kHz spinning frequency using a recycle delay of 20 s. For all the samples, a presaturation sequence was applied to ensure equivalent conditions for each transient.
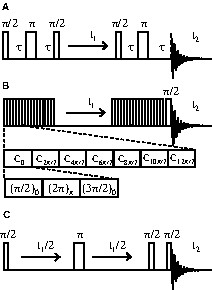
(a) Refocused INADEQUATE pulse sequence [12] used for the 2D through-bond SQ–DQ correlation experiment; (b) POSTC7 pulse sequence [7] used for the 2D through-space SQ–DQ correlation experiment; (c) z-filtered spin-echo pulse sequence [26] used for the 2D J-resolved experiment.
The 2D 31P through-space SQ–DQ MAS NMR correlation spectrum of ZrP2O7 was acquired at 10 kHz spinning frequency. The POSTC7 recoupling sequence [7] (Fig. 1b) was used to reintroduce the 31P–31P homonuclear dipolar interaction and to achieve the excitation and reconversion of double quantum coherences. The rf-field strength was ~70 kHz. Short excitation and reconversion periods of 600 μs were used. 80 t1 increments, with 16 transients each, were collected using a recycle delay of 30 s and a presaturation sequence.
The 2D homonuclear J-resolved MAS NMR spectra were recorded using a z-filtered spin-echo sequence (Fig. 1c) [25–26]. The π/2 pulse duration was 2.3 μs. For the ZrP2O7 sample, the spinning frequency was 12 kHz and 64 t1 increments with 32 transients each were collected using a recycle delay of 20 s. For the PbOx(P2O5)1–x glasses, 70 t1 increments, with 32 transients each, were recorded at 14 kHz spinning frequency, with a recycle delay of 20 s. In both cases, a presaturation sequence was applied to ensure equivalent conditions for each transient.
All the two-dimensional pure absorption phase spectra were obtained using a hypercomplex acquisition [27]. To avoid spinning sidebands in the ω1 dimension, the t1 time increment was synchronized with the rotor period. The spinning frequency was stabilized to ±10 Hz for all experiments. 31P chemical shifts were referenced relative to 85% H3PO4 aqueous solution using the high-frequency positive convention. Simulations of the 1D MAS and 2D MAS correlation spectra were performed using the dmfit software [28].
3 Results and discussion
3.1 Space-group selection in MP2O7 crystalline compounds
The 31P 1D MAS NMR spectra of ZrP2O7 obtained at different spinning rates are shown in Fig. 2 (isotropic region only). These spectra exhibit at least 13 partially overlapping 31P isotropic resonances, indicating that the asymmetric unit of ZrP2O7 contains at least 13 crystallographically different P sites. This allows the exclusion of the possibility of a cubic symmetry with space group, for which only 11 independent phosphorus sites are expected. It can be noted that the positions and linewidths of these resonances vary to different extents with the spinning frequency. In particular, the single resonance located at –44.8 ppm for a spinning frequency of 10 kHz gives rise to a doublet at lower spinning rate, with a spinning-frequency-dependent splitting. Such unusual line shape effects have first been observed in 31P MAS NMR spectra of homonuclear spin pairs with two chemically equivalent spins [29–32]. They are due to the 31P–31P homonuclear dipolar interaction, which lead to a second-order perturbation term in the average Hamiltonian, inversely proportional to the spinning frequency [29–32]. More recently, similar lineshape behaviours have been reported for P2O7 units with two inequivalent P sites having a weak isotropic chemical shift difference, as in the case of TiP2O7 [19] and Cd2P2O7 [33].

31P MAS NMR spectra of ZrP2O7 recorded at spinning frequencies of 3, 5 and 10 kHz (isotropic region only).
