

1 Introduction
Environmental acceptable manufacturing processes, including chemical synthesis, have been the recent focus of a wave of new technology commonly called ‘green chemistry and engineering’ [1]. Among the technologies addressing this problem is supercritical fluid technology and its many variants, namely supercritical fluid extraction (SFE), supercritical fluid fractionation (SFF), supercritical fluid chromatography (SFC) and supercritical fluid reactions (SFR). In addition it is important to note that the nature of critical fluid processing has also changed in the last 30 years. No longer is supercritical fluid extraction (SFE) the only method used for the isolation of specific components from natural matrices. As shown in Fig. 1, fractionation and reaction modes utilizing supercritical fluids are becoming more common, and can be coupled with SFE to produce a customized end product or fraction. Whereas the traditional first step in Fig. 1 was usually extraction-based (i.e., the removal of caffeine from coffee to produce a decaffeinated product, or isolation of a hops extract for flavoring), today's processes can utilize a supercritical fluid fractionation (SFF) process, either initially or coupled with SFE, to produce value-enhanced products. Recently, the recognition that critical fluids offer a unique processing media in which to conduct reactions [2] opens but yet another option to the food and agro-product processor.
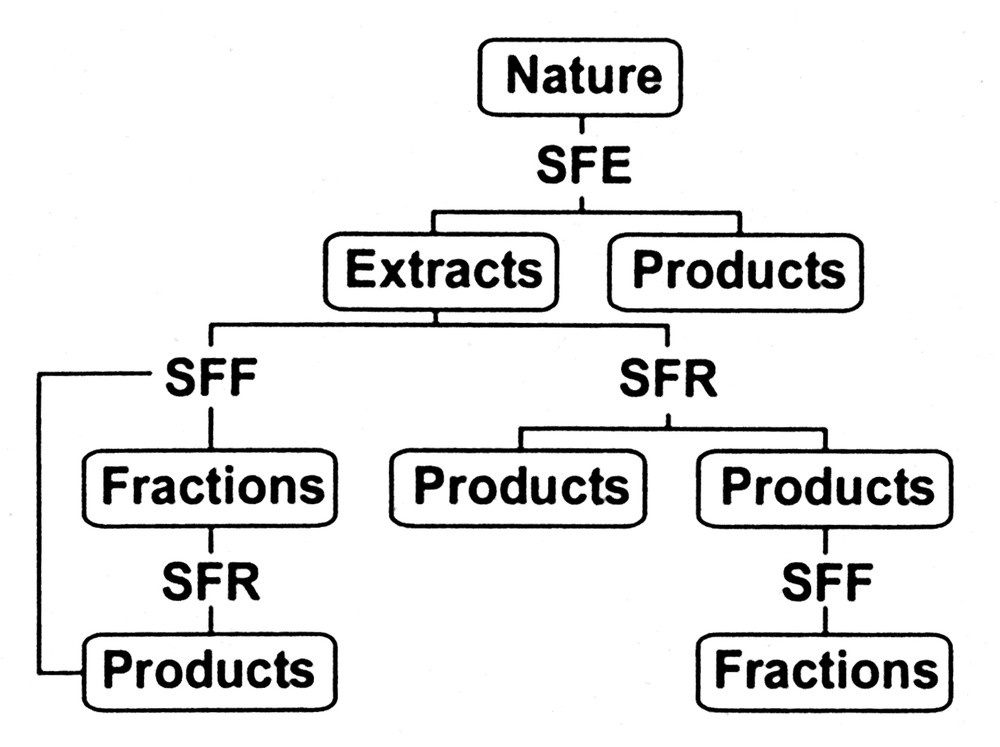
Process sequences utilizing supercritical fluid media for isolating or synthesizing desired end products.
The application of supercritical fluids (SF) and similar media for the processing of agricultural or natural products has traditionally focused on the extraction mode utilizing carbon dioxide in its supercritical (SC-CO2) or liquid (LCO2) state. Beginning in the mid-1980s, options other than varying the extraction fluid’s density in the SFE mode were developed, such as columnar and chromatographic techniques, which facilitated SF-derived extracts or products having more specific composition and properties. This development was followed by the advent of conducting reactions (SFR) in the presence of SFs as documented in the literature [3,4]. Further examination of alternative fluids, such as subcritical water have expanded the ‘natural’ fluid base available to the processor of agriculturally- derived products. Therefore it should be possible to process natural product matrices utilizing a series of pressurized fluids such as suggested by the sequence below:
The above sequence suggests that some degree of selective solvation should be possible, with SC-CO2 or LCO2 extracting non-polar solutes followed by the enhanced solubilization of more polar moieties via the addition of ethanol to the SF. Processing with pressurized water, i.e., subcritical H2O (sub-H2O) expands the range of extractable solutes into the polar solute range with selectivity being controlled by the temperature of extraction or addition of ethanol. Depending on the composition or morphology of the natural product being extracted, there is no reason in theory or practice that the above process could not be done in the reverse order. When these fluid media are combined with the processing options mentioned previously, a powerful array of possibilities result that can be used for enriching higher value chemicals/phytonutrients from natural product substrates. This combination of process options and environmentally benign fluids has been previously noted by King et al. [5].
The review describes how supercritical fluid processing can be applied to great advantage in the conversion of seed-derived oils to useful industrial chemicals. A major topic is the utilization of enzymes for converting oils to useful derivatives. We have also found in certain instances that high pressure CO2 can act as a catalytic agent, thereby avoiding the use of heterogeneous catalysts that must be removed from the product mixture after completion of a reaction. Enrichment of specific components in a product mixture can also be affected by SFF, and we have applied a thermal gradient column to enrich the monoglyceride content of a synthetic glyceride mixture. SFC and SFF techniques can also be employed to enrich target nutraceutical moieties from complex natural product matrices.
2 Fractionation with critical fluids
Prior to the mid-1980s, critical fluid-processed products were largely derived using SFE; either by selecting a given fluid density that would yield the desired product, altering the extraction fluid density as a function of processing time, or in some cases selectively decreasing the pressure after the extraction stage to achieve the desired extract. Useful separations have been attained using the above techniques [6], but largely between compounds differing significant in their physicochemical properties, e.g., molecular weight, vapor pressure, polarity.
Recently, fractionation processes utilizing critical fluid media has been improved by combining principles utilized in supercritical extraction with other separation techniques. These improved methods often make use of fractionating columns or chromatography to yield improvements in the resolution of molecular species. The fractionating column or tower approach is somewhat analogous to operating a distillation column, but there are differences when using critical fluid media. For an understanding of the fundamentals involved in using this technique, one should consult the books by Clifford [7] and Brunner [8].
Fig. 2 illustrates the components and principles involved in fractionating using a very simple column. In this case, SC-CO2 is brought to the desired pressure and then directed to flow upward inside the fractionating column, which usually contains a packing to encourage contact between the SC-CO2 and the components being separated. The components to be separated are injected with a pump into the flowing SC-CO2, prior to its entry into the column. This fluid-solute mixture than enters the first heated zone in the vertical fractionating column and the separation process is initiated. The SC-CO2–solute mixture than ascends the column, usually encountering zones of increasing temperature, which facilitate the separation of solutes based on their relative solubilities in SC-CO2 and their respective vapor pressures. In effect, the column is operating under a density gradient since the fluid is kept isobaric. The described fractionating column can be operated in either the batch or semi-continuous mode with co-current flow of the solute and supercritical fluid streams. Using this approach, we and others have demonstrated the enrichment of monoglycerides from a concentration of 45 weight% in a mixed glyceride starting material to a 95 weight% content in the extract taken off the top of the column [9].
