

1 Introduction
Reaction of a solvated transition-metal ion with a multidentate ligand generally results in coordination of all suitably-disposed ligand donor atoms to form a metal complex containing the maximum possible number of chelate rings. Thus, ligands such as ethylenediamine (1,2-ethanediamine) and 2,2′-bipyridine almost invariably coordinate to transition metals in a bidentate fashion and the number of metal complexes containing these chelated ligands is legion. This propensity for coordination of all donor atoms is usually rationalised in terms of the Chelate Effect, a subject on which much has been written [1]. However, there is a small but steadily growing number of complexes in which multidentate ligands are coordinated through fewer than the maximum number of donor atoms. Although such complexes have been known for decades, it is only relatively recently that the word ‘hypodentate’ has been coined by Constable to describe a ligand “in which fewer than the maximum number of donor atoms are involved in interactions with metal centres” [2], and this terminology is now being increasingly used in the chemical literature1. Although the term “hypodentate” was originally introduced to refer to a particular mode of ligand binding, its use has also extended to complexes containing hypodentate ligands, and thus it is common to see reference to a ‘hypodentate complex’. Both usages will be found herein. Hypodentate coordination of multidentate ligands is more common than might at first be thought; monodentate coordination of potentially chelating oxoanion ligands such as carbonate, phosphate, sulfate and any number of carboxylates is not at all uncommon, and such ligands lie outside the scope of this discussion. Even amongst amine ligands, hypodentate coordination is encountered surprisingly frequently, and thus the following article is restricted to complexes containing common multidentate amine ligands bound in a hypodentate fashion. Important factors in the rational syntheses of such complexes are also detailed.
2 Hypodentate complexes
2.1 Nomenclature
The nomenclature of hypodentate coordination merits comment, as a variety of methods have appeared in the literature, and a consistent approach to the naming of such complexes is obviously to be preferred. As an example of the confusion that can occur, protonated monodentate ethylenediamine is often abbreviated as enH (for example, in the complex cation [Co(NH3)5enH]4+), but ambiguity can arise when the monodentate ethylenediamine is not protonated; the complex fac-[Re(CO)3(en)2](O2PPh2) (section 2.2) contains both chelated and monodentate ethylenediamine ligands, a fact not immediately obvious from this simple formula. Thus, a more complete notation is required in such cases and IUPAC recommends one of two possible unambiguous nomenclature schemes [3]. The first involves specifying the bound donor atoms of the ligand in italics; in this case, monodentate ethylenediamine would be designated (using the correct IUPAC naming) as 1,2-ethanediamine-N, whereas the chelated ligand would be 1,2-ethanediamine-N,N′. The kappa convention is recommended for more complicated ligands; the italicised donor atom is in this case preceded by κ, and is placed immediately following the portion of the ligand to which it directly applies. Thus the two possible tridentate modes of binding of triethylenetetramine (Fig. 5) would be designated as N-(2-amino-κN-ethyl)-N′-(2-aminoethyl)-1,2-ethanediamine-κ2N,N′ (bound through one primary and two secondary N atoms) and N,N′-bis(2-amino-κN-ethyl)-1,2-ethanediamine-κN (bound through one secondary and two primary N atoms). Even for simple ligands, such notation often becomes unwieldy and cumbersome (the Re complex above, for example, would be fac-[Re(CO)3(1,2-ethanediamine-N,N′)(1,2-ethanediamine-N)](O2PPh2) or fac-[Re(CO)3(1,2-ethanediamine-κ2N,N′)(1,2-ethanediamine-κN)](O2PPh2) under the two systems), and, in many cases, a picture is worth (literally) a thousand words.
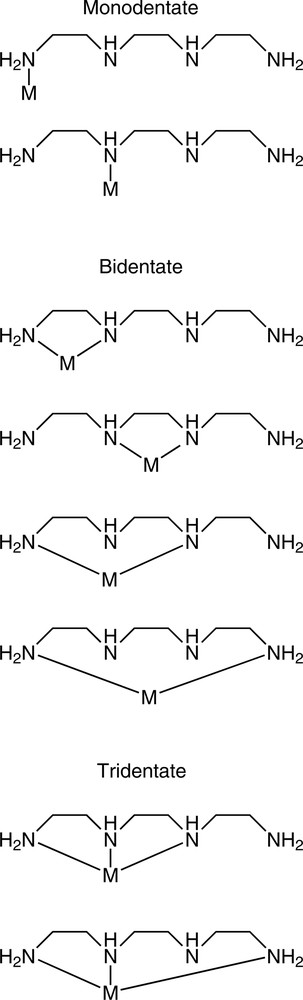
The possible hypodentate binding modes of trien to a single metal ion.
2.2 Hypodentate aliphatic amine ligands
Every undergraduate textbook uses ethylenediamine as the archetypal example of a chelating ligand, and the possibility that it can bind in a hypodentate fashion as anything other than a short-lived intermediate in the chelation process is rarely, if ever, mentioned. However, there is a large number of examples of hypodentate coordination of ethylenediamine in stable, well-characterised complexes and some of these are detailed below.
The first complex containing hypodentate ethylenediamine may well have been prepared by Werner. In a 1916 manuscript unpublished in his lifetime, he reported the synthesis of a chromium complex having the formula H[Cr(en)2Cl4] which he formulated as an 8-coordinate species [4–6]. He then used this as a starting material to prepare [Cr(en)2(OH2)Cl3], H[Cr(en)2(ClO4)Cl3] and H2[Cr(en)2(C2O4)Cl3], all of which he also formulated as eight-coordinate species. It has been suggested that H[Cr(en)2Cl4] is in fact [Cr(en-N,N′)(enH-N)Cl3]Cl, a six-coordinate complex containing one chelating and one monodentate ethylenediamine ligand, the latter protonated on the uncoordinated nitrogen atom. While this formulation is somewhat at odds with Werner's observation that addition of silver ion to an aqueous solution of this complex did not give immediate precipitation of AgCl, this could be explained by coordination of the added silver ion to the uncoordinated amine nitrogen atom. The available data are also consistent with a structure containing two monodentate ethylenediamine ligands, one of which is protonated, and four coordinated chloride ligands. The exact identity of this complex remains unknown and a reinvestigation of this system using modern characterisation techniques would surely be of interest.
Although the existence of complexes containing hypodentate ethylenediamine had been postulated in the late 1950s [7,8], the first characterised example of a hypodentate ethylenediamine complex was reported in 1968, with the isolation of [Cr(enH)(H2O)5]4+ from the hydrolysis of the chelate complex [Cr(en)(H2O)4]3+ [9]. The hypodentate complex was characterised on the basis of elemental analysis, its behaviour as a 4+ ion during ion-exchange chromatography, and its subsequent hydrolysis to form [Cr(H2O)6]3+. Further study of this system in 1975 gave evidence for the production of a number of hypodentate complexes in the hydrolysis reactions of both [Cr(en)(H2O)4]3+ and [Cr(en)3]3+, including the bis-hypodentate complex cis-[Cr(enH)2(OH2)4]5+, although again, no X-ray structural data were reported [10].
The synthesis of the diamagnetic complex cis-[Co(en-N,N′)2(enH-N)Cl]Cl3·H2O in 1970 allowed characterisation of hypodentate coordination by NMR for the first time [11]. The complex was obtained from the room-temperature reaction of trans-[Co(en)2Cl2]Cl with ethylenediamine in MeOH, with purification achieved by cation exchange chromatography. The 1H NMR spectrum of this complex showed two broad peaks at 3.3 ppm (2 H) and 2.9 ppm (10 H), with the former being assigned to the methylene protons of the monodentate enH ligand adjacent to the uncoordinated nitrogen atom. Measurement of the pH of a half-neutralised solution of the complex suggested a pKa around 7.1. Base hydrolysis of the complex in aqueous solution at pH 8.5 surprisingly resulted in little (< 5%) formation of the tris-chelate [Co(en)3]3+, with the major red-orange product formulated as [Co(en-N,N′)2(en-N)(OH)]2+. This presumably implies that chloride release from the cis-[Co(en-N,N′)2(enH-N)Cl]3+ cation in alkaline solution does not occur via a concerted process involving nucleophilic attack of the uncoordinated N atom, as perhaps might be expected, but rather results through the intermediacy of a five-coordinate species that shows a distinct preference for entry of bulk solvent over the free amino N atom. However, dissolution of the complex in Me2SO and addition of piperidine gave complete formation of [Co(en)3]3+. The different behaviour in the two solvent systems is intriguing, and certainly warrants further investigation.
[Co(NH3)5(enH)]Br4 was first prepared in 1975 in low yield, from the reaction of [Co(NH3)5(OH2)](ClO4)3 with ethylenediamine in Me2SO at 85 °C [12]. Cation-exchange chromatography was used to separate this complex from a number of other chelated mono, bis and tris ethylenediamine species. Potentiometric titration of the complex gave a pKa of 7.53, and the complex could be deprotonated in aqueous ammonia solution to give [Co(NH3)5(en)]3+, which was isolated as the bromide salt following addition of NaBr. Higher yielding preparations of this complex as both the chloride ([Co(NH3)5(enH)]Cl4·0.5 H2O) and bromide ([Co(NH3)5(enH)]Br4) salts have since been reported which utilise the labile starting materials [Co(NH3)5(OSO2CF3)](CF3SO3)2 and [Co(NH3)5(Me2SO)](ClO4)3 respectively [13] [14], and [Co(NH3)5(enH)]Br4 has been structurally characterised (Fig. 1) [14].