As well as the number of individual 31P sites, the number of P2O7 groups in the asymmetric unit can be used for space group assignment. To characterize the number of unique P2O7 groups in the ZrP2O7 structure, we have used the refocused INADEQUATE experiment, which allows determination of the through-bond P–O–P connectivities in crystalline and disordered phosphates [13]. The 2D 31P through-bond SQ–DQ correlation MAS spectrum of ZrP2O7, obtained using the refocused INADEQUATE sequence [12], is depicted in Fig. 3. This 2D spectrum clearly exhibits 11 pairs of resolved cross-correlation peaks (A to K) that reflect the through-bond intra-P2O7 connectivity. The paired cross-peaks A show significantly broader linewidths and integrated intensities about three times higher than those of the remaining correlations (B to J), suggesting the presence of several overlapping contributions. In contrast, the paired cross-peaks K exhibit strongly reduced intensities, as expected from the overlap and the weak chemical shift difference of the coupled resonances. This 2D refocused INADEQUATE spectrum thus indicates that the room temperature structure of ZrP2O7 contains more than 11 P2O7 groups with two inequivalent P sites. This makes unlikely the possibility of P213 space group, for which only 11 P2O7 groups with two inequivalent P sites are expected and suggests that the most probable symmetry of ZrP2O7 is Pbca, which gives rise to 13 P2O7 groups with two inequivalent P sites. It should be remarked that, for Pbca symmetry, a single P2O7 groups with two crystallographically equivalent P sites related by a centre of inversion is also expected (see Table 1). Such P2O7 groups with two magnetically equivalent 31P nuclei are not observable in a through-bond correlation spectrum, but they give rise to diagonal auto-correlation peaks in a through-space dipolar correlation spectrum. The 2D through-space SQ–DQ correlation spectrum of ZrP2O7 obtained using the POSTC7 sequence [7] is shown in Fig. 4 [34]. In addition to the 11 pairs of intense cross-correlation peaks characteristic of P2O7 groups with two inequivalent P sites previously/proved, this spectrum clearly shows a single intense auto-correlation peak along the diagonal (L), arising from a P2O74– group containing two equivalent P sites. It should be noted that this spectrum, obtained using short excitation and reconversion periods, displays some additional correlation peaks of weaker intensities, due to the presence of weaker longer-range dipolar couplings. These weak-intensity peaks reflect the inter-P2O7 spatial proximities, providing additional structural information, but they can damage the spectral resolution in more complex cases [35]. Analysis of the peak intensities in Fig. 4 shows again that the paired cross-peaks A have approximately triple integrated intensities, suggesting the presence of three overlapping cross-correlation peaks corresponding to three P2O74– groups. Good fits (not shown) of the quantitative 1D MAS and 2D correlation spectra were obtained with 27 distinct 31P resonances of approximately equal intensity corresponding to 13 P2O74– units (A to K) with two inequivalent P sites and one P2O74– group (L) with two equivalent P sites. This indicates that the room temperature structure of ZrP2O7 adopts Pbca space-group symmetry, which requires 13 P2O74– groups in general positions and one lying on an inversion centre.
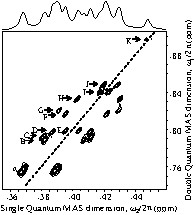
31P 2D through-bond double quantum MAS correlation spectrum of ZrP2O7 obtained at 12 kHz spinning frequency using the refocused INADEQUATE experiment. The dashed line indicates the diagonal of the spectrum. The 1D MAS spectrum is shown above the 2D spectrum.

31P 2D through-space double quantum MAS correlation spectrum of ZrP2O7 obtained using the POSTC7 sequence (10 kHz spinning frequency). The dashed line indicates the diagonal of the spectrum. The 1D MAS spectrum is shown above the 2D spectrum. Adapted from reference [34].
As observed for other crystalline phosphates, the weak through-bond 2J(P–P) coupling that drives the coherence transfer in the refocused INADEQUATE experiment is significantly smaller than the line width of the 31P resonances and is not resolved in 1D MAS and 2D correlation spectra. This through-bond J coupling can be clearly proved from the 2D J-resolved experiment [25], which has been previously used to measure weak 2J(P–P) couplings in metal phosphine complexes [9,36]. The 2D J-resolved MAS spectrum of ZrP2O7, recorded using a z-filtered spin-echo sequence [26], is depicted in Fig. 5. In this 2D spectrum, the characteristic doublets due to the weak 2J(P–P) couplings are clearly resolved in the indirect ω1 dimension and are correlated to the individual 31P resonances in the ω2 dimension. From the observed splitting, the magnitude of the intra-P2O7 2J(P–P) coupling constants is measured to range between 10 and 14 Hz. As expected, the resonance associated with the P2O7 group with two P sites related by a centre of inversion (located at –42.1 ppm in the ω2 dimension) gives rise to a single peak in the ω1 dimension, since there is no observable J-coupling for magnetically equivalent spins. It should be mentioned that the magnitude of the coupling constant measured here is slightly smaller than those reported for other pyrophosphates which range between about 16 and 25 Hz [18,19,33,35,37] and that the absolute sign of the 2J(P–P) coupling in P–O–P units is known to be negative [37].