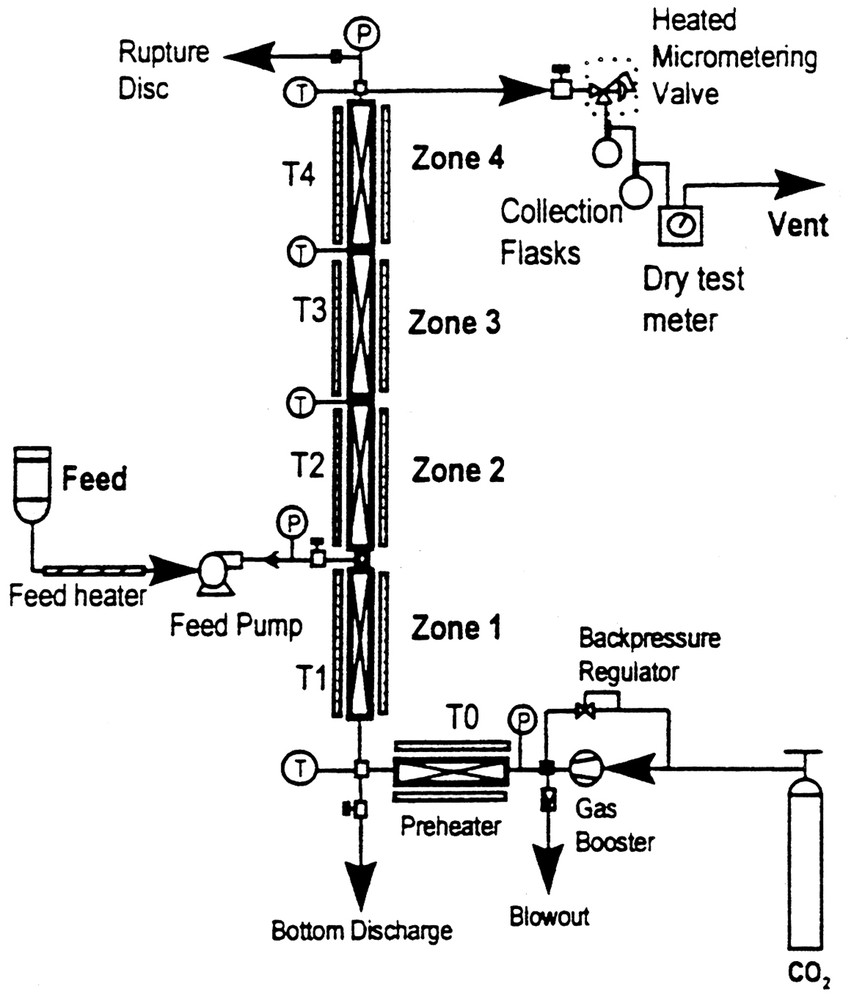
Schematic of the packed column thermal gradient fractionation system.
The use of a packed tower fractionation scheme does not necessarily require prohibitively high processing pressures to obtain significant enrichment of the monoglyceride component in these feed mixtures. Enrichment of the monoglyceride (MG) component relative to the diglyceride (DG) and triacylglyceride (TG) components can be achieved at pressures as low as 17.5 MPa. Yield of the top product from the fractionating tower is approximately linear with respect to pressure. In these experiments [9], a packed 56 cm long column (1.43 cm in diameter) was used at pressures between 20–35 MPa, incorporating a thermal gradient ranging from 65–95 °C (see Fig. 2) to enrich the MGs in reaction mixtures containing mixed glyceride species (i.e., glycerolysis products). Fractionating efficiency is an acute function of glyceride throughput and pressure, the optimal fractionation occurring at lower pressures (17.5 MPa).
This indicates that there is a tradeoff between separation efficiency and product yield as is so often the case in separation technology [10]. Interestingly, significant MG enrichment can be achieved by using an isothermal column temperature (90 °C), which indicates the role that solute vapor pressure plays in achieving the reported fractionation. However, maximum top product yields were achieved at an isothermal column temperature of 65 °C, indicating also the importance of CO2 density on the fractionation process. The achieved MG, DG and TG separation factors appear to be independent of CO2 flow rates from 2–12 l min–1 (decompressed), while top product yield per kilogram of CO2 was found to decrease as the expanded CO2 flow rate increased.
Similarly, in recent studies designed to achieve an enriched nutraceutical-rich extract [11], contaminant free fatty acids were removed from vegetable oil deodorizer distillates to enrich the free sterols and steryl ester content of the raffinate as shown in Table 1. Note that for both rice bran and soybean oils, the starting material (the feed into the column) contained between 32–38 weight% free fatty acid and 13–18 weight% sterols, while the raffinate collected after the fractionation, had a free fatty acid content of between 5–8 weight% and sterol-steryl ester enrichments of 27–34 weight%. These results could undoubtedly be improved upon by employing an even longer fractionating column or by operating in the countercurrent mode as demonstrated by investigators in Germany [12] and Italy [13].
Raffinate compositions (weight%) from fractionation tower separation of vegetable oil deodorizer distillates (DD) using SC-CO2
| Component | Rice Bran DD | Soybean DD |
| Free fatty acids (FFA) | 5 | 8 |
| Sterols | 20 | 31 |
| Steryl esters (StE) | 7 | 3 |
| Triglycerides (TG) | 38 | 30 |
Another SFF option is to employ chromatography in the preparative or production mode, in its own right, or coupled with a preliminary SFE enrichment stage. Our research has shown that it is possible to enrich target compounds (e.g., tocopherols) to levels that were not possible by using SFE alone. Fig. 3 illustrates the principle involved in this form of fractionation when using SC-CO2, both with and without an organic cosolvent, in either the SFE or supercritical fluid chromatography (SFC) stages. Here a preliminary optimized SFE step is utilized to achieve the best possible enrichment of solutes prior to introduction of the extract to the chromatographic column. Then, using inexpensive commodity adsorbents whenever possible, the extract from the extraction stage is diverted onto the top of the chromatographic column, where it is further fractionated using either neat SC-CO2, or SC-CO2 with ethanol and water as eluents.
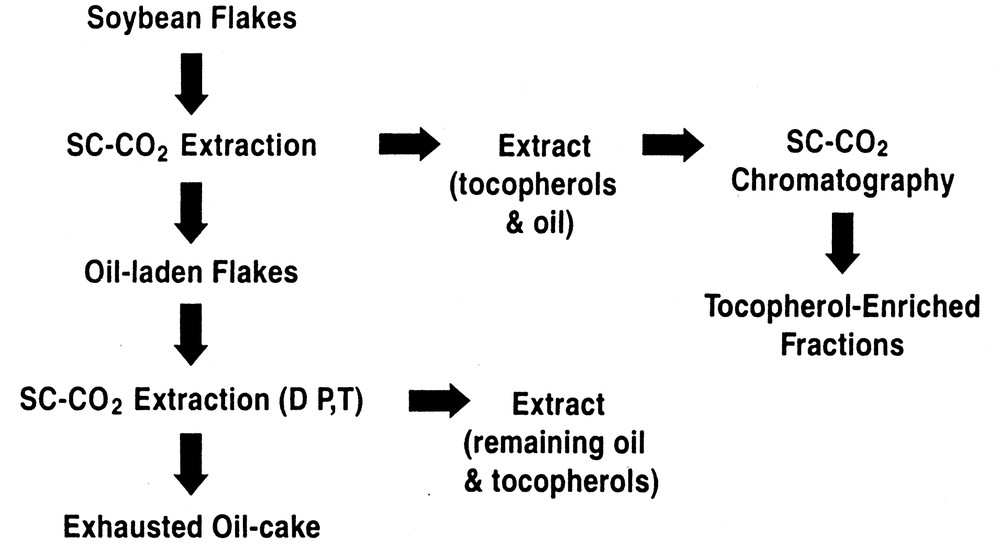
Process for the enrichment of tocopherols by SFE/SFC.
