

1 Introduction
Chlorinated volatile organic compounds (ClVOCs) are generated in large amounts (approximately 1.5 millions tons per year) by chemical plants, dry cleaning and degreasing processes [1]. Catalytic oxidation is one of the most promising technologies for the destruction of ClVOCs in gaseous effluents [2–7]. The destruction should occur at low temperatures and short residence times and selectively into CO2, H2O and HCl. The formation of toxic by-products (CO, Cl2, COCl2, etc.) even in traces should be avoided.
This paper deals with the destruction of dichloromethane (DCM) in low amounts (1000 ppm) in wet air over PtFAU zeolite catalysts. It was first confirmed that this destruction occurs through a bifunctional pathway involving successively DCM hydrolysis over the zeolite support with formation of HCl and formaldehyde then oxidation of formaldehyde over Pt. Afterward, hydrolysis of DCM was carried out over a series of FAU samples with different Si/Al ratios and with protons or alkaline cations. A general reaction mechanism is proposed and the role of zeolite acidobasicity on the reaction rate is specified.
2 Experimental
The NaY and NaX samples were supplied by IFP and USHY (HFAU(5)) by PQ zeolites. The various cationic zeolites (LiY, KY and CsY) were prepared by exchange of NaY with corresponding nitrate salt. The degree of exchange was 43% with Li, 91% with K and 64% with Cs. No protonic sites could be observed on all the alkaline FAU samples [8]. The Si/Al ratio of all the Y and X samples was respectively equal to 2.4 and 1.3. The denomination of the zeolite samples was as follows: cation or proton followed by the name of zeolite structure: FAU with the Si/Al ratio into brackets.
Pt catalysts were prepared by ion exchange with Pt(NH3)4Cl2 in competition with NH4+, followed by calcination under dry air flow at 500 °C for 6 h. The dispersion of Pt was determined by CO adsorption followed by IR spectroscopy.
A Setaram DSC 111 calorimeter equipped with a volumetric vacuum line was used for microcalorimetric measurements. Each sample (100 mg) was pretreated overnight at 200 °C under vacuum (10–3 Pa). Adsorption was carried out at 25 °C by admitting successive doses of DCM and recording the thermal effect.
DCM transformation was carried out at constant temperature in a fixed bed reactor containing 140 mg of catalyst. A fresh catalyst sample was used for each experiment. The standard feed (with 2.7 mol.% water) was obtained from the mixture of reconstituted air (nitrogen + oxygen only) containing 3000 ppm of DCM and of reconstituted air having passed in a saturator containing water at 25 °C. The space velocity (volume of feed introduced per volume of the catalyst bed and per hour) was always fixed to 20,000 h–1 with respect to the catalyst. The effluent gases were analysed on line by using two GC apparatus; the first one was equipped with two columns and two detectors: a CPSil 5 capillary column linked to a FID detector for the quantitative analysis of organic reactant and products, a Porapak Q column linked to a TCD detector for the quantitative determination of CO2; the second one, equipped with a 13X molecular sieve column and a TCD detector, was used to determine the production of CO. Hydrochloric acid was recovered in water and its amount estimated by pH measurement. Mass spectrometry (MS) and specific Dräger tubes were used for detecting the eventual production of Cl2 and of phosgene (never observed in this work). Furthermore, GC-MS analysis allowed us to confirm the formation of formaldehyde.
3 Results and discussion
3.1 Comparison of PtHFAU(5) and PtNaFAU(2.4) catalysts
Both catalysts (after a short deactivation period in the case of PtNaFAU) are very stable. The reaction products are formaldehyde and hydrochloric acid in a 1/2 molar ratio and CO2. We have checked through MS analysis that neither organic products other than formaldehyde nor Cl2 and COCl2 (not detected also with Dräger tubes) were present in the effluents [6,7]. PtHFAU(5) samples are much less active than PtNaFAU(2.4) samples: indeed whereas DCM can be converted at 60% at 380 °C only, it is the case at 300 °C with PtNaFAU(2.4). Furthermore, Pt has no effect on the catalyst activity but changes significantly the product distribution with an increase in CO2 conversion at the expense of formaldehyde. Therefore, the following bifunctional scheme can be proposed to explain CO2 formation [7].
This successive reaction scheme is confirmed by the effect of temperature on DCM conversion and product formation. This is shown as an example in Fig. 1a for a 0.5PtNaFAU(2.4) sample. At 280 °C, DCM is only transformed into formaldehyde (and hydrochloric acid), platinum being not enough active to oxidise formaldehyde; above 340 °C, the formaldehyde intermediate is completely oxidised (Fig. 1a).