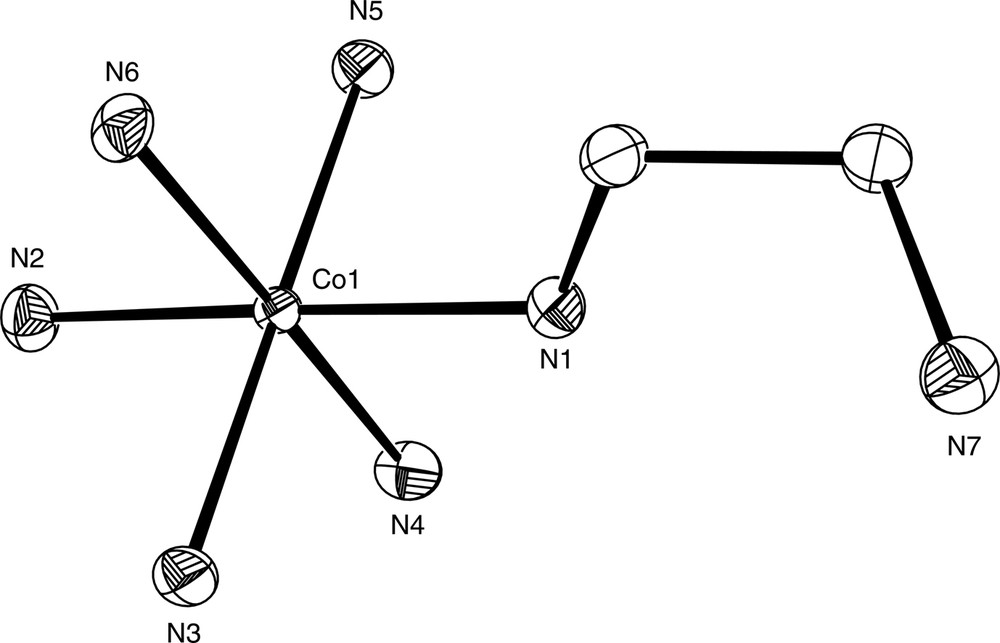
Diagram of the [Co(NH3)5(enH-N)]4+ cation in the structurally characterised complex [Co(NH3)5(enH-N)]Cl4.
The rhenium carbonyl complex fac-[Re(CO)3(en-N,N′)(en-N)](O2PPh2), reported in 1980, was the first complex containing hypodentate ethylenediamine to be structurally characterised by X-ray crystallography [15]. Since this time, crystal structures of a small number of discrete molecular complexes containing hypodentate ethylenediamine have been obtained and these are listed in Table 1. Hypodentate ethylenediamine has also been observed in a number of crystallographically-characterised polymeric metal-phosphate systems; see for example [16]. fac-[Re(CO)3(en-N,N′)(en-N)](O2PPh2) was obtained from treatment of polymeric [μ-Ph2PO2Re(CO)3]n with excess ethylenediamine and heating at 110 °C for 24 h. The crystal structure of the complex confirmed the presence of a hypodentate ethylenediamine ligand in each of the two independent cations in the asymmetric unit (Fig. 2). The hypodentate ligands have no apparent conformational preference about the C-C bond; one displays a near anti conformation (N–C–C–N = 171.93°) while the other exhibits a gauche conformation (N–C–C–N = 66.60°). This conformational freedom appears to be a feature of all structurally characterised complexes containing hypodentate ethylenediamine, as can be seen from the values of the N–C–C–N torsion angles for these listed in Table 1. M–N bond lengths to the hypodentate ligand in these complexes show no significant deviation from average values, and appear unaffected by the protonation state of the terminal N atom; Ir–N distances to the hypodentate ligand in mer-[IrCl3(en-N,N′)(enH-N)]Cl·H2O and mer-[IrCl3(en-N,N′)(en-N)] are 2.103 Å and 2.101 Å respectively [17].
Structurally characterised discrete molecular complexes containing hypodentate ethylenediamine
| Complex | Dihedral / ° | Reference |
| fac-[Re(CO)3(en-N,N′)(en-N)](O2PPh2) | 171.93, 66.60 | [15] |
| [Mo2(OAc)2(en-N,N′)2(en-N)2,(OAc)2]·en | 99.3, 70.4 | [63] |
| fac-(CO)3W(en-N,N′)(en-N)[2,2,2-crypt-K]4[1-M(CO)3(η4-Pb9)]·2.5 en | 60.56 | [64] |
| fac-(CO)3Mo(en-N,N′)(en-N)[2,2,2-crypt-K]4[1-M(CO)3(η4-Pb9)]·2.5 en | 58.33 | [64] |
| (Ph3PNPPh3) trans-[Mo(O)(en-N)(CN)4] | 49.57 | [65] |
| mer-[IrCl3(en-N,N′)(en-N)] | 24.72 | [17] |
| [Pt(enH-N)Cl5]·H2O | 180.00 | [66] |
| [Pt(enH-N)Cl3] | 73.27 | [67] |
| cis-[Co(en-N,N′)2(enH-N)Cl](ZnCl4)Cl | 176.45 | [68] |
| cis-[Cr(en-N,N′)2(enH-N)Cl](HgCl4)Cl | 176.67 | [22] |
| [OsCl(en-N,N′)(enH-N)(C(CH3)C(CH3)C(CH3)CHCH2)]Cl2·0.5 C2H5OH | 87.37 | [69] |
| mer-[IrCl3(en-N,N′)(enH-N)]Cl·H2O | 161.28 | [17] |
| [Co(NH3)5(enH-N)]Cl4 | 78.41 | [14] |
| cis-[Co(en-N,N′)2(enH-N)(imH)]Br4 | 78.46 | [14] |
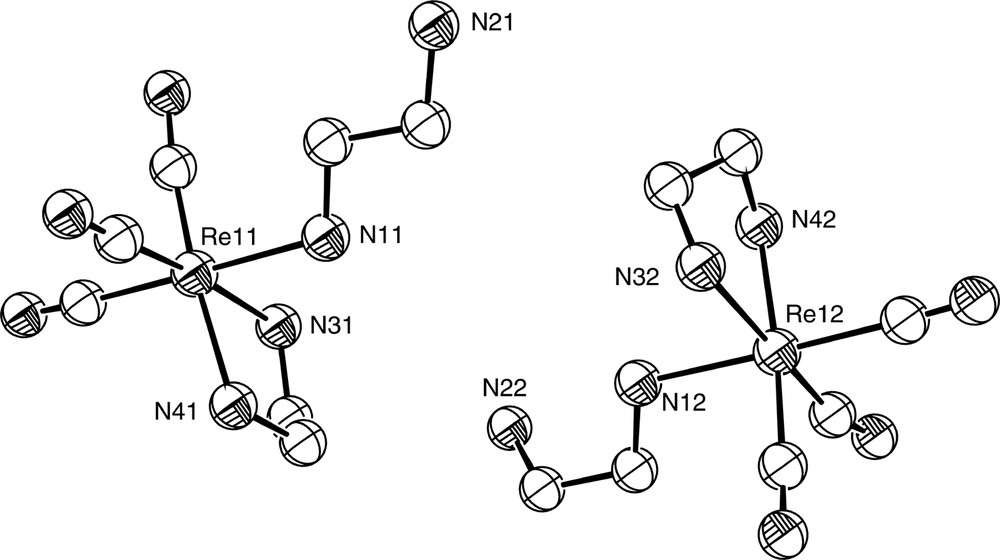
Diagram of the two independent fac-[Re(CO)3(en-N,N′)(en-N)]+ cations in the unit cell of fac-[Re(CO)3(en-N,N′)(en-N)](O2PPh2).
The nominally tridentate ligand diethylenetriamine (dien) has more than one possible hypodentate binding mode available, being able to coordinate to a single transition-metal ion in the two monodentate and two bidentate fashions shown in Fig. 3. Examples of monodentate coordination are extremely rare, with only [Co(EDTRA)(dienH2-N)]Cl2 [18] and [Co(NH3)5dienH2-N)]Cl5 [14] having been isolated, both of which feature coordination of the dien ligand to cobalt through a primary nitrogen atom. Monodentate coordination of dien through the secondary N atom remains unknown and is an interesting synthetic challenge. Of the two possible bidentate coordination modes, that resulting in the formation of a five-membered chelate ring is by far the most common and the X-ray structurally characterised examples of this are [Pt(CN)2(dienH-N,N′)]2[Pt(CN)6] [19], [PtCl2(dienH-N,N′)]Cl [20], [PtCl2(dienH-N,N′)]2(PtCl4)·H2O [21], s-fac-[Cr(dien-N,N′,N″)(dienH-N,N′)Cl](Hg2Cl7) [22] and mer-[RuBr3(NO)(dienH-N,N′)]Br·H2O [23]. Evidence for the formation of the square planar bis bidentate chelate complex [Pd(dienH-N,N′)2]4+ in solution has been obtained [24], and the octahedral rhenium bis bidentate chelate complex trans-[ReO2(dien-N,N′)2]I has also been prepared [25]. DFT calculations on the latter complex suggest the free aminoethyl arms are situated cis with respect to each other [26]. The tris bidentate chelate complexes [Co(dien-N,N′)3]X3 (X = BPh4, B(Bu)Ph3, BBu4) have been reported in the patent literature [27–29]. While there are no X-ray structurally-characterised complexes displaying bidentate coordination through the terminal N atoms of dien, the square planar Pt(II) complex trans-[Pt(NH3)2(dienH-N,N″)]Cl3·2 H2O (Fig. 4) provides a remarkable example of dien acting as a trans spanning ligand [30]. The protonated central N atom must presumably be situated in an axial position above the Pt ion and an X-ray structural investigation of this complex would be of great interest. Multidentate ligands containing three or more donor atoms also have the possibility of hypodentate coordination to more than one metal ion. Dien could thus potentially bridge two metal ions through either both primary N atoms, or a primary and the secondary N atoms, and both modes have been reported [31,32].