31P 2D J-resolved MAS spectrum of ZrP2O7 recorded at 12 kHz spinning frequency using a z-filtered spin-echo sequence. The 1D MAS spectrum is shown above the 2D spectrum.
The use of the through-bond SQ–DQ correlation NMR spectra for space-group selection in MP2O7 compounds has also been recently illustrated in more complex cases. As an example, the refocused INADEQUATE spectrum obtained for SnP2O7 [35] is shown in Fig. 6. This 2D spectrum clearly exhibits a spectacular gain in resolution relative to the 1D MAS spectrum, and shows 49 pairs of distinct cross-correlation peaks (some of them having significantly stronger intensities than others). In this case, this shows that the asymmetric unit contains at least 49 P2O7 units with two inequivalent P sites and suggests that the room-temperature structure of SnP2O7 is monoclinic, with P21 or Pc symmetry [35].
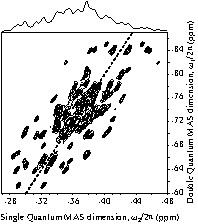
31P 2D through-bond double quantum MAS correlation spectrum of SnP2O7 obtained using the refocused INADEQUATE experiment (10 kHz spinning frequency). The dashed line indicates the diagonal of the spectrum. The 1D MAS spectrum is shown above the 2D spectrum. Adapted from reference [35].
3.2 Through-bond connectivities in PbOx–P2O5 glasses
The 31P 1D MAS NMR spectra of the PbOx(P2O5)1–x glasses (x = 0.52, 0.59, 0.66) are shown in Fig. 7. These MAS NMR spectra exhibit several broad isotropic resonances that can be attributed to the different types of PO4 tetrahedra forming the glass network, corresponding to the 31P isotropic chemical shift range in crystalline lead phosphates (Qn units, with n the number of bridging oxygen atoms) [23]. In agreement with previous work [23,38], the MAS spectrum of the glass with x = 0.52 shows an intense isotropic peak at about –24 ppm, which corresponds to Q2 middle-chain groups involved in long phosphate chains or cycles, and a weaker intensity peak at about –9 ppm assigned to Q1 end-chain groups. As the PbO content increases up to x = 0.65, the intensity of the Q1 resonance increases, indicating a progressive depolymerisation of the phosphate network with PbO addition. For the glass with x = 0.66, the MAS spectrum exhibits a third isotropic resonance at 1.4 ppm, indicating the presence of Q0 units. For each Qn unit, the broadening of the resonance comes from a large distribution of 31P chemical shifts that reflect the structural disorder in the glass, such as bond angles, bond-length variations and higher coordination-sphere disorder.
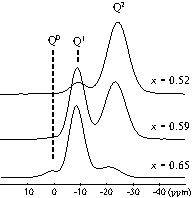
31P MAS NMR spectra of the PbOx(P2O5)1–x glasses recorded at a spinning frequency of 14 kHz (isotropic region only).
Although the quantitative analysis of these 1D MAS spectra provides a determination of the Qn unit distribution as a function of the glass composition, information about pair connectivities between the Qn units remains highly desirable for a better description of the disordered glassy network. Over the last few years, several homonuclear dipolar recoupling MAS NMR methods have been used to probe these Qn pair connectivities [21–23]. These experiments allow the determination of nearest-neighbour spatial proximities between Qn units in 2D through-space NMR correlation spectra, from which information about chemical connectivities is derived. A more convenient way to determine these Qn connectivities is to use the weak through-bond 2J(P-P) coupling. Through-bond homonuclear correlation NMR spectra have already been obtained in disordered solids [12,39] and we have recently shown that the refocused INADEQUATE experiment applies very efficiently to glassy phosphates [13].