Influence of reaction temperature on the conversion of dichloromethane (XDCM) and on the yield in CO2 and formaldehyde (HCHO) after 2 h reaction: (a) 0.5PtNaFAU(2.4) and (b) 0.4PtSiO2/NaFAU(2.4) mixture.
It is well known that the first step of Pt introduction in NaY, i.e. the exchange of the zeolite with Pt(NH3)42+ in presence of NH4+ as competition ions, causes the elimination of Na cations (38% in the 0.5PtNaFAU(2.4)) with as a consequence the development of acidic OH-groups after calcination. Thus 66 μmol g–1 of protonic sites able to protonate pyridine at 150 °C were found on the catalyst. To determine the actual activity of a PtNaFAU samples without protonic sites, 0.5PtNaFAU(2.4) was substituted by a mixture of 0.4PtSiO2 and NaY (Fig. 1b) having the same concentration of Pt site per g of catalyst as 0.5PtNaFAU(2.4), i.e. 2.2 × 1018 Pt sites per g. The initial activity (per g of zeolite) of this catalyst was much higher than that of 0.5PtNaFAU(2.4): initial conversion of 100% at 300 °C instead of 50%. This difference in activity is a confirmation of the higher activity of the NaFAU zeolite for DCM hydrolysis. However, a deactivation can be observed during the first 20 min. Afterward, the DCM conversion is approximately of 60%, hence still greater than on 0.5PtNaFAU(2.4). Moreover from the comparison of Figs. 1a and 1b, it can be concluded that while the PtSiO2/NaY mixture is more active, the complete disappearance of formaldehyde occurs at a higher temperature (380 °C instead of 340 °C with PtNaY). This observation, which was already made with other bifunctional transformation [9,10], can be related to the longer distance between the zeolite and Pt sites.
From these experiments, it can be concluded that over PtFAU zeolites, DCM transformation occurs through a bifunctional pathway with hydrolysis of DCM over the zeolite followed by oxidation of the formaldehyde intermediate over the Pt sites. Bifunctional

catalysts with a NaFAU zeolite are more active than those with a protonated zeolite. To specify the reaction mechanism and the active sites, experiments were carried out with pure zeolites.
3.2 Hydrolysis of DCM over various FAU zeolites
DCM transformation was carried out under standard conditions over the acidic HFAU(5) zeolite, over MFAU(2.4) zeolites (Y type) where M is an alkaline atom Li, Na, K, Cs and with a NaFAU(1.3) sample (X).
3.2.1 Influence of time on stream (TOS)
With HFAU(5), there is neither change in DCM conversion with TOS nor change in the product distribution: only HCl and formaldehyde products in the 2/1 molar ratio at temperatures lower than 380 °C, appearance of other products: CO, CO2 and methyl chloride (CH3Cl) at higher temperatures. With cationic zeolites, an initial period of deactivation can be observed. During this period, there is formation of formaldehyde and CO2. As an example, the effect of TOS on DCM conversion and on formaldehyde and CO2 formation is reported in Fig. 2a for NaFAU(1.3) at 220 °C and in Fig. 2b for NaFAU(2.4) at 300 °C, because of its lower activity (at 300 °C DCM conversion is equal to 100% over stabilised NaFAU(1.3)). The difference in activity of fresh zeolites is very significant: thus, DCM is totally transformed on fresh NaFAU(1.3) at 220 °C whereas on fresh NaFAU(2.4) the conversion of DCM is only of 80% at 300 °C; yields of 40% and 14% in CO2 are respectively obtained on fresh NaFAU(1.3) at 220 °C and NaFAU(2.4) at 300 °C (Fig. 2).