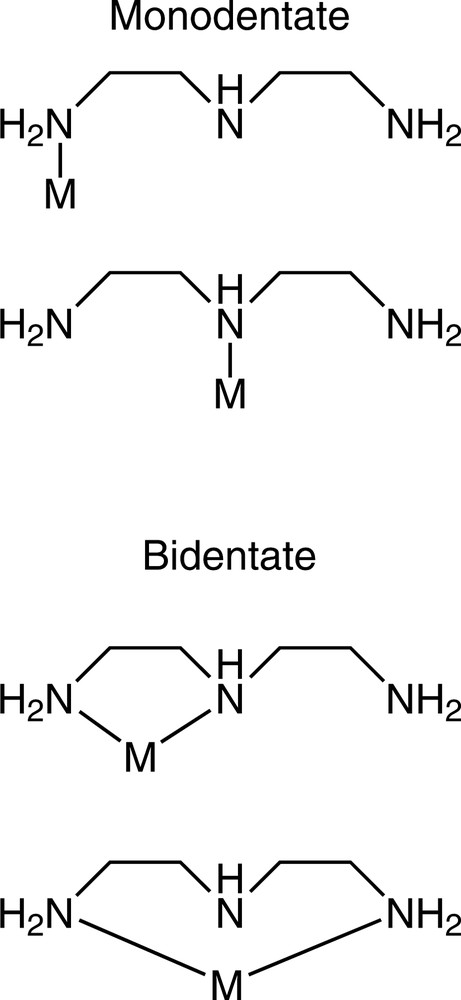
The possible hypodentate binding modes of dien to a single metal ion.
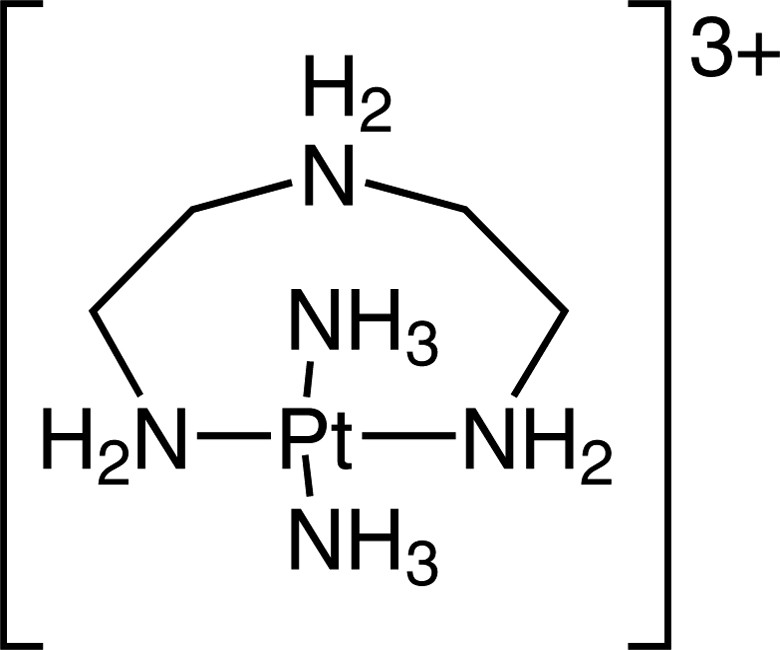
The trans-[Pt(NH3)2(dienH-N,N″)]3+ cation, in which dien acts as a hypodentate trans-spanning ligand.
Obviously, the greater the number of donor atoms in a multidentate ligand, the more hypodentate bonding modes are available. Thus trien has the eight possible monodentate, bidentate and tridentate hypodentate binding modes to a single transition-metal ion outlined in Fig. 5, and in addition can potentially bridge two or three transition-metal ions in a hypodentate fashion. The tridentate form can also bind in mer and fac configurations in an octahedral complex and the configuration of the protons on the secondary nitrogen atoms can give rise to still further isomeric possibilities. While there are no examples of monodentate trien, both bidentate (through N1 and N4) [25] and tridentate (through N1, N4 and N7) [33] modes of binding have been documented. Surprisingly, there appear to be no examples of trien acting as a hypodentate bridging ligand to either two or three metal ions. The branched chain tetraamine ligand tren affords fewer hypodentate modes to a single metal ion (six in total) and monodentate (primary N) [14], bidentate (primary and tertiary N) [34] and tridentate (tertiary N and two primary N) [35,36] modes have been reported. A crystallographically characterised example of tren acting as a triply bridging ligand has also been described [37]. As the number of N-donor atoms in a linear ligand increases, the possibility of hypodentate coordination to a single metal ion necessarily increases, although there appear to be few, if any studies of the coordination behaviour of ligands having seven or more N atoms. An example of the normally hexadentate ligand linpen binding in a hypodentate fashion is provided by the peroxo-bridged dimer [(linpenH-N,N′,N″,N‴,N′′′′)CoO2Co(linpenH-N,N′,N″,N‴,N′′′′)](ClO4)6·3 H2O in which the amine ligands bind in a pentadentate manner with the terminal protonated N atom remaining unbound [38].
The preceding examples have concentrated solely on ligands in which the N donor atoms are separated by two-carbon units and which thus form five-membered chelate rings on coordination. As the carbon chain length between the donor atoms increases, the propensity of a multidentate ligand to bind as a chelate decreases markedly, due to both entropic and ring strain effects. This is nicely illustrated by data from the Cambridge Structural Database [39] which show 1473 transition metal complex structures containing chelated ethylenediamine, 194 containing chelated propylenediamine, 17 containing chelated butanediamine and none containing chelated pentanediamine. Thus it might be expected that hypodentate coordination of α,ω-diamine ligands containing long methylene chains would be favoured over chelation and this has been borne out to a certain extent by the work of Ogino who made a systematic study of the products obtained on reaction of [Co(NH3)5OH2]3+ with the aliphatic diamine ligands H2N(CH2)nNH2 (n = 2,3,4,5,7,8,10,12,14) in Me2SO. In addition to substantial amounts of [Co(NH3)6]3+ in all cases, the chelate complexes [Co(NH3)4(NH2(CH2)nNH2-N,N′)]3+ were formed for n = 2,3,4,12 and 14, while monodentate coordination was observed for the remaining ligands to give hypodentate [Co(NH3)5(NH2(CH2)nNH2-N)]3+ species. The syntheses of the bis-hypodentate complexes [Co(NH3)4(NH2(CH2)4NH3-N)2]X5 (X = ClO4, I) and [Co(en)2(NH2(CH2)4NH3-N)2]Br5·4 H2O were also reported [40]. A subsequent paper reported pKa values for a number of [Co(NH3)5(H2N(CH2)nNH3-N)]4+ (n = 2–8, 10) complexes. [41] These were found to largely mirror the first pKa values for the free diprotonated diamines, with an increase in pKa observed as the methylene chain length increased. Increasing the length of a pendant arm in a polydentate ligand has also been found to encourage hypodentate coordination. Thus, whereas tren and baep coordinate to Cu2+ as tetradentate ligands, abba apba and ahba (Fig. 6) all coordinate as tridentate ligands with the longest arm remaining uncoordinated [42], as can been seen in the crystal structure of [Cu(abbaH-N,N′,N″)Cl2]Cl·CH3OH·2 H2O (Fig. 7).