Using this approach, we have been able to enrich moieties such as tocopherols, phospholipids, or steryl esters from vegetable oils, seeds, and by-products of the milling or vegetable oil refining processes. In a previously reported study King et al. [14] have shown that by careful control of SFE parameters it is possible to produce an enrichment of tocopherol components in soybean oil extract. This is achieved due to the differential extraction rates of tocopherols relative to the triglycerides in the oil at 25 MPa and 80 °C, which produces enrichment factors relative to their concentration in soybean ranging from 1.8–4.3. To provide further enrichment, these researchers employed preparative scale supercritical fluid chromatography (SFC) on the extracts from the SFE stage using a commodity silica gel. After screening assorted of extraction conditions, further SFC enrichment was affected by conducting SFC at 25 MPa and 40 °C. After SFC was performed, enrichment factors rose from 2.5 to 31 depending on the individual tocopherol.
Fig. 4 is for the specific case for the separation, enrichment, and fractionation of phospholipids (PLs) from vegetable oil or seeds [15]. Here soybean flakes are initially extracted with SC-CO2 to remove the oil from flakes, followed by extraction of the PLs from the de-oiled flakes with a SC-CO2/ethanol mixture. The second extraction step produces an extract enriched in PLs since PLs are not appreciably soluble in neat SC-CO2, but can be selectively removed from the flake matrix with the aid of ethanol as a cosolvent. As shown in Table 2, the second SFE using SC-CO2/ethanol produces an extract containing a total of 43.7% by weight of PLs. This is a considerable enrichment relative to the concentration of the PLs in the starting oil or seed matrix. Further PL enrichment is facilitated as noted above, by transferring this extract enriched in PLs to an alumina preparative SFC column, where SC-CO2 modified with a 5–30 vol.%, 9:1 ethanol:water eluent is used to elute and fractionate the PLs. In the case of the SFC enrichment step, eluent fractions can be collected as a function of time and their PL content quantitated. As indicated by the data given in Table 2, collection of discrete fractions during the SFC process can produce purities in excess of 75% for the individual PLs, phosphatidylcholine and phosphatidylethanolamine. It should be noted in the described process, that in the SFC steps, that only GRAS (Generally Regarded As Safe) solvents are being used for the enrichment process.
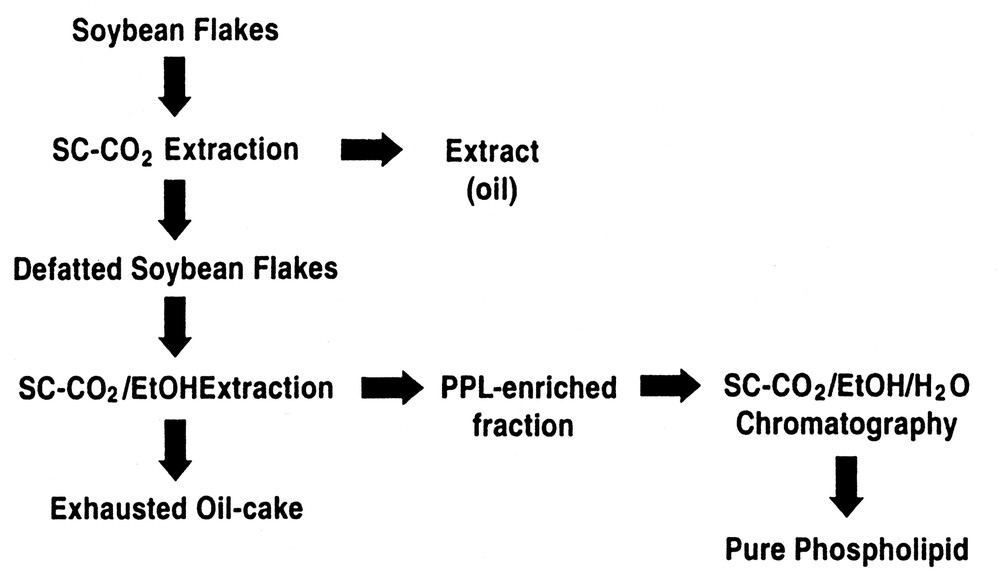
Process for the enrichment of phospholipids by SFE/SFC.
Percentage amounts of PLs in extracts derived from SFE and SFC processing stages*
| Phospholipid type | SFE Stage | SFC Stage |
| Phosphatidylethanolamine | 16.1 | 74.9 |
| Phosphatidylinositol | 9.2 | 20.8 |
| Phosphatidic acid | 2.8 | 55.8 |
| Phosphatidylcholine | 15.6 | 76.8 |
* Relative to other eluting constituents (oil and unidentified peaks).
Recently, a similar SFE/SFC process has been used to isolate sterols and phytosterol esters from agricultural by-products such as corn bran and fiber [16]. For example, by using both SFE and SFC, it has been possible to isolate a fraction containing up to 53% by weight of a cholesterol-lowering agent, ferulate phytosterol esters (FPE), from corn fiber oil. Sterols and steryl esters can also be produced by SFE/SFC from a similar substrate, corn bran. The enrichment of these types of nutraceutically-active compounds with natural agents such as CO2, ethanol, or water, creates an attractive additive system for producing a unique functional food composition as well as demonstrating the potential of SFF.
3 Applied reaction chemistry in critical fluids
There is a historical precedent for conducting specific reactions under supercritical conditions, such as the polymerization of ethylene or isomerization of olefins. In these reactions, it is often just the application of pressure as a thermodynamic variable that permits the reaction equilibria to be shifted, yielding the desired end products. However since the early 1990s, there has been an increasing awareness that by conducting reactions in critical fluid media, one can control not only the equilibrium position of a reaction, but the product distribution and end properties (i.e., color or morphology).
For example, mass transfer rates of reactants and products are substantially improved in supercritical fluids, resulting in transport properties (i.e., diffusion coefficients) and numbers that are more attractive relative to those found in the liquid state. Alteration of the density of the fluid also allows subtle control for the reactant or product solubility in the dense transport media and control of the product distribution. It is also possible in some specific cases to conduct reactions at low temperatures in a non-oxidative environment (CO2), thereby protecting compounds that would be altered in more severe thermal environments. Reaction catalysts can also be regenerated with the aid of supercritical fluids (6) after completion of a SFR.