The possibility of directly determining the P–O–P through-bond connectivities between Qn units in disordered phosphate networks is clearly illustrated in Fig. 8, which shows the through-bond SQ–DQ correlation spectra of the PbOx(P2O5)1–x glasses obtained using the refocused INADEQUATE experiment. These correlation spectra clearly show several broad correlation peaks that reflect the through-bond connectivities between the Qn units. As shown in Fig. 8a, the refocused INADEQUATE spectrum of the glass with x = 0.52 exhibits an intense Q2–Q2 auto-correlation peak and Q1–Q2 cross-correlation peaks of weaker intensities, revealing the Q2–Q2 and Q2–Q1 connectivities involved in long phosphate chains. For the glass with x = 0.59, the 2D correlation spectrum shows the intense Q1–Q2 and Q2–Q2 correlations expected for phosphate chains of moderate length and an additional Q1–Q1 auto-correlation peak. This Q1–Q1 correlation peak gives evidence of the through-bond connectivity between two Q1 end-chain units and is characteristic of P2O7 groups. This spectrum thus reveals the presence of a chain-length distribution in the glass network and confirms the results previously obtained from the interpretation of through-space dipolar correlation spectra [23]. It should be noted that, in a dipolar correlation experiment, the auto-correlation characteristic of P2O7 groups and the correlations between nearby (but not bonded) Q1 groups of different chains overlap strongly, making the interpretation of the 2D spectrum more ambiguous. The 2D correlation spectrum of the glass with x = 0.66 is mainly composed of Q1–Q1 and Q1–Q2 correlations (Fig. 8c). As expected, the Q0 resonance corresponding to isolated [PO4]3– groups, for which there is clear evidence in the 1D MAS spectrum, is filtered out of the 2D through-bond correlation spectrum.
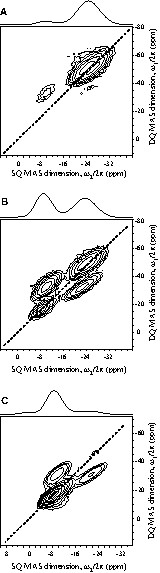
31P 2D through-bond correlation MAS NMR spectra of the PbOx(P2O5)1–x glasses obtained using the refocused INADEQUATE experiment (14 kHz spinning frequency): (a) x = 0.52, (b) x = 0.59, (c) x = 0.66. The dashed line indicates the diagonal of the 2D spectra. The 1D MAS spectra are shown above the 2D spectra.
A notable feature revealed by these SQ–DQ through-bond correlation spectra is that the auto- and cross-correlation peaks of each Qn unit appear at significantly different frequencies in the single-quantum dimension. For example, the centres of gravity of the broad auto- and cross-correlation peaks of the Q1 resonance are located at about –8.4 and –9.7 ppm in the SQ dimension of the 2D maps. This was first observed from through-space dipolar correlation spectra [21–23] and suggests that the Qn units linked to different types of PO4 tetrahedra can be distinguished by their average isotropic chemical shift. As suggested by Witter et al. [22], the Qn species are classified according to their connectivities, Qn,ij, where the additional superscripts describe the type of the bonded PO4 groups. Following this notation, the number of additional superscripts depends on n. For example, the Q1 units forming P2O7 groups are labelled Q1,1 and the Q1 end-chain groups linked to Q2 units are labelled Q1,2, while a Q2 group bonded to two Q1 units (forming a trimer) is labelled Q2,11. From the simulation of the 2D refocused INADEQUATE spectra, assuming a Gaussian line shape for each Qn,ij group, we have determined the 31P isotropic chemical shifts and linewidths of the various Qn,ij units. To reduce the number of fitting parameters, the experimental SQ–DQ correlation spectra were sheared along the ω1 dimension to obtain symmetric SQ–SQ correlation spectra with two equivalent dimensions [25]. The simulation of the 2D correlation spectrum obtained for the PbO0.59(P2O5)0.41 glass is shown in Fig. 9a. For each glass composition, the relative concentrations of the Qn,ij units were obtained from the fit of the quantitative 1D MAS spectrum (Fig. 9b), using the parameters determined from the 2D spectra and an additional intensity constraint, [Q1,2] = 2[Q2,11] + [Q2,21], required by the stoichiometry. The 31P isotropic chemical shifts, linewidths and relative intensities of the Qn,ij groups obtained from these simulations are reported in Table 2. As pointed out previously [22], the phosphate chain-length distribution can be obtained from the Qn,ij concentrations for glasses with low P2O5 content.

(a) Sheared and symmetrized 31P 2D refocused INADEQUATE spectrum of the PbO0.59(P2O5)0.41 glass and its simulation with the parameters reported in Table 2. (b) Fit of the quantitative 31P MAS spectra with the Qn,ij contributions as determined from (a).