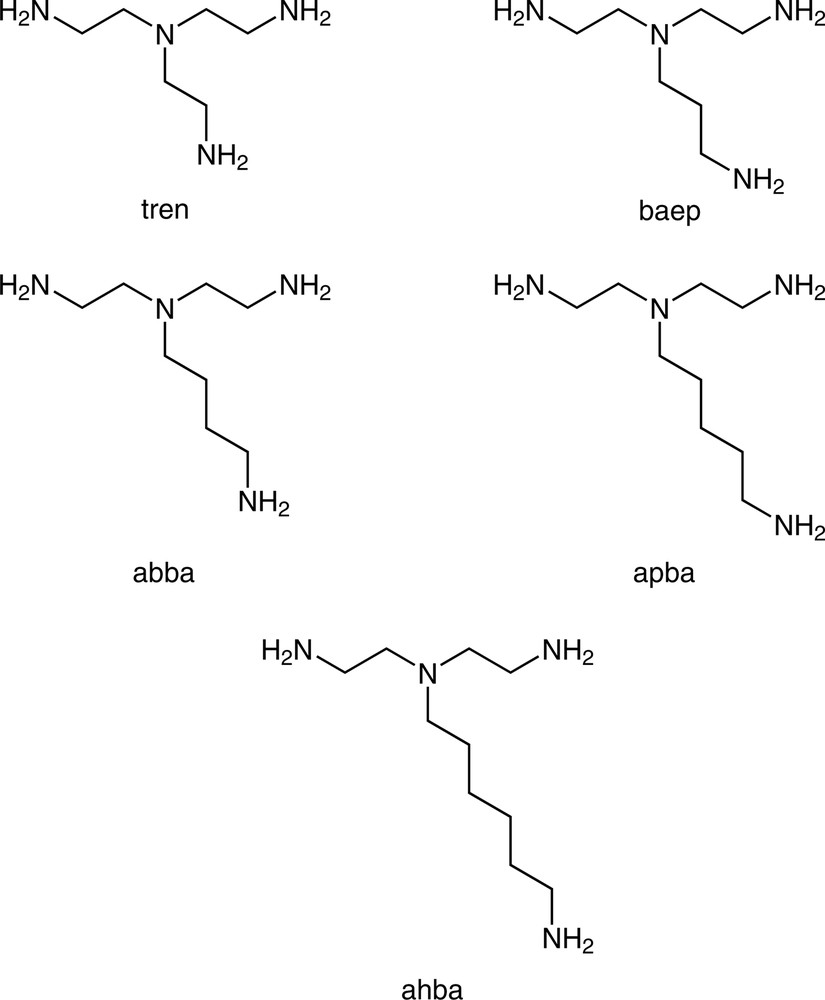
An homologous series of tripodal tetraamine ligands, in which the increasing length of the unique pendant arm encourages hypodentate coordination.
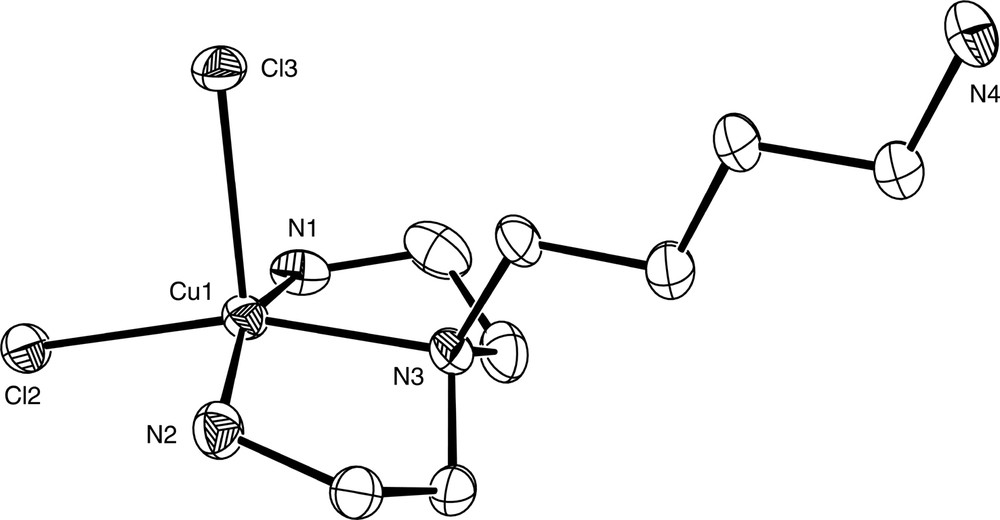
Diagram of the [Cu(abbaH-N,N′,N″)Cl2]+ cation in the structurally characterised complex [Cu(abbaH-N,N′,N″)Cl2]Cl·CH3OH·2 H2O.
2.3 Hypodentate alicyclic amine ligands
The constrained geometric nature of macrocyclic ligands and the preorganised arrangement of their donor atoms make them less likely than their open chain counterparts to display hypodentate coordination, and such examples are indeed relatively rare. The reaction of equimolar amounts of tacn and K2PtCl4 in aqueous solution at 80–90 °C affords a number of hypodentate complexes, the nature of which depends on the pH of the reaction mixture (Fig. 8). At pH 5, the complex [Pt(H2tacn-N)Br3]Br crystallises from solution, and this provides the only example of tacn acting as a monodentate ligand. At pH 7, the protonated bidentate complex [Pt(Htacn-N,N′)Br2]Br is obtained, while at pH 9–10, bidentate [Pt(tacn-N,N′)Br2] is isolated. When excess tacn is used, the bis bidentate complex [Pt(tacn-N,N′)2]Br2·2 H2O is isolated, and a crystal structure of this (Fig. 9) shows that the unbound N atoms of the tacn ligand do not interact with the metal ion [43]. Interestingly, all examples of tacn acting as a bidentate ligand involve square planar complexes of the d8 metals Pt(II) and Pd(II). There are no examples of tacn acting as a hypodentate bridging ligand between two metal ions.
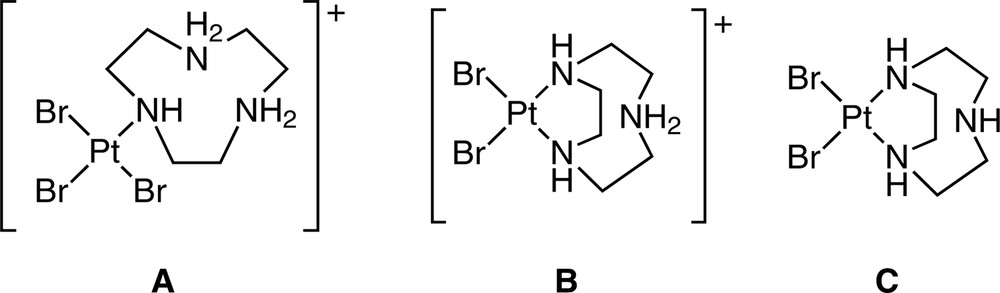
Hypodentate complexes isolated from the reaction of tacn with K2PtCl4 at various pH values. A: The cation of [Pt(H2tacn-N)Br3]Br (pH 5). B: The cation of [Pt(Htacn-N,N′)Br2]Br (pH 7). C: [Pt(tacn-N,N′)Br2] (pH 9–10).
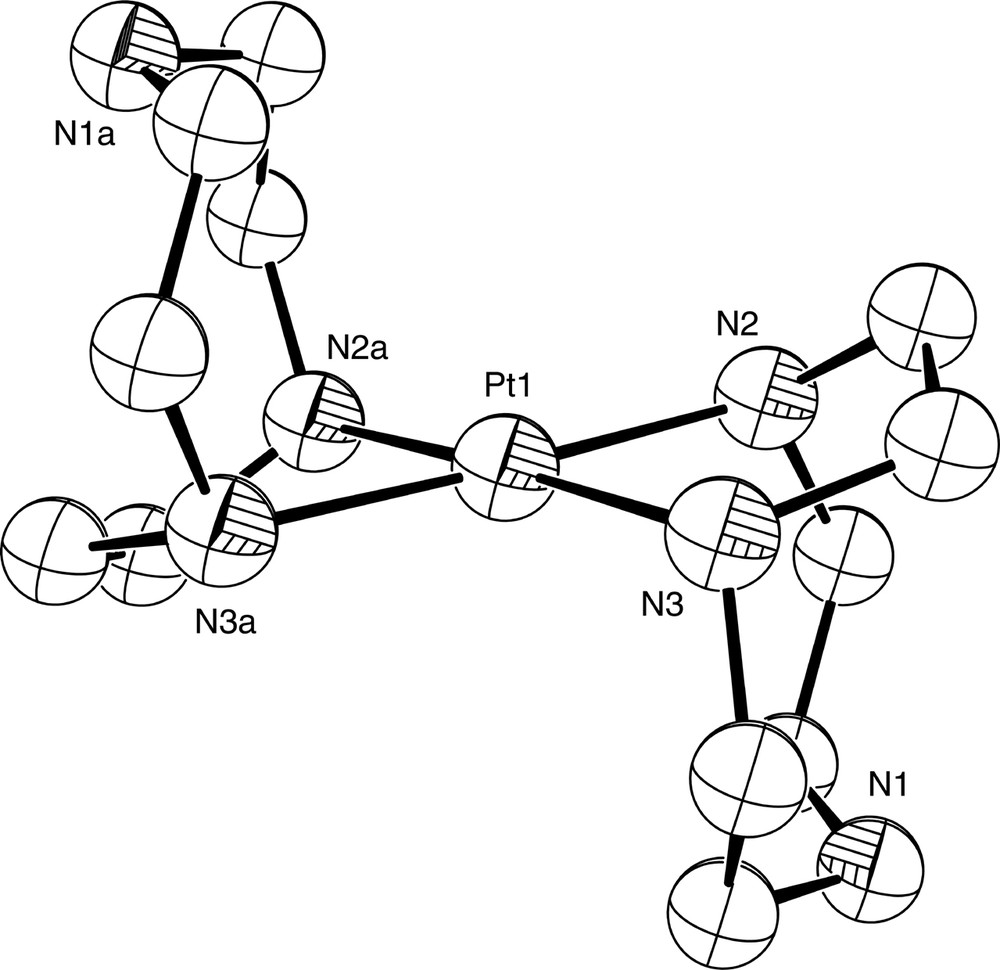
Diagram of the [Pt(tacn-N,N′)2]2+ cation in the structurally characterised complex [Pt(tacn-N,N′)2]Br2·2 H2O.