Two types of reactions that we have studied and have implications in the oleochemical industry are enzymatic-catalyzed and hydrogenation reactions. The coupling of enzyme catalysis with critical fluids as CO2 is particularly attractive, since both are ‘natural’ agents, allowing lipid food components to be manufactured without introducing a solvent or catalyst residues into the final product. In some cases, SC-CO2 can act as a reactant in a process or alternatively as a catalyst. An example of the latter case is the effect of SC-CO2 on conversion of vegetable oils to yield mixed glyceride compositions enriched in monoglyceride content. Temelli et al. [17] have shown that by subjecting a glycerolysis reaction to a pressure of 21 MPa at 250 °C that mixtures of mono-, di- and triglycerides, equivalent to those obtained using alkali metal catalysts, can be synthesized. However absence of the metal catalysts in the SC-CO2-initiated process eliminates the troublesome filtration step associated with the currently used industrial processes. Further, the produced mixed glyceride compositions are much lighter in color then those synthesized using metal catalysts. These end products represent the lower cost products for the glyceride-based emulsifier market.
Table 3 shows the synthesis of monoglyceride-containing mixtures from several commodity vegetable oils employing the above reaction conditions. The monoglyceride component ranges from 49.2 wt% for the glycerides derived from glycerolysis of soybean oil to 41.1 wt% in the case of cottonseed oil. These results were attained at a reaction temperature of 250 °C, a reactor pressure of 20.7 MPa, using a glycerol-to-oil ratio of 25 and the addition of 4% water to the stirred autoclave. Such results suggest that SC-CO2 has an autocatalytic effect on the glycerolysis.
Weight percent composition of glycerolysis products obtained from different vegetable oils during glycerolysis in the presence of SC-CO2
| Oil type | Monoglycerides | Diglycerides | Triglycerides | Fatty acids |
| Soybean | 49.2 | 26.6 | 10.1 | 14.0 |
| Peanut | 46.6 | 32.1 | 12.5 | 8.8 |
| Cottonseed | 41.1 | 35.0 | 12.6 | 11.3 |
| Corn | 45.6 | 32.3 | 13.0 | 9.2 |
| Canola | 41.7 | 33.0 | 16.0 | 9.3 |
The above transformations can be conducted in the presence of a lipase, but particular attention must be paid to the temperature, pressure, and presence of water in the supercritical fluid system. Reactions of this type can be conducted in either batch or flow systems, the latter being attractive for the continual production of the desired product. Of particular note is the use of Novozym 435, a lipase derived from C. antarctica, as a catalyst supported on a polyacrylate resin. This enzyme has proven particularly effective catalyst in the presence of SC-CO2, although the need for more pressure- and temperature-resistant lipases would be a welcomed addition for use with critical fluid media.
An example that parallels the above-cited glycerolysis reaction and demonstrates the versatility offered by using enzymatic catalysis in the presence of flowing SC-CO2 is the glycerolysis of soybean oil with Novozym 435. The described process has been patented [18], but details are available in a related publication [19]. To initiate the glycerolysis in SC-CO2, the vegetable oil is fed into the SC-CO2 by a pump, where it is dissolved in the compressed fluid along with an excess of glycerol containing a small quantity of water. This mixture is then passed over the Novozym 435 supported enzyme bed in the reactor vessel, where total or partial glycerolysis takes place to yield glyceride mixtures. The overall yield and composition of the resultant glyceride mixture is dependent on a number of adjustable variables, such as flow rate of reactants or water content of the glycerol. As shown in Table 4, one can change the glyceride product distribution obtained from the reaction, by varying the water content of the reactants. Table 4 indicates that by using a relatively low water content, a glyceride mixture having a high monoglyceride content (84%) can be obtained, however by increasing the water content in the glycerol reactant solution to 4.2% the monoglyceride content in the final product can be reduced to 67% by weight. The resultant glyceride mixtures also tend to be much paler in color when using the SC-CO2-based synthesis method, a fact confirmed in a related study involving glyceride synthesis in the batch mode [17].
Effect of water content of glycerol (weight%) on the product distribution from soybean oil glycerolysis using SC-CO2a
| Components | 0.7% H2Ob | 4.2% H2Ob |
| Free fatty acids | N.D. | 1 |
| Monoglycerides | 84.0 | 67.0 |
| Diglycerides | 15.4 | 28.9 |
| Triglycerides | 0.6 | 3.1 |
a Conditions: corn oil oil and glycerol flows 10 μl min–1, 70 °C, 27.6 MPa.
b Product distributions in weight%.
Initial screening studies conducted on the Isco SFX-2–10 extractor system indicated that corn, cottonseed, olive and soybean oils underwent glycerolysis readily at pressures between 20.7–34.5 MPa and temperatures of 40–70 °C. These conditions were optimized for soybean oil at 27.6 MPa and 70 °C. It is important when utilizing a lipase during a glycerolysis reaction that the water level in the reactor be controlled and the moisture level in the reactants be assessed. Karl Fischer titration on the Novozym 435 preparation revealed a 1.4 w/w% moisture level, while the moisture level on different lots of glycerol employed in the glycerolysis experiments ranged between 0.7–4.2 wt%. Disadvantages of having a high water level in the reactor are twofold: excessive moisture inhibits the activity of the enzyme (i.e., the glycerol containing 4.2 w/w% reduced the enzyme’s activity to only 60% of the activity exhibited in the presence of 0.7 w/w% water), and in addition, the presence of water promotes the competitive reaction of hydrolysis, thereby facilitating the formation of fatty acids. A kinetic time lag in the reaction may also be exhibited until the enzyme’s water content is partially removed by the flowing SC-CO2 [19].
The reaction of soybean oil with various alcohols was also compared in this study and found to parallel the solubility of the alcohols in SC-CO2. Curiously, it was found that the reaction with alcohols such as 1,2-propanediol and glycerol occurred, despite feed rates of these alcohols and soybean oil that exceeded their solubility in CO2. This suggests that a multi-phase system is present in the fluid phase that is not inhibitory with respect to conducting the glycerolysis. Similar results were also obtained in reacting soybean oil with ethylene glycol, but the presence of ethylene glycol also tended to denature the enzyme. The relative activity of Novozym 435 towards alcoholic reactants was 100, 90.6, 53.4, and 2.2 for reactants methanol, 1,2 propanediol, ethylene glycol, glycerol, respectively.
Along a similar theme, Jackson et al [20] demonstrated that the above lipase could also effectively randomize vegetable oils to yield potential products for incorporation into margarine formulations. The degree of randomization attained on the vegetable oils dissolved in SC-CO2 was a function of extraction/reaction pressure, the flow rate of the SC-CO2, and the quantity of enzyme utilized. Dropping point and solid fat index (SFI) data of the products randomized in SC-CO2 were compared to oils randomized by conventional methods and the agreement between these two differently synthesized products was encouraging. As shown in Fig. 5, randomized palm olein (PO) and genetic-engineering soybean oil, randomized in SC-CO2 (HS-1), had similar SFI as a function of temperature to hydrogenated blends of vegetable oils. It was further found that the fat dropping point was dependent on the catalyst concentration, yielding an inverse dependence on the throughput of the dissolved oil across the supported lipase bed. High performance liquid chromatographic (HPLC) analysis of the randomized and initial oils showed only small changes in the tri-unsaturated, di-unsaturated, di-saturated, and tri-saturated glyceride species.

Solid fat content of palm olein (PO) and HS-1 soybean oil before and after randomization.