Conversion of dichloromethane (XDCM), yield in formaldehyde (H2CO) and in CO2 as a function of TOS: (A) NaFAU(1.3) and (B) NaFAU(2.4) zeolites.
In recent papers [6,11], the initial deactivation of NaFAU samples was explained by their transformation into HNaFAU owing to a partial protonic exchange. This protonic exchange causes a decrease in the heat of DCM adsorption, this decrease being more pronounced with NaFAU(1.3) than with NaFAU(2.4) (Fig. 3). It should be remarked that both on fresh and stabilised samples, the heat of DCM adsorption is selectively greater on NaFAU(1.3) than on NaFAU(2.4). These differences can be explained by considering that hydrochlorocarbons interact on M zeolites both by hydrogen bonding with basic framework oxygen and by interaction of chlorine atoms with M cations [12–14]. The stronger adsorption of DCM on NaFAU(1.3) would be due to the increase in the basic strength with decreasing zeolite Si/Al ratio. However, large differences could also exist in the distance, hence in the interaction between Cl atoms of DCM and Na atoms of NaX and NaY [13].

Differential heat of DCM adsorption as a function of the number of DCM molecules adsorbed per supercage over (A) NaFAU(1.3) and (B) NaFAU(2.4) zeolites.
3.2.2 Relations between activity and heat of DCM adsorption on MFAU zeolites
As indicated above, adsorption of hydrochlorocarbons over FAU zeolites involves interaction between chlorine atoms and cations (or H+) and hydrogen bonding with the framework oxygens (adsorbed species I).
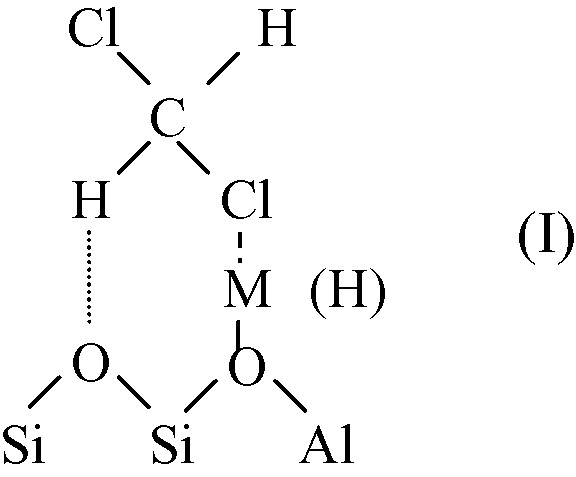
This acidobasic interaction depends on the cation and on the Si/Al ratio of zeolite. The negative charge on the zeolite framework oxygen (hence the basicity) increases with decreasing the Si/Al ratio and the electronegativity of the cation [15–17].
The heat of DCM adsorption (–ΔH) over all the studied FAU samples was plotted in Fig. 4 as a function of the number of molecules adsorbed per supercage. Whatever the sample, there is an initial decrease in (–ΔH) followed by a plateau. The higher the calculated [17] negative partial charge (δO) on the zeolite framework oxygen, the higher the (–ΔH) value. A quasi-linear correlation is furthermore found (Fig. 5) between –ΔH and δO.

Differential heat of DCM adsorption as a function of the number of DCM molecules adsorbed per supercage over various cationic Y zeolites studied. (♦) HY, (■) LiY, (○) NaY, (□) KY, (Δ) CsY.