The potentially tetradentate ligand cyclen has not been reported to act as either a monodentate or bidentate ligand, but there are several examples of coordination as a tridentate ligand, which will be discussed further below. Again, as the number of N donor atoms in the ring increases beyond six, the chances of hypodentate coordination to a single metal ion increase, but the concomitant increase in the size of the macrocycle means that coordination of more than one metal ion becomes more likely. While the seven nitrogen donor ligand [21]aneN7 is large enough to accommodate three Pd2+ ions [44], it can also coordinate as a hexadentate ligand in the monomeric complex [Ni([21]ane7)](ClO4)2 (Fig. 10), with the uncoordinated N atom lying 3.075 Å from the nickel ion [45]. The notable exception amongst the alicyclic amine ligands with respect to hypodentate coordination is piperazine, which is in fact found more often as a monodentate ligand than as a chelate, presumably due to the favoured chair conformation of the ring which results in a positioning of the nitrogen lone pairs that is not optimal for chelation.
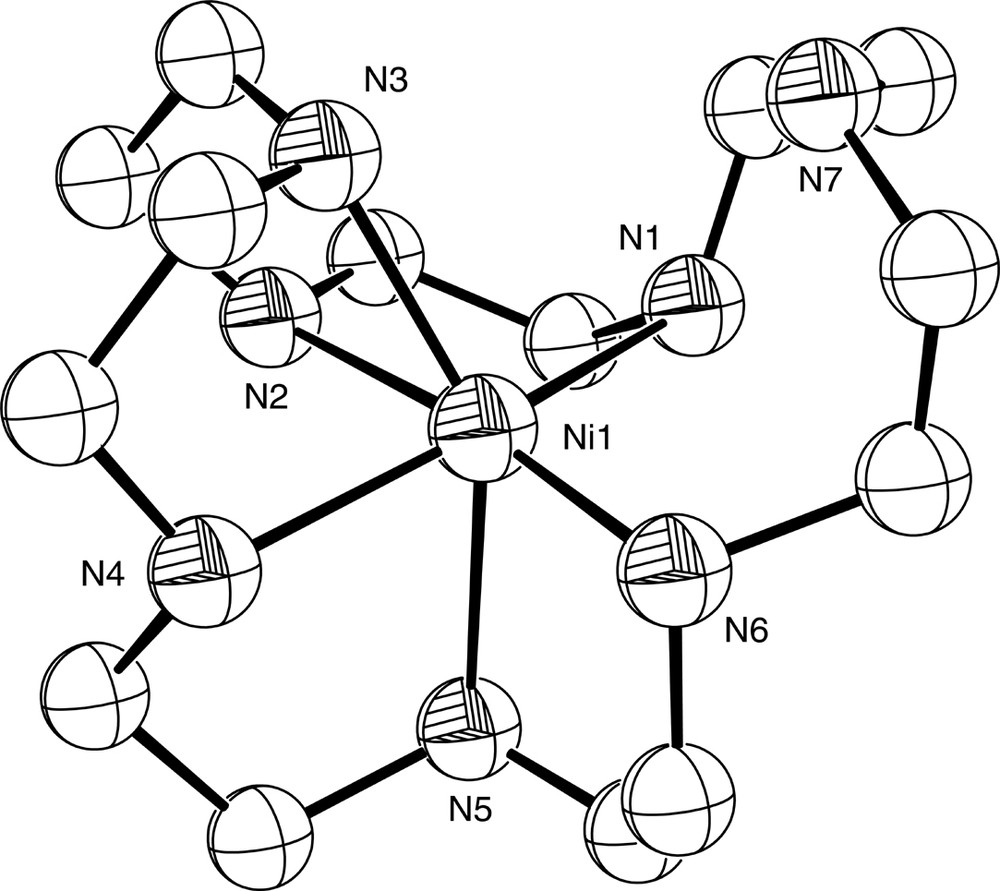
Diagram of the [Ni( [21]ane7)]2+ cation in the structurally characterised complex [Ni( [21]ane7)](ClO4)2. N7 is not coordinated to the Ni2+ ion.
2.4 Hypodentate aromatic amine ligands
There are no crystallographically-characterised complexes containing monodentate 2,2′-bipyridine (bipy). A search of the CSD reveals two complexes purportedly containing monodentate 2,2′-bipyridine (ref. codes RIFLAT and RULBUV). However, the first of these contains bipy bound to Cr in a normal bidentate fashion, while the second contains bipy bonded to Hg in a manner best described as asymmetric bidentate [46]. The apparent paucity of such complexes is presumably due to the relatively constrained nature of the bipy ligand, where coordination of one nitrogen atom almost of necessity brings the other into close proximity to the metal ion. This behaviour is in contrast to ethylenediamine, the acyclic congener of bipy, where free rotation about the C–C and C–N bonds can position the second N atom remote from the metal ion even after the first N atom is bound. While chelation of bipy can be rendered impossible simply by rotation about the interannular bond such that the nitrogen atoms become trans with respect to this, such rotation generally results in bipy binding either as a cyclometallated chelate through C3′ or as a bridging ligand, neither of which can be classified as hypodentate in the strictest sense of the word. A salutary example of the difficulty of confirming hypodentate coordination in the absence of crystal structural data is provided by the cyclometallated complex [Ir(bipy)2(bipy-C3,N′)](ClO4)2·⅓ H2O. This was originally proposed to contain the [Ir(bipy-N,N′)2(bipy-N)(OH2)]3+ cation, with one monodentate and two bidentate bipy ligands, on the basis of elemental analysis and spectral data, and the observed pKa of 3.0 was ascribed to deprotonation of the coordinated water ligand [47]. However, subsequent crystal structural determinations of protonated yellow [Ir(bipy)2(Hbipy-C3,N′)](ClO4)3·H2O [48,49] and deprotonated red [Ir(bipy)2(bipy-C3,N′)](ClO4)2·⅓ H2O [50] confirmed binding of the unique bipy ligand as a chelate through N1 and C3′, with the observed acid/base behaviour being due to reaction at the unbound nitrogen atom.
The 1,10-phenanthroline ligand lacks the rotational freedom of 2,2′-bipyridine, and as a result, examples of hypodentate binding of this ligand are rare, if not non-existent. The crystallographically characterised complex [Pd(PPh3)2(phen)Cl]BF4·Me2CO contains phen bound in a highly asymmetric manner, with Pd–N distances of 2.09(5) Å and 2.68(4) Å. The latter is longer than any other Pd–N bond in a crystallographically-characterised complex and this species might perhaps be best described as 4-coordinate square planar with a very weak axial interaction. More convincing examples of hypodentate coordination of polypyridine ligands are found in complexes of the normally tridentate ligand 2,2′:6′,2″-terpyridine (terpy) and Table 2 lists the crystallographically-characterised examples of these. The majority contain terpy bound as a bidentate ligand, with the third ring twisted with respect to the plane of the other two, thus situating the unbound N atom remote from the metal ion. An example of this can be seen in the crystal structure of [Ru(bipy-N,N′)2(terpy-N,N′)](PF6)2 shown in Fig. 11. The complex [Au(terpy)(CN)2Br] contains terpy bound in an extremely asymmetric bidentate fashion, with Au–N bond lengths of 2.078 Å and 2.839 Å, and this complex is possibly better formulated as containing monodentate terpy [51]. An authentic example of monodentate terpy is found in [Rh2(OAc)4(terpy-N)2], the crystal structure of which is shown in Fig. 12, in which monodentate terpy ligands are coordinated to the Rh2(OAc)4 dimer unit through the N atoms of the terminal pyridine rings [52]. The formally tetradentate ligand 2,2′:6′,2″:6″,2‴-quaterpyridine (qtpy) coordinates in a hypodentate fashion to Fe(II) in the bis ligand complex [Fe(qtpy)2](ClO4)2. This complex displays an extremely distorted octahedral geometry about the iron ion with each qtpy ligand binding through three N atoms (Fig. 13) [53]. Higher polypyridine ligands such as 2,2′:6′,2″:6″,2‴:6‴,2′′′′-quinquepyridine and 2,2′:6′,2″:6″,2‴:6‴,2′′′′:6′′′′,2′′′′′-sexipyridine appear predisposed to the formation of helical assemblies containing more than one metal ion, although some examples of hypodentate coordination have been reported [54].