As shown in Fig. 6, there is a change in relative composition of the glyceride components for palm olein, particularly in the increase of the LOP, SOO and POO triglycerides in randomized palm olein, relative to the levels found in palm olein. Fig. 7 compares the HPLC profiles for the high stearate soybean oil with the randomized product after passage through the lipase reactor bed. Again, there are subtle changes in the triglyceride composition that have a considerable impact on the physical properties of the oil before and after interesterification. Both of the above oils show losses in the di-saturated glyceride along with a concomitant increase in tri-and di-unsaturated triglycerides. The loss of disaturate functionality has been shown to aid in fat crystallization [21]. This nicely illustrates how a customized oleochemical-derived product can be synthesized using a reaction conducted in supercritical fluid media.
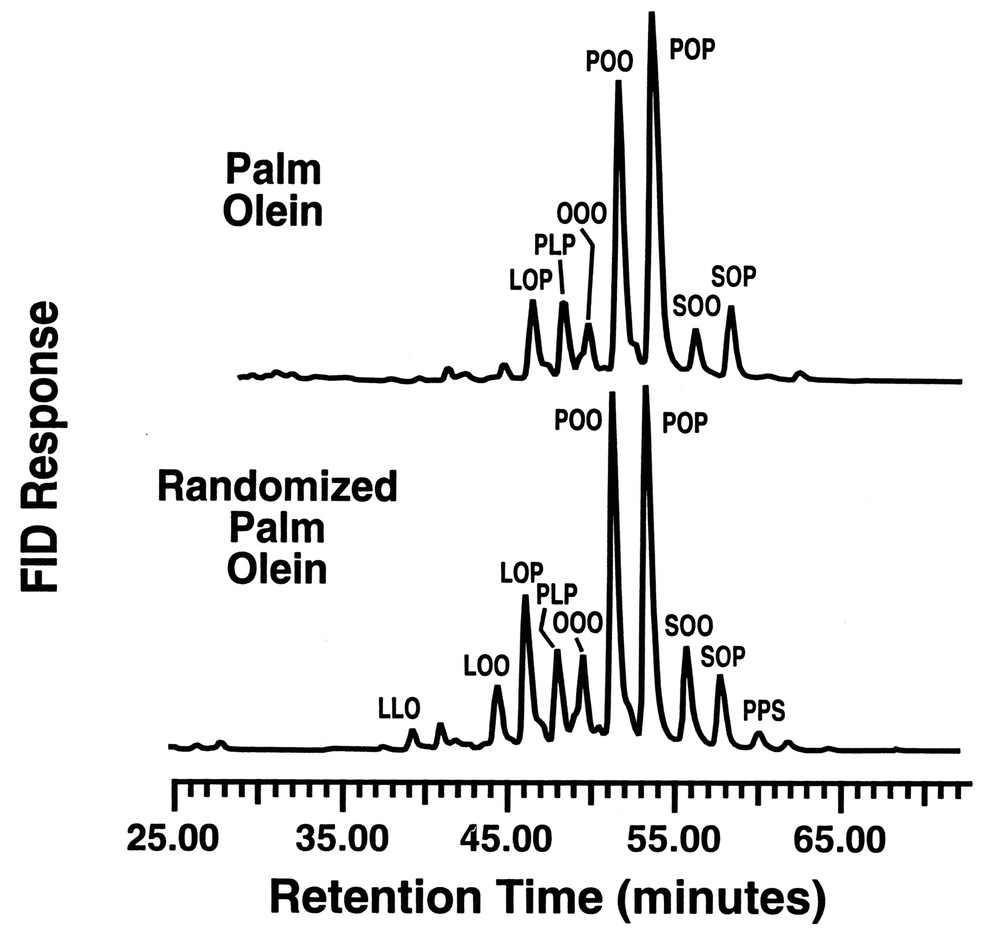
RP–HPLC of palm olein fraction before and after randomization.
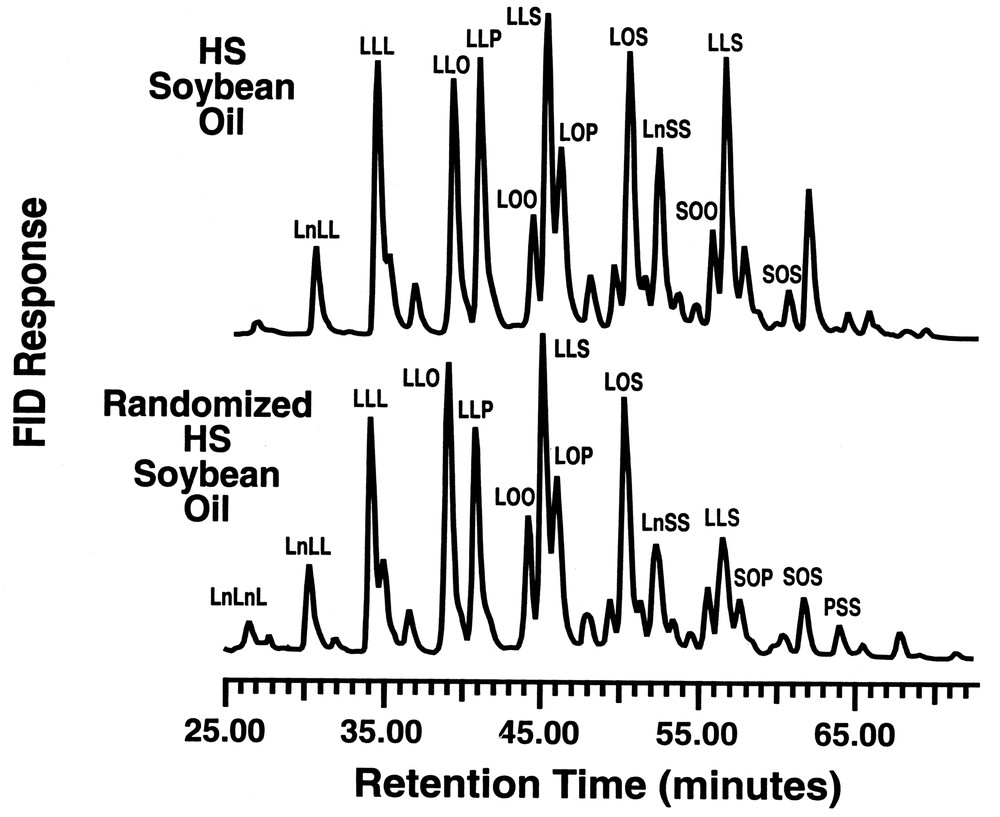
RP–HPLC of high stearate soybean oil before and after randomization.
Research by Jackson and King [22] has demonstrated that vegetable oils could be readily converted to their methyl esters after SFE of the seed moiety via transesterification over the supported Novozym SP 435 catalyst in the presence of flowing SC-CO2 containing dissolved methanol. The synthesized methyl esters were identical in composition to those obtained by alkali metal-initiated methanolysis. Again the advantage of the supercritical fluid synthesis becomes apparent since it eliminates catalyst filtration after completion of the reaction. The highly reproducible quantitative conversion of soybean and corn oils to their methyl esters via the enzymatic-catalyzed reaction in flowing SC-CO2 has also found uses in analytical chemistry [23]. Typical conditions for the methanolysis of soybean oil are a level of 0.1 volume percent of water in SC-CO2 at 24 MPa and 50 °C which proves sufficient to facilitate the reaction, yet prevent denaturization of the enzyme; however it should be noted that additional water can prove inhibitory to facilitating esterifications or transesterifications in the presence of SC-CO2.