Differential heat of DCM adsorption over various cationic Y zeolites as a function of the negative partial charge on the zeolite framework oxygen (δO).
The activity of the stabilised FAU samples at 300 and 260 °C was plotted in Fig. 6 as function of (–ΔH) at the plateau. The greater the (–ΔH) value hence the stronger the DCM adsorption, the higher the activity. Furthermore, this figure shows that a minimum value of 50 kJ mol–1 is necessary for the zeolite to be active at both temperatures. The HFAU(5) zeolite for which (–ΔH) at the plateau was equal to 45 kJ.mol–1, is inactive at 260 and 300 °C. From Figs. 5 and 6, it can be concluded that the zeolite basicity plays a significant role in both DCM adsorption and transformation.
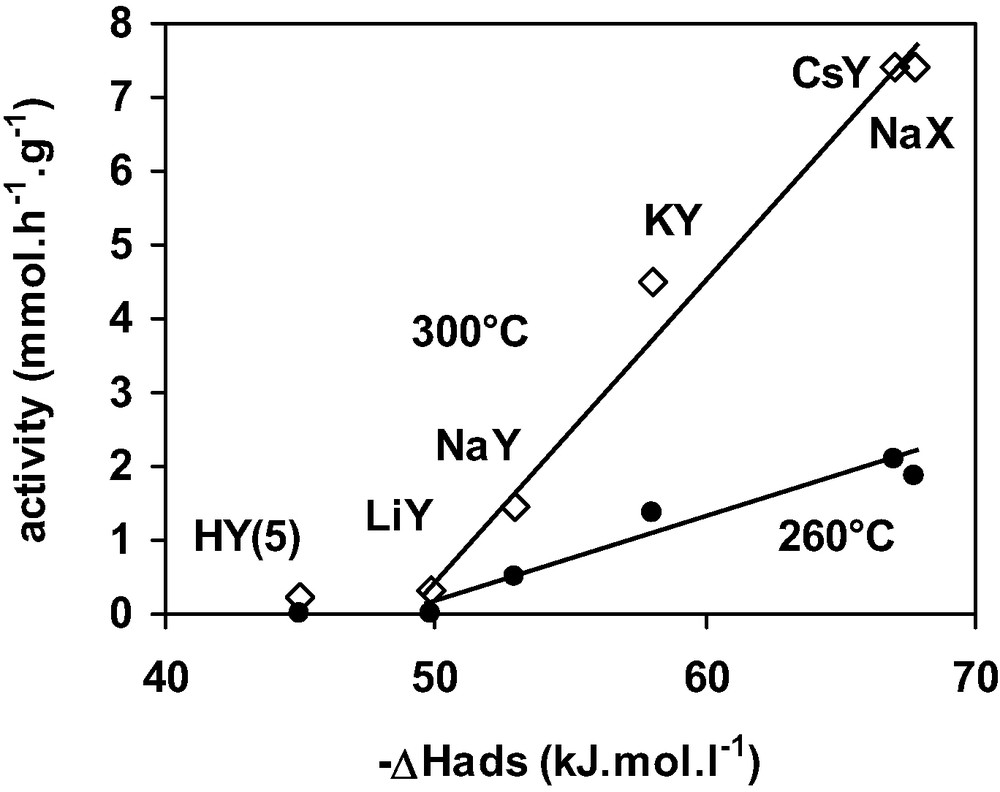
Activity of cationic Y zeolites for DCM transformation taken after 2 h reaction (260 and 300 °C) as a function of the differential heat of DCM adsorption.
3.2.3 Mechanism of DCM hydrolysis
Chloromethoxy species (II) were found to be formed by interaction of DCM with a ZnY zeolite [18]. Alkoxy species are furthermore currently invoked as intermediates in zeolite catalysis.

These chloromethoxy species result from elimination of MCl (or HCl) from DCM adsorbed species I.