Structurally characterised discrete molecular complexes containing hypodentate terpy
| Complex | Reference |
| [Ru(terpy-N,N′)(CO)2Cl2] | [70] |
| [Ru(terpy-N,N′)(CO)2(mnt)] | [71] |
| [Ru(terpy-N,N′)(CO)2(tdt)] | [71] |
| [Au(terpy-N,N′)(CN)2Br] | [51] |
| [PtIMe3(terpy-N,N′)]·CH2Cl2 | [72] |
| fac-[Re(CO)3(terpy-N,N′)Cl] | [73] |
| [Ru(bpy)2(terpy-N,N′)](PF6)2 | [59] |
| [Ru(bipy)(terpy-N,N′)(CO)(C(O)OH)]PF6 | [74] |
| [Ru(bipy)(terpy-N,N′)(CO)2](PF6)2 | [75] |
| [Ru(bipy)(terpy-N,N′)(CO)2](PF6)2·0.75 CH3OCH3 | [75] |
| [Ru(4,4′-Me2bipy)(terpy-N,N′)(CO)2](PF6)2·CH3C(O)CH3 | [75] |
| [Ru(bipy)(terpy-N,N′)(CO)(CH2OH)]PF6·0.5 CH3CN | [75] |
| [Ru(9S3)(terpy-N,N′)Cl]PF6 | [76] |
| [Pd(η3-C3H5)(terpy-N,N′)]ClO4 | [77] |
| fac-[Re(terpy-N,N′)(CO)3Br] | [78] |
| [Rh2(OAc)4(terpy-N)2] | [52] |
| fac-[Re(CO)3(terpy-N,N′)Cl]·H2O | [79,80] |
| [Pd(C6F5)2(terpy-N,N′)] | [81] |
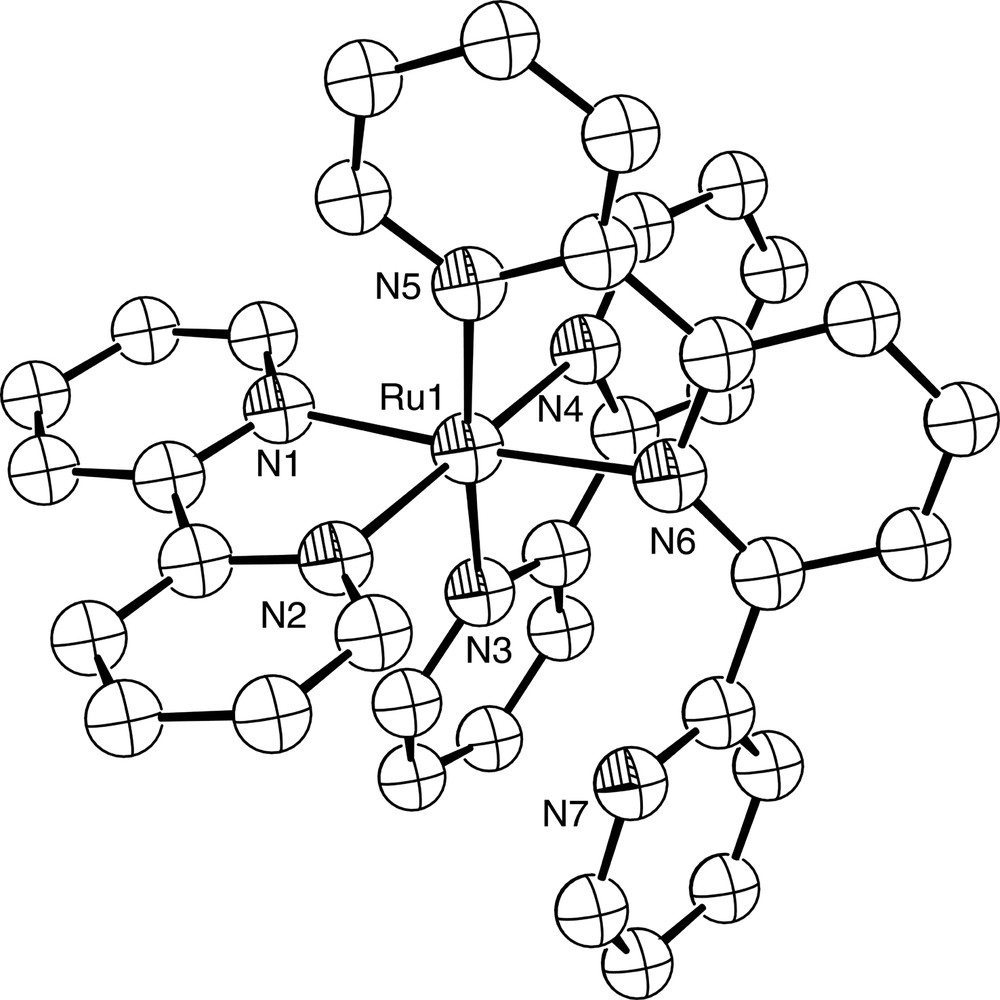
Diagram of the [Ru(bpy)2(terpy-N,N′)]2+ cation in the structurally characterised complex [Ru(bpy)2(terpy-N,N′)](PF6)2. N7 is not coordinated to the Ru2+ ion.
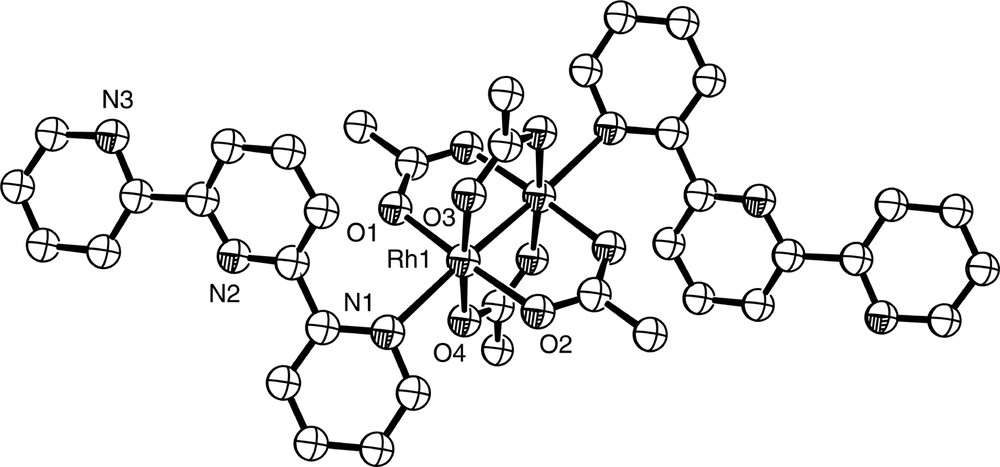
Diagram of structurally characterised [Rh2(OAc)4(terpy-N)2], a rare example of monodentate terpy.
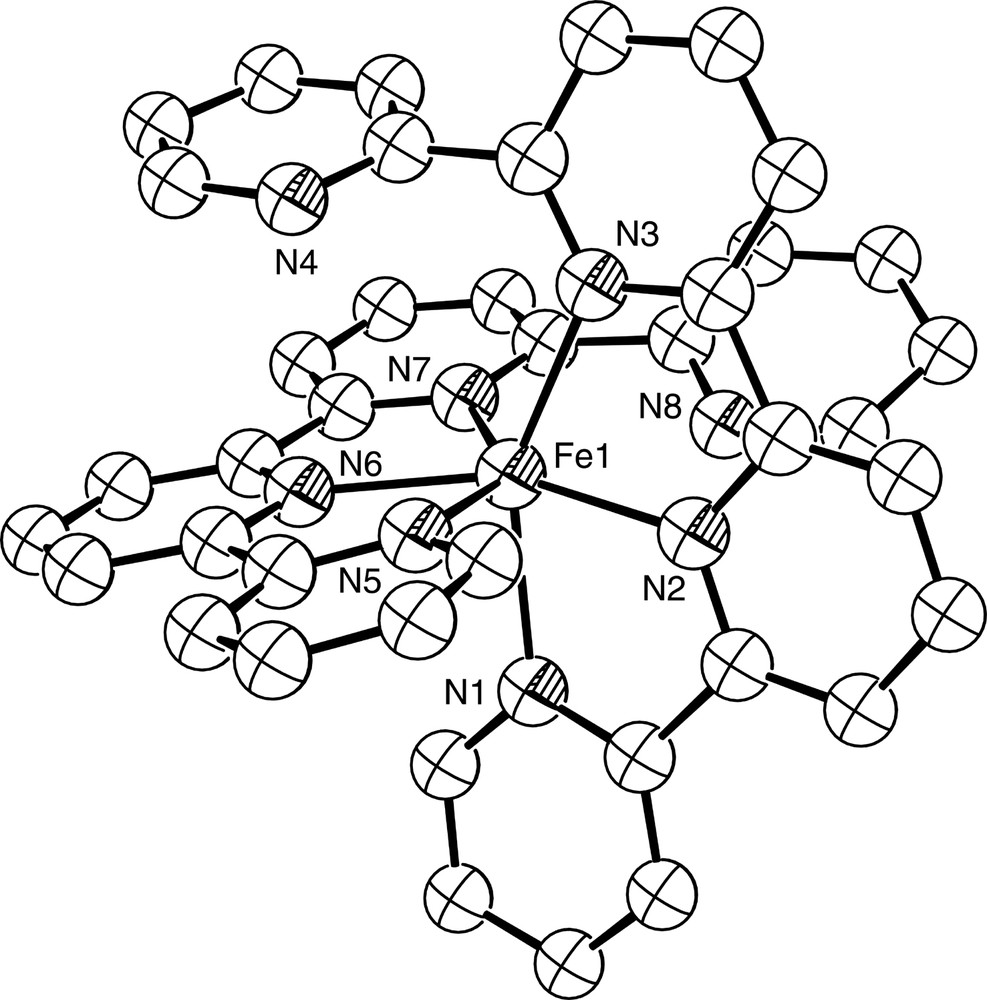
Diagram of the [Fe(qtpy)2]2+ cation in the structurally characterised complex [Fe(qtpy)2](ClO4)2. N4 and N8 are not coordinated to the Fe2+ ion.