Simple esters such as propyl laurate can also be made in less than 25 minutes at nearly 100% yield using Novozym SP 435. Recently, the production of sterol esters has been accomplished using Chirazyme L-1 with SC-CO2. As illustrated in Fig. 8, fatty acids of various chain lengths can be reacted with sterols in the presence of SC-CO2. Ester yields were consistently above 90% for C8–C18 saturated fatty acids reacting with the sterols at 27.6 MPa and 50 °C. Of particular interest were the > 98% yields achieved for the C16 and C18 sitostanol esters which can be utilized in functional food margarine formulations to lower cholesterol levels in humans.
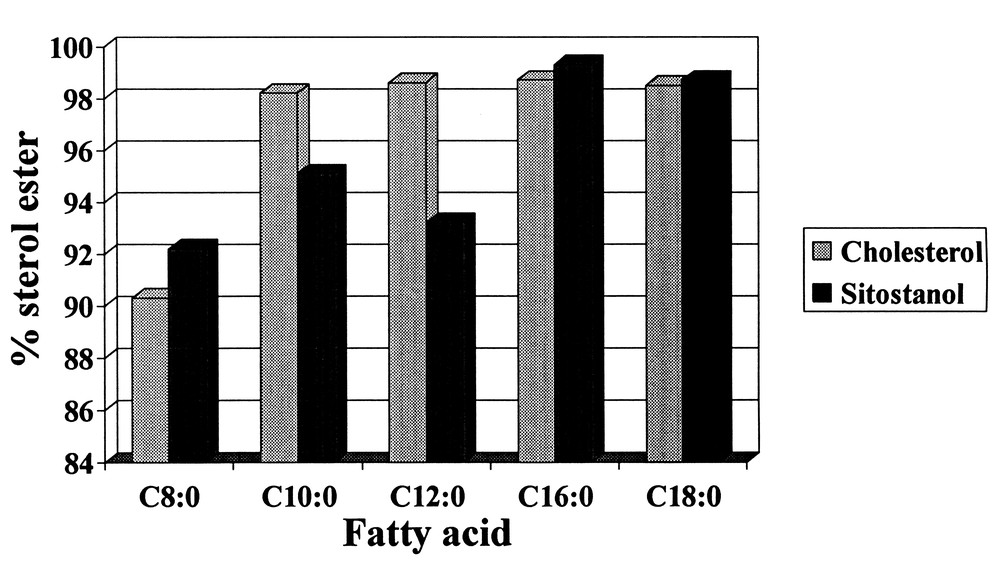
Product yields for fatty acids reacting with cholesterol and sitostanol using Chiarzyme L-1 as a catalyst.
Hydrogenation in supercritical fluid media has attracted considerable interest, based to a large extent, on the superior mass transfer characteristics achieved when using hydrogen mixed with SC-CO2. Due to several factors, contact of the dissolved hydrogen with catalyst and substrates is facilitated in both flow and batch reactor systems using a H2/SC-CO2 mixture. Recent studies by Harrod and colleagues [24] and others [25] have demonstrated the increased hydrogenation rates are possible when synthesizing fatty alcohols from fatty acid methyl ester substrates, as well as for the hydrogenation of vegetable oils.
Recently we have conducted experiments in batch reactor systems at slightly elevated pressure (up to 13.8 MPa) in which hydrogen is admixed with carbon dioxide in a vessel containing a Ni commercial catalyst and soybean oil. Using mixtures of H2 and CO2 at equal pressures and temperatures between 120–140 °C, a variety of hydrogenated oils can be produced that have varying iodine values (IV), % trans fatty acid and SFI. It is particularly interesting to note that the SFI are relatively invariant with respect to temperature and can be adjusted by varying the ratio of H2 to CO2 pressure. It is possible using this technology to produce equivalent or better-hydrogenated oil base stocks for use in margarine or shortenings, relative to those obtained using conventional technology. As shown in Table 5, oils with lower trans fatty acids are possible using the above technique, relative to an equivalent commercial product of like IV value, produced by conventional hydrogenation. Thus potential useful margarine and shortening basestocks can be produced by this method having the dropping point (DP) characteristics stated in Table 6.
Properties of potential vegetable oil-derived margarine/shortening using hydrogen/carbon dioxide mixtures during hydrogenation
| Oil property | Margarine basestock (D.P. 32–39 °C) | Shortening basestock (D.P. 45–52 °C) | ||
| Conventional | Experimental | Conventional | Experimental | |
| % Stearic acid | 6–9 | 7–11 | 11–13 | 13–24 |
| % Trans | 11–30 | 1–3 | 15–20 | 3–8 |
| IV value | 90–110 | 108–114 | 85–90 | 88–102 |
Comparison of dropping points of various fats and oils before and after randomization in flowing supercritical carbon dioxide
| Sample | Initial dropping point (°C) | Final dropping point (°C) |
| RBD soybean Oila | 0.9 | 10.7 |
| HS-1 soybean oilb | 18.3 | 27.6 |
| A-6,3 soybean oil | 20.5 | 30.4 |
| Palm olein | 21.7 | 37.7 |
| Cocoa butter | 32.0 | 41.6 |
| Tallow | 41.3 | 38.8 |
| 20% tristearin/80% SBO | 60.4 | 37.1 |
a Refined, bleached, deodorized soybean oil.
b High stearate soybean oil.
4 Sub-critical and pressurized water processing
The success in applying both SC-CO2 and liquid CO2 (L-CO2) as benign solvents for the extraction of naturally derived products has catalyzed the search for additional extraction media that can be used in food or nutraceutical processing. Over the past five years, subcritical water, that is H2O held in its hot pressurized liquid state above its normal boiling point, has been used as reported by Hawthorne and colleagues [26]. Initially, one may question the use of this hot medium to extract biological or organic materials, due to concerns of its reactivity, resulting in the subsequent degradation of solutes under elevated temperatures (note that this is not the technique of supercritical water oxidation which is usually conducted above the critical temperature of water, 375 °C, for the expressed purpose of degrading organic wastes, etc.). However, recently this medium has been applied successfully, over a restricted temperature range, to the extraction of food materials and natural products.
The solvent power of subcritical water is largely varied by changing temperature and applying enough external pressure to eliminate vaporizing the water when used above its normal boiling point. Hence, by varying the temperature of water over a range of 25–250 °C (see Fig. 9), its dielectric constant can be changed from 78 to less than 30, thereby attaining solvent polarities normally associated with polar organic solvents. Thus in theory, subcritical water is a perfect compliment to SC-CO2 for processing natural products, thereby avoiding the use of organic solvents such as ethanol. The potential reactivity of solutes under subcritical water conditions, i.e., conditions slightly above the boiling point of water, suggests that temperatures between 100–175 °C are optimal for most food and natural product applications. Degradation of thermally labile solutes, such as anthocyanins from fruit berries can be minimized by optimizing extraction kinetics to minimize the transient time of such solutes in the extraction stage [27]. It should be noted however, that certain hydrolysis reactions can be conducted in subcritical water [28], and that the generation of flavor precursors under conditions akin to those obtained in aqueous-based cooking processes is also possible.