Isotropic 31P chemical shifts (δISO) and linewidths (full width at half-maximum: fwhm) of the Qn,ij units in the PbOx(P2O5)1–x glasses obtained from the simulations of the 2D refocused INADEQUATE spectra (δISO ± 0.2 ppm, fwhm ± 0.5 ppm). The relative intensities (I) of the Qn,ij units were obtained from the fit of the quantitative 1D MAS spectra using the same parameters (I ± 7%).
| Qn,ij unit | Q0 | Q1,1 | Q1,2 | Q2,11 | Q2,21 | Q2,22 |
| δISO (ppm), | 1.4 | –8.3 | –9.7 | –20.8 | –22.8 | –24.9 |
| Fwhm (ppm) | 6.2 | 6.2 | 6.6 | 8.5 | 8.8 | 9.2 |
| Glass Pb0.52(P2O5)0.48, intensity (%) | — | — | 11.0 | — | 11.0 | 78.0 |
| Glass Pb0.59(P2O5)0.41, intensity (%) | — | 14.0 | 31.5 | 6 | 19.5 | 29.0 |
| Glass Pb0.66(P2O5)0.34, intensity (%) | 5.5 | 52.0 | 27 | 11.5 | 4.0 | — |
As in the case of crystalline samples, the very weak isotropic 2J(P–P) coupling used in the through-bond correlation experiment can be demonstrated using the 2D J-resolved experiment. As shown in Fig. 10a, the 2D J-resolved spectrum of the PbO0.66(P2O5)0.34 glass allows the clear resolution of characteristic multiplet patterns due to the 2J(P–P) couplings. As expected, the Q1 and Q2 resonances are correlated in the indirect ω1 dimension to doublet and triplet structures, respectively. A single resonance at zero-frequency is observed for the Q0 resonance, since there is no 2J(P–P) coupling. The cross-sections extracted along the indirect ω1 dimension at the centres of gravity of the various Qn resonances are depicted in Fig. 10b. Clearly, this 2D experiment provides an unambiguous attribution of the various Qn resonances in glasses according to their J-multiplet structures and confirms the previous assignments based on the 31P isotropic chemical shift range. Importantly, this 2D spectrum reveals a strong correlation between the distributions of 31P isotropic chemical shifts and the distribution of 2J(P–P) coupling constants of the Qn resonances. We observe that, as the 31P isotropic chemical shift varies from –6 to –11 ppm across the broad Q1 resonance, the magnitude of the 2J(P–P) coupling constants determined from the observed splitting increases continuously from 17 to 20.5 Hz. It should be noted that such a strong correlation between isotropic chemical shifts and J-coupling values is not observed for crystalline lead phosphates or for the MP2O7 compounds (see Fig. 5). Further experiments, allowing the measurement of the distributions of 2J(P–P) coupling constants for each Qn,ij units from 3D NMR spectra, are in progress.
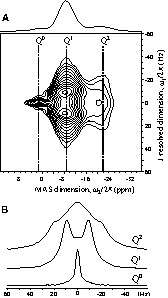
(a) 31P 2D J-resolved MAS spectrum of the PbO0.66(P2O5)0.34 glass recorded at 14 kHz spinning frequency using a z-filtered spin-echo sequence. The 1D MAS spectrum is shown above the 2D spectrum. (b) Cross sections of the 2D J-resolved spectrum at –22 ppm for the Q2 units (top), at –8.7 ppm for the Q1 units (middle) and at 1.4 ppm for the Q0 units (bottom).
4 Conclusion
In this work, through-bond correlation NMR experiments have been used to characterize the structure of crystalline and disordered phosphates. In the case of MP2O7 crystalline compounds, we show that the refocused INADEQUATE experiment provides evidence of the number of distinct P sites and the number of P2O7 group in the asymmetric unit, allowing discrimination between the 12 possible space group symmetries of the room temperature structure. The results suggest that ZrP2O7 adopts the space group symmetry Pbca at room temperature. For lead phosphate glasses, we show that the refocused INADEQUATE experiment can be efficiently used to determine the P–O–P through-bond connectivities of the various Qn units and to obtain intermediate-range order information in the disordered network. In these solids, the weak through-bond 2J(P–P) couplings are clearly shown by using the 2D J-resolved experiment. For the lead phosphates glasses, this allows the assignment of the Qn resonances according to their multiplet patterns. A strong correlation between the distribution of 2J(P–P) couplings and the distribution of 31P isotropic chemical shifts of the Qn resonances is experimentally observed in these disordered systems.
Acknowledgements
FF, DM, IJK and RKH acknowledge support by European Community Contract HPRI-CT-1999-00042 and HPMT-CT-2000-00169. JSOE acknowledges support from the EPSRC under GR/N00524 and GR/M35222/01; RKH thanks the EPSRC for Grant GR/M88341.