3 Synthetic methods for the preparation of hypodentate complexes
It is probably not untrue to say that the majority of complexes containing hypodentate ligands have been prepared by accident. However, rational methods of synthesis of such complexes have been developed and the important factors that appear to govern such syntheses are summarised below.
The majority of well-characterised complexes containing hypodentate ligands contain metal ions that are substitution inert. This can be rationalised when one considers in detail the act of coordination of a multidentate ligand to a metal ion. This process does not involve simultaneous coordination of all donor atoms to the metal ion, but in fact results from initial coordination of a single donor atom to form a hypodentate species whose lifetime is then dependent on, amongst other things, the lability of the metal ion. Formation of this hypodentate species then usually facilitates coordination of the other donor atoms in the same ligand, as the initial coordination constrains these to the vicinity of the metal ion. Hence, if the metal ion is labile, then rapid ligand exchange increases the chances of coordination of all donor atoms within the multidentate ligand, and such a process is also usually entropically favourable. Conversely, the slow ligand exchange rates found for Co(III) and second and third row transition-metal ions can allow hypodentate species to be isolated provided that appropriate reaction conditions are chosen. An excellent example of this is found in Rh(III) complexes of the potentially hexadentate ligand tpen (Fig. 14), where the very slow rate of ligand exchange at the Rh(III) centre allows isolation of two hypodentate complexes, cis-[RhCl2(tpen-N,N′,N″,N‴)]ClO4, in which the ligand is tetradentate with two uncoordinated pyridines, and [RhCl(tpen-N,N′,N″,N‴,N′′′′′)](PF6)2·CH3CN, in which the ligand is pentadentate with one uncoordinated pyridine, from the reaction of RhCl3 with tpen. Surprisingly, the complex containing the fully coordinated ligand could not be prepared [55].
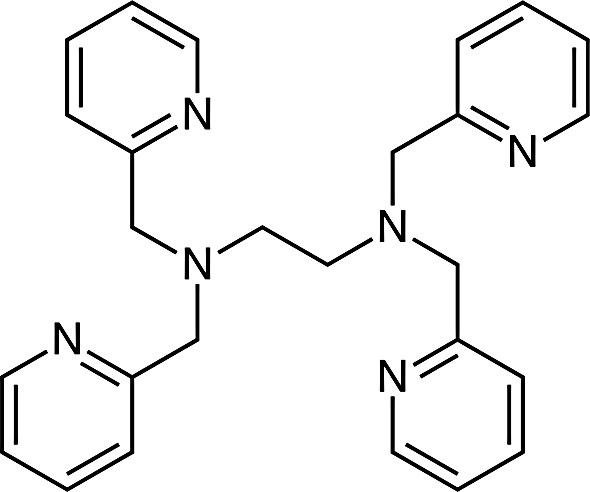
The tpen ligand (N,N,N′,N′-tetrakis(2-pyridylmethyl)ethylenediamine).
The stereochemical and electronic preference of the metal ion can play an important role in the formation and isolation of hypodentate species. An elegant example of this is found in the synthesis of hypodentate polyamine complexes of Cr(0), Mo(0) and W(0) [56–58]. Reaction of either cyclen or cyclam with [M(CO)6] in refluxing n-butyl ether gives the fac-[M(CO)3(L-N,N′,N″)] complexes in which the tetraamine ligand is bound through only three of the four donor atoms, due to the great stability of the fac-[M(CO)3] fragment (Fig. 15). This mode of binding allows the macrocycle to be selectively monoalkylated and monoacylated at the unbound N atom in high yield on reaction with alkyl halides and acid chlorides respectively. Similar reactivity is observed with trien and 1,5,8,12-tetraazadodecane, the linear tetradentate counterparts of cyclen and cyclam respectively, to give [M(CO)3(L-N,N′,N″)] complexes which can then be monofunctionalised at the unbound primary N atom. These complexes offer substantial synthetic utility, as it is difficult, if not impossible, to directly monofunctionalise multidentate amine ligands. Similarly, it has proven impossible to isolate Pt2+ complexes containing either tridentate TACN or tetradentate cyclen or tren, owing to the extraordinary preference of this d8 ion for square planar geometry. Thus attempts to prepare such complexes under ambient conditions invariably result in the formation of hypodentate Pt2+ complexes, or oxidation of the metal ion to octahedral Pt4+, which can then coordinate all donor atoms of the ligand.
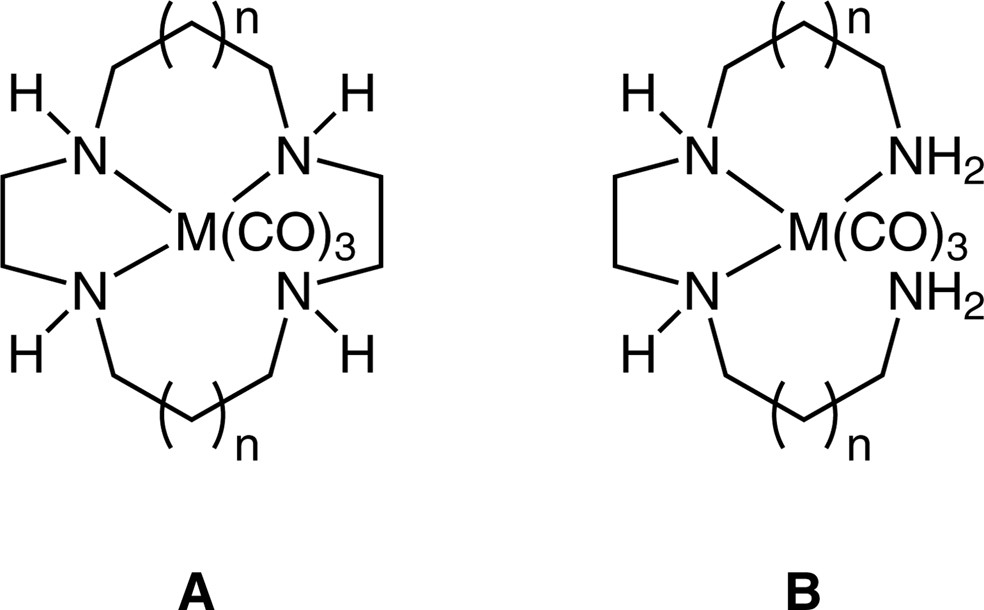
Hypodentate coordination of cyclen (A, n = 0) cyclam (A, n = 1), trien (B, n = 0) and 1,5,8,12-tetraazadodecane (B, n = 1) in fac-[M(CO)3(L-N,N′,N″)] complexes (M = Cr, Mo, W).
The choice of ancillary ligands about the metal ion can also be crucial to the isolation of hypodentate species. Ideally, a starting material for the formation of a hypodentate complex should be coordinatively saturated and contain one or more fewer relatively labile ligands than there are donor atoms in the multidentate ligand to be coordinated. For example, the ammine ligands in [Co(NH3)5X]n+ (X = Me2SO, n = 3; X = CF3SO3, n = 2) undergo replacement only under extremely forcing conditions, and thus treatment of these starting materials with a multidentate ligand at mild temperature can give replacement of the single labile Me2SO or CF3SO3- ligand only, resulting in formation of hypodentate species. Similar strategies have been employed in the synthesis of hypodentate ruthenium terpy complexes, where only the two chloride ligands of the [Ru(bipy)2Cl2] starting material can be easily replaced by two N donor atoms of the normally tridentate terpy ligand [59].
Chemical modification of a ligand by the introduction of bulky groups can influence its coordination properties markedly, and in some cases can encourage hypodentate coordination. Thus, whereas there are no crystallographically-characterised examples of monodentate bipy itself, the introduction of bulky substituents at the appropriate ring positions can force the pyridine rings of the bipy skeleton to be non-coplanar, thus leading to hypodentate binding. This is exemplified by the mercury(II) complex [Hg(3,3′Me2bipy-N)(CH3)]NO3 which contains the ligand 3,3′-dimethyl-2,2′-bipyridine. The crystal structure of this complex (Fig. 16) shows that the ligand coordinates in a monodentate fashion to a single Hg(II) ion, with the pyridyl rings twisted at an angle of 100.5° relative to each other due to the both the steric interaction between the two methyl groups, and the inability of the ligand to cyclometallate through C3′ [60]. Similar behaviour is found in the gold(III) complex of 3,3′-dimethyl-2,2′-biquinoline [Au(3,3′Me2-2,2′-biq-N)Br3]·0.5 HOCH2CH2OH, where the rings of the monodentate biquinoline ligand are at an angle of 109.4° to each other [61] and the gold ion adopts an essentially square planar geometry (Fig. 17). Likewise, the Pd(II) complex of 6-methyl-2,2′-bipyridine [Pd(6-Mebipy-N,N′)(6-Mebipy-N)Cl]BF4 contains one monodentate 6-Mebipy ligand (Fig. 18); the square planar [Pd(6-Mebipy-N,N′)2]2+ ion is not formed due to potential steric clashes between the methyl groups [62].