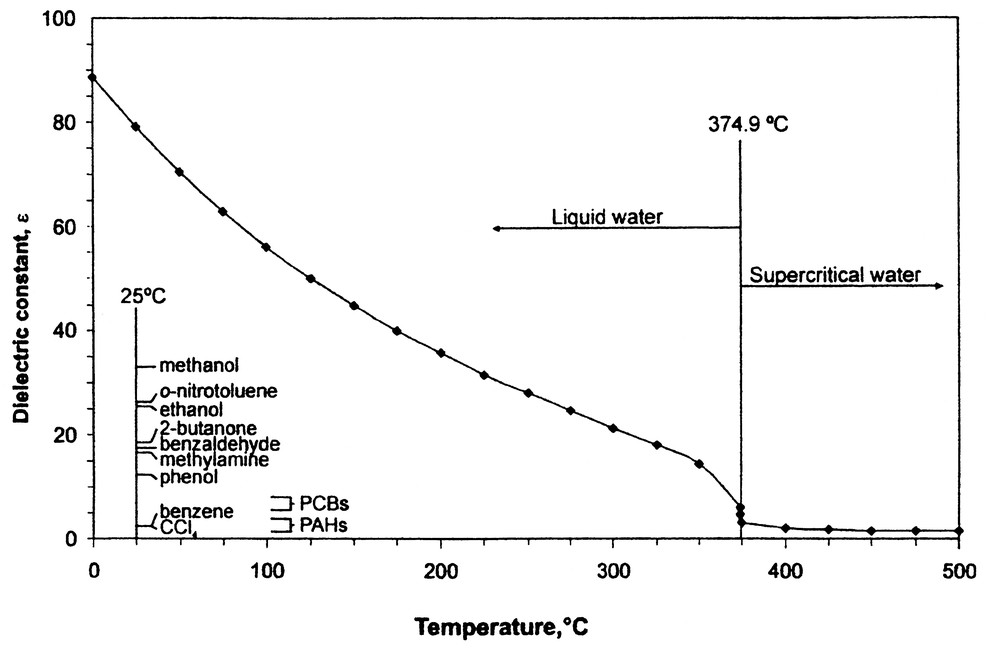
Dielectric constant of water versus temperature up to its critical temperature.
As opposed to some of the more optimal conditions for conducting SC-CO2 extraction, subcritical water extraction does not require drying of the substrate before the extraction step, and can be optimized to avoid the extraction of waxes and other lipid material from plant matter. Clifford et al. [29] have shown the utility of subcritical water as an extraction medium for the removal of oxygenated compounds from rosemary, clove buds, and the deterpenation of lemon oil. As shown in Table 7, the yields of various components in milligrams from a 4-g sample of rosemary, are better using pressurized hot water extraction at 150 °C, than by applying steam distillation. Other factors, such as the selectivity for oxygenated solutes over monoterpenes and the higher heat recovery versus steam distillation, make subcritical H2O an attractive processing technique. Recently, countercurrent deterpenation of citrus oils has been demonstrated on a continuous basis using subcritical water.
Comparison of yields from rosemary extraction by steam distillation and subcritical water extraction at 150 °C*
| Compound | Steam distillation | Subcritical water extraction |
| α-Pinene | 10.9 | 4.0 |
| Camphene | 2.0 | 1.5 |
| Limonene | 1.4 | 1.3 |
| 1,8-Cineole | 2.7 | 5.3 |
| Camphor | 3.4 | 6.4 |
| Borneol | 2.1 | 3.5 |
| Verbenone | 5.0 | 6.3 |
| Isobornyl acetate | 1.0 | 1.5 |
| Unidentified sesquiterpene | 0.7 | 0.01 |
| Total | 50.7 | 54.2 |
* Yields in mg per 4 g of sample.
Reference was made earlier to the use of subcritical water as a medium for processing lipid materials. Historically, subcritical water (not referred to by that name) has been used in the classical fat splitting procedures, such as the Twitchell and Colgate-Emery processes. The temperatures employed for the splitting of fats is quite high due to the need to assure miscibility of the fat with the hot subcritical water. Under these conditions, a pressure head is applied to the water to keep it in its liquid state, i.e., subcritical state, between its normally atmospheric boiling point and its critical temperature. We have exploited these properties recently for conducting the hydrolysis of model lipid compounds and vegetable oils in subcritical water between 250–340 °C without the aid of a catalyst. Using a flow reactor consisting of 316–stainless steel, rapid hydrolysis rates and high conversion of soybean oil to fatty acids have been achieved. Fatty acid yields exceeding 98% have been achieved in 10–15 min at 340 °C and 13 MPa, using water-to-oil ratios of 5–2.5:1. Under these conditions, the water/lipid mixture is in one phase, as detected with the aid of high-pressure flow sight glass.
GC-FAME analyses were performed on a number of the resultant fatty acid mixtures to see if any chemical decomposition or conversion had occurred on these moieties at these temperatures. Both saturated and unsaturated fatty acids remain relatively unaffected below 300 °C, although linolenic acid was slightly altered and converted from its original cis,cis,cis isomer form. Extreme temperatures, such as 375 °C, close to the critical temperature of water, produced a dark brown mixture. GC-FAME ester analysis of the oil revealed the presence of severe decomposition and/or polymerization of the constituent fatty acids.
5 Integrated supercritical fluid-based processing
As remarked in the introduction to this review, the coupling of discrete unit processes such as SFE, fractionation (SFF), or SFR in various combinations with a matrix of extraction fluids, offer a number of unique processing options that can yield unique products as shown below by combining the following:
A more detailed example of the above coupled-processing concept is shown below for the case of processing citrus oils using pressurized fluids. Here the individual processes are listed followed by combinations that can be used to extract or modify the starting citrus/essential oil:
- Processes:
- (1) SFE (SC-CO2)
- (2) SFF (SC-CO2) – pressure reduction
- (3) SFF (SC-CO2) – column deterpenation
- (4) SFC (SC-CO2/cosolvent)
- (5) SFF – (subcritical H2O deterpenation)
- (6) SFM – (aqueous extract/SC–CO2)
- Combinations: (1) + (2)
- (1) + (2) + (3)
- (1) + (4)
- (1) + (5)
- (1) + (5) + (6)
Here six discrete unit processes are listed which include conventional SFE with SC-CO2, SFF employing stage-wise pressure reduction, SFF using fractionation column-based deterpenation (3), supercritical fluid chromatography (SFC), an additional variant of SFF called subcritical water deterpenation (4), and utilization of a SC-CO2 or LCO2 with a permselective membrane. For example, a combination of processes (1) and (2) listed below could be combined to yield a more specific extract composition. Unit process (1) if conducted by sequentially increasing the extraction density when coupled with a sequence of let down pressures (unit process 2) can amplify the SFF effect. Likewise, by combining unit process (1) using SC-CO2 followed by application of unit process (2) that utilizes subcritical H2O to deterpenate the extract from unit process (1), will yield a more specific final product from the starting citrus oil. To obtain a more enriched and/or concentrated product from the latter process, one could add on unit process (6), a supercritical fluid membrane-based separation of the aqueous extract/ fractions from unit process (5).