In the presence of water, chloromethoxy species undergo a SN2 substitution leading to HCl and to hydroxymethoxy species III.
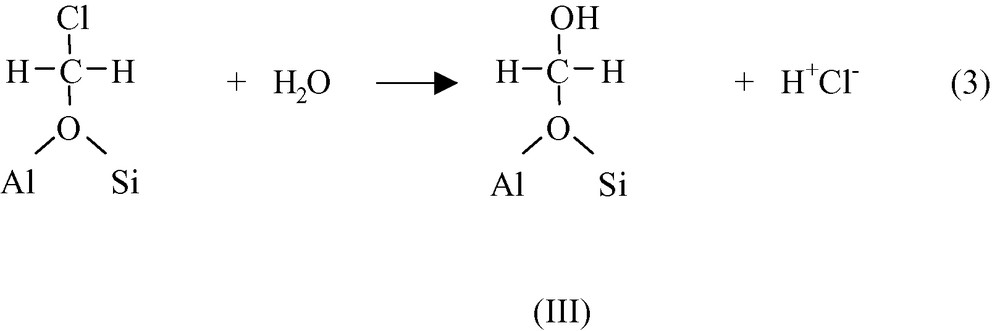
These hydroxymethoxy species III, which correspond to formaldehyde molecules adsorbed on protonic sites of zeolites (hemiacetal-like species), decompose into formaldehyde and bridging hydroxyl groups.
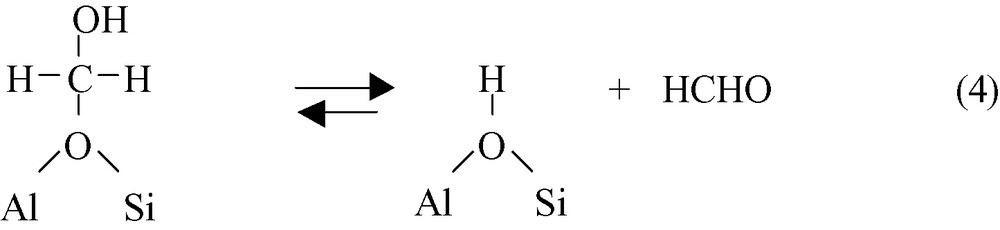
Whereas only adsorption of DCM and surface steps 2–4 occur in the catalytic hydrolysis of DCM over protonated zeolites, an additional step is necessary in the case of cationic zeolites in order to recover the cationic sites. Most likely this step (Reaction 5) occurs between bridging OH groups and MCl released by Reaction (2).

To explain the initial deactivation of cationic zeolites and the simultaneous creation of hydroxyl groups, it was proposed that on fresh basic zeolites, thermodynamic equilibrium will be favoured in the reverse direction with as a consequence a stoechiometric transformation of DCM (Reaction 6).
CH2Cl2 + H2O + MOZ = HCHO + HCl + MCl + HOZ (6)
4 Conclusions
The catalytic destruction of DCM in low amount in wet air, can be carried out over Pt/H or /alkali FAU zeolites (MFAU); CO2, H2O and HCl are the only products. This transformation involves two successive reactions: the first one catalysed on the acidobasic sites of the zeolite is DCM hydrolysis into a 1/2 molar mixture of formaldehyde and HCl; the second one is the oxidation of formaldehyde over Pt sites. The most basic FAU zeolites (CsFAU(2.4) or NaFAU(1.3)) used in this study were found to be the most active catalysts. These catalysts are furthermore more active than PtAl2O3 catalysts.
A mechanism is proposed for DCM hydrolysis. This mechanism involves four (HFAU) or five (MFAU) successive steps:
- (1) adsorption of DCM with interaction of a chlorine atom with the protons or the cations of the zeolite and of an hydrogen atom with a framework oxygen atom;
- (2) transformation of the adsorbed DCM species into a chloromethoxy species with elimination of a molecule of hydrochloric acid or of cation chloride;
- (3) SN2 substitution of Cl by OH from water with formation of hydrochloric acid and hydroxymethoxy species, which correspond to formaldehyde adsorbed on bridging OH groups;
- (4) desorption of formaldehyde with formation of bridging hydroxyl groups;
- (5) recovery (in the case of MFAU zeolites) of the OM group by exchange of the bridging hydroxyl groups with the cation chloride formed in step 2.
With the most basic zeolites, this last step only occurs after partial elimination of M cations during an initial stoechiometric hydrolysis of DCM, hence after a decrease in the zeolite basicity.
Acknowledgements
L. Pinard gratefully acknowledges the ‘Agence de l’environnement et de la maîtrise de l’énergie’ (ADEME) and the ‘Région Poitou-Charentes’ for a scholarship.