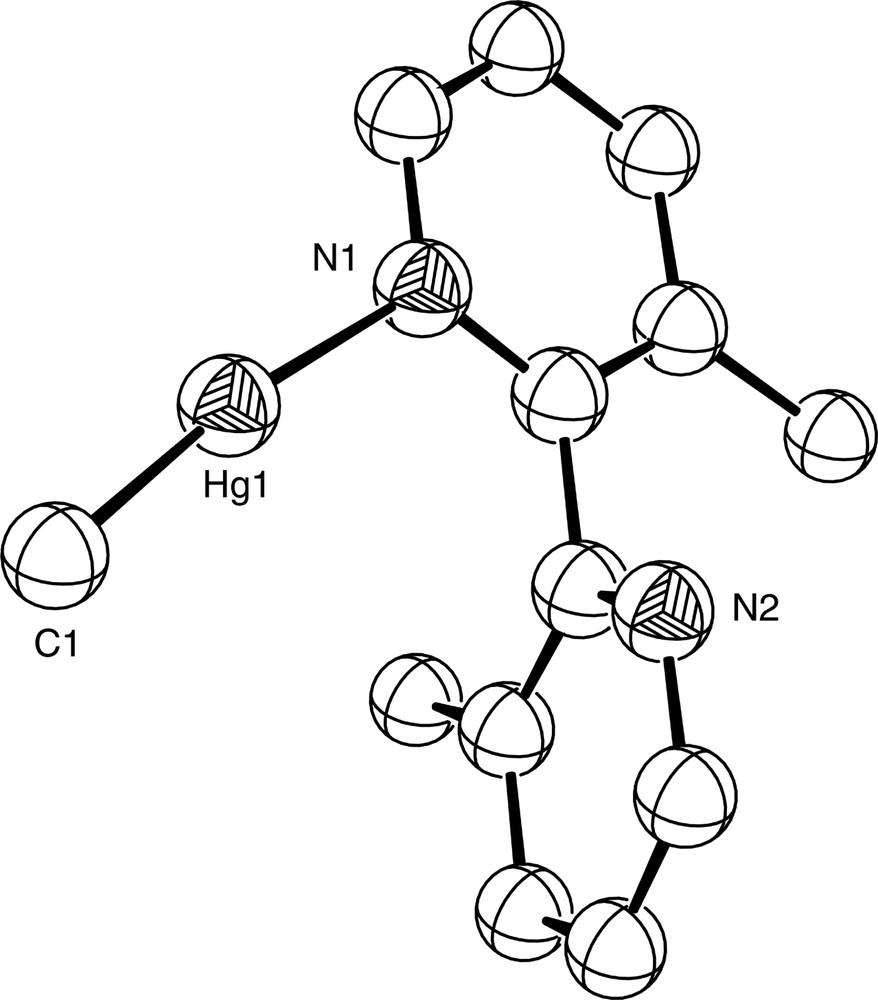
Diagram of the [Hg(3,3′Me2bipy-N)(CH3)]+ cation in the structurally characterised complex [Hg(3,3′Me2bipy-N)(CH3)]NO3.
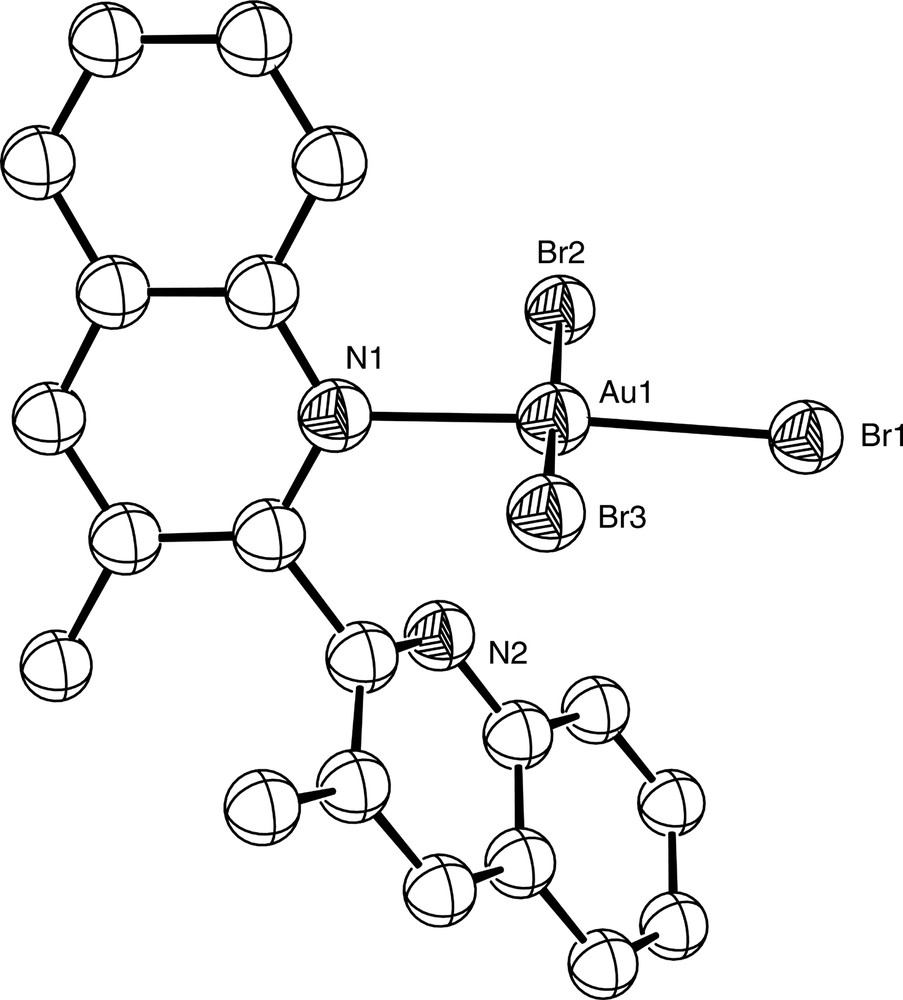
Diagram of [Au(3,3′Me2-2,2′-biq-N)Br3] (solvate molecules removed for clarity), showing monodentate coordination of the biquinoline ligand.
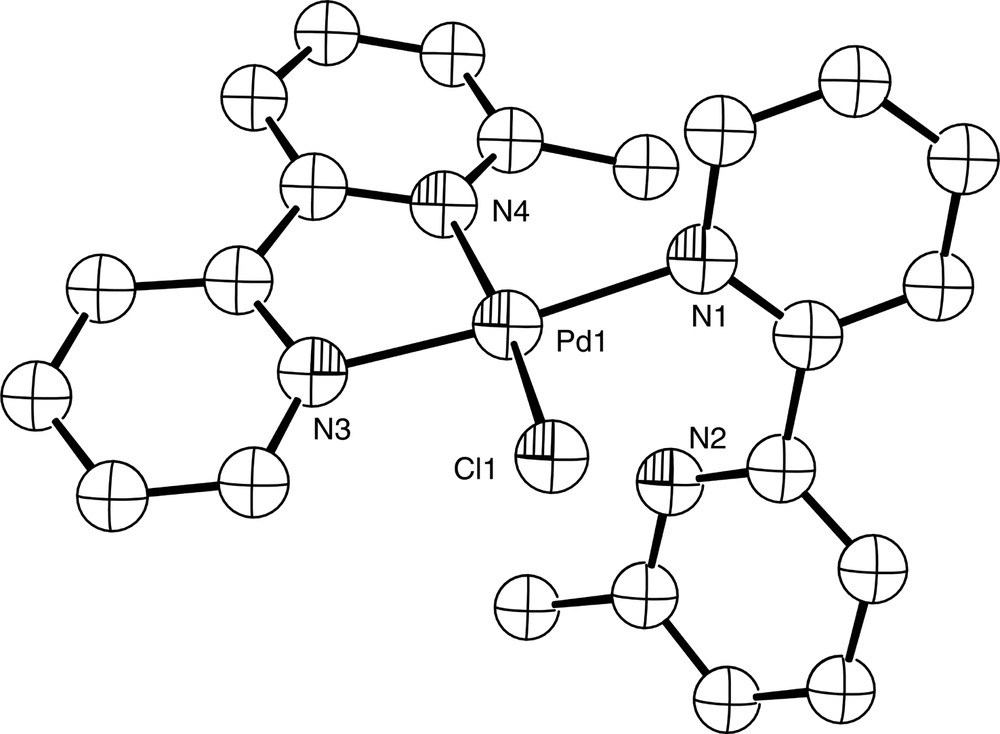
Diagram of the [Pd(6-Mebipy-N,N′)(6-Mebipy-N)Cl]+ cation in the structurally characterised complex [Pd(6-Mebipy-N,N′)(6-Mebipy-N)Cl]BF4.
4 Conclusions
This article, as stated in the introduction, is by no means meant to be a comprehensive or definitive review of hypodentate coordination of multidentate ligands. Rather, I have aimed to introduce and briefly summarise a concept within coordination chemistry that has not received much attention in the chemical literature as a subject within itself. A brief literature search will reveal many more examples of hypodentate coordination than are detailed herein and it is to be hoped that this cursory introduction to the topic will have piqued the interest of more than a few readers in what is still a relatively unexplored, but nevertheless fascinating, area of coordination chemistry.
1 He also proposes the term sundentate to describe a ligand in which all donor atoms are coordinated to the metal, although this does not appear to have yet been used in the chemical literature.