To further illustrate the advantage of coupling critical fluid-based processes for isolating and purifying natural products, we cite our results for obtaining enriched-phospholipid (PL) extracts and fractions starting from seed oil sources, such as soybean [30]. As shown below, comminuted soybeans can be initially extracted with either hexane or SC-CO2, and then countercurrently contacted with SC-CO2 in a packed fractionating tower to yield a degummed vegetable oil (VO) while isolating a very enriched lecithin (PL) fraction:
The lecithin fraction can than be further fractionated by applying preparative SFC in which a stepwise gradient program facilitates isolation of PLs, such as phosphatidylcholine (PC) or phosphatidylethanol amine at 75% purity levels. Taylor et al. [15] have demonstrated the efficacy of the above process for both enriched lecithin fractions as well as starting from soybeans. It is interesting to note that several SC-CO2 processed PL extracts are available today as commercial products.
A method has also been developed which permits the production of fish oil esters using the following sequence of steps:
This process has been practiced at a plant located in Tarragona, Spain where fish stocks from European waters have been chemically-esterified to their ethyl esters before applying a preliminary SFE step to enrich the polyunsaturated fatty acid (PUFAs) ester fraction. Large-scale production SFC is then applied to enrich the various omega fish oil ester fractions using chemically bonded silica-based packings, yielding a commercial PUFA product of 95% purity. This is an excellent example of a combined SFE-SFC process. It is worth noting that the above chemical esterification could have been conducted in the presence of SC-CO2 to form the desired ester [22], and thereby creating a SFR–SFE–SFC coupled processing scheme.
As previously noted, hydrogenation of oils and lipids can also be achieved under supercritical fluid conditions, i.e., conditions in which the oil or lipid is dissolved in a binary fluid mixture consisting of H2 + SC-CO2 or H2 + SC-propane. Harrod and coworkers at Göteborg University (Sweden) [31] have shown that very high yields can be achieved in hydrogenating oils or oleochemicals at high space velocities through catalytically-active, packed bed reactors. We have taken their approach one step further by combining a transesterification reaction with a hydrogenation reaction using packed bed reactors coupled consecutively, using either SC-CO2 or SC-C3H8 as a reaction and transport medium. Transesterification conditions similar to those quoted previously have been used to produce the resultant FAME distribution expected from soybean oil. These FAMEs are then introduced into either a SC-CO2 or SC-C3H8 stream with a varying molar concentration of H2 in order to conduct exhaustive hydrogenation of the FAMEs to fatty alcohols. Fig. 10 shows the binary reactor system in which a coupling of enzymatic-initiated conversion to FAMES with exhaustive hydrogenation of the FAMES to fatty alcohols is accomplished. A maximum yield of fatty alcohols (C16 + C18 saturated alcohols from soybean oil) is attained at 250 °C and 25 mol% H2 using a non-Cr-containing catalyst. Equivalent yields can be achieved using less H2 in SC-C3H8 or by employing Cr-based catalysts and/or lower reaction temperatures. Gas chromatographic (GC) analysis of the hydrogenated products along with supercritical fluid chromatography (SFC) verified that the transesterification and hydrogenation processes were completed. This is an excellent example of how a two-step synthesis process can be conducted in supercritical fluid media, that is environmentally-benign by permitting reuse of the critical fluid media as well as the reaction by-product from the hydrogenation step, methanol.
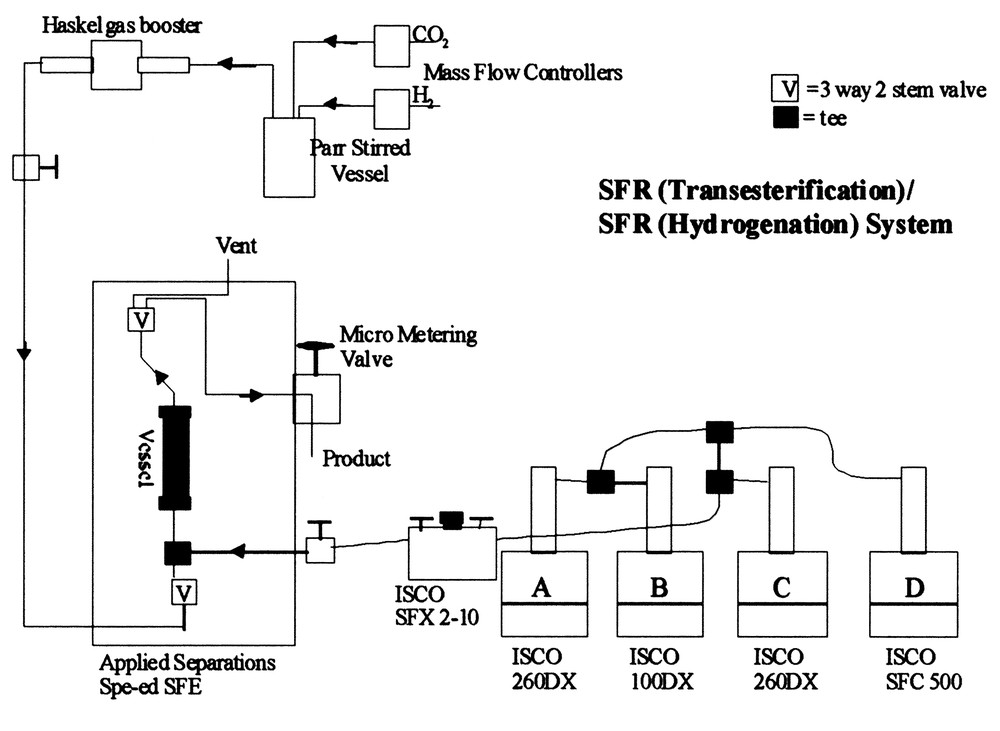
Two-step reaction system for converting vegetable oils to fatty alcohol mixtures.
6 Conclusions
In this brief review, we have attempted to document some of the current trends in critical fluid technology as applied to the processing of natural products and agriculturally derived feedstocks. The use of critical fluid media from various regions of the phase diagram is becoming more prevalent as illustrated by the use of SC-CO2, LCO2, subcritical water and selected cosolvents that are compatible with the environment and end use in functional foods. Although simplistic, the author has reason to believe that many natural product extractions and separation problems can be solved by the combination of carbon dioxide, ethanol, and water; an all-green trio of solvents. Supercritical processing is also beginning to merge with high pressure hydrostatic processing [32] as the prophylactic benefits of a residual CO2 atmosphere in SFE-derived products has been documented [33]. Undoubtedly the coupling of membrane technologies with critical fluid media will see application in the near future.
General References for Critical Fluids
Y. Arai, T. Sako, Y. Takebayashi (Eds.), Supercritical Fluids, Springer-Verlag, Berlin, 2002.
J.M. DeSimone and W. Tumas (Eds.), Green Chemistry using Liquid and Supercritical Carbon Dioxide, Oxford University Press, Oxford, UK, 2003.
A. Gopalan, C. Wai, H.J. Jacobs (Eds.), Supercritical Carbon Dioxide: Separations and Processes, American Chemical Society, Washington DC, 2003.
E. Kiran, P.G. Debenedetti, C.J. Peters (eds.), Supercritical Fluids – Fundamentals for Applications, Kluwer Academic Publishers, Dordrecht, The Netherlands, 2000.
M.B. King, T.R. Bott (eds.), Extraction of Natural Products Using Near Critical Solvents, Blackie Academic, London, 1993.
M. Mukhopadhyay, Natural Extracts Using Supercritical Carbon Dioxide, CRC Press, Boca Raton, Florida, 2000.
E. Stahl, K.-W. Quinn, D. Gerard, Dense Gases for Extraction and Refining, Springer-Verlag, Berlin, 1986.