

1 Introduction
The field of zeolite synthesis has gone through several phases, each leading to breakthroughs in chemical understanding and discovery of new materials. Much of the structure of the field comes from research over 50 years ago emanating from the Union Carbide labs and those of Professor Richard M. Barrer [1]. Based upon a geological understanding of zeolite deposit growth, work led to a number of new aluminosilicate phases from hydrothermal reactions [2–4]. Products were rich in aluminum (by comparison with what was to come) with corresponding arrays of counterbalancing cations [5].
Very different types of zeolites, rich in silica, arose from the introduction of organo-cations into the synthesis media. These organo-cations, later to be termed templates or structure-directing agents (SDA), were found as guest molecules within the void regions of the crystalline host lattice. Smaller organo-cations like tetramethylammonium could lead to a variety of products and often served a charge counterbalancing function as well. The use of the guest molecules as bases in their hydroxide form also allowed for a diminution of alkali cations which normally accompanied the use of hydroxide sources. This change also opened up new avenues for finding novel high silica zeolite structures [6–10].
Another departure was seen about 20 years ago as Wilson et al. [11] introduced molecular sieves based upon alternating Al and P tetrahedral in oxide lattices. A wide array of chemistry variants followed in this system [12]. Eventually some of the novel properties of these molecular sieves allowed them to emerge as commercial winners [13,14].
At the same time F anions were being used as mineralizing agents replacing the necessity for OH in the syntheses. Flanigen had introduced a route to an all-silica MFI using this chemistry [15], and an effort was begun at Mulhouse, led by Professors Guth and Kessler with early results leading to an understanding that (1) zeolites could be crystallized in the pH 6–8 regime, (2) unusually large crystals could be grown, and (3) the products were remarkably low in lattice defects [16,17]. Continued research by these talented chemists led to such spectacular discoveries as the cloverite crystal in the GaPO4/HF chemistry system [18]. This discovery ushered in a whole new era of inorganic synthesis of crystalline molecular sieves.
In the last decade another important breakthrough has emerged, this time from the labs of Professor Avelino Corma in Valencia Spain. The synthesis work from these labs showed a novel approach of using HF and organo-cations in silicate media, but at remarkably high concentrations compared with zeolite syntheses previously reported in the literature. For the first time all-silica products were obtained that also possessed low-density frameworks. This had never been seen before. This led not only to the syntheses of structures that combined both features but also to the discovery of entirely novel framework structures [19–22]. Previously, the only low-density zeolite solids were those associated with high lattice substitution for silica (Al, Zn, Be, B). Fig. 1 shows a general trend for the effect of lattice substitution on the types of products crystallized in a conventional OH-based synthesis [23], and then Fig. 2 gives a somewhat parallel picture in the HF-based synthesis where concentration emerges as the key determinant for low-density products. In both systems the low-density products tend to be richer in 4-ring sub-units and we will later discuss why each reaction medium may afford that. Much of the proliferation of new materials via the HF-route with variable concentration can be attributed to the successful collaborations of a number of researchers at Valencia in the 1990s. A number of ITQ (Institute of Technologia and Quimica) materials were described [24].
Lattice substitution versus size of organo-cation as factors influencing the crystallizing zeotype.
ITQ studies on low H2O/SiO2 runs [9].
The synergy of using both F-containing reactions and the high concentration reactions was further contrasted by the observation that other pioneers in the use of low water reactions (for different applied chemistry goals) were typically not encountering new phases in systems lacking F [25,26].
Over the years the zeolite synthesis programs at Caltech and Chevron (now ChevronTexaco) Technology Center have synthesized a wide array of SDA molecules. In recent years new zeolite structures have been found via use of these SDA in borosilicate systems CIT-1 (CON) [27], SSZ-55 (ATS) [28], and SSZ-53 (SFT) [29]. Building upon the experiences at Mulhouse and Valencia in the HF chemistry, this medium seemed to offer opportunities for applying our collection of SDA to the task. In this contribution we report our results for a broad range of synthesis screening reactions using the SDA largely developed in our labs. While not every reaction successfully produced a guest/host crystalline product, and molecular sieves like beta (*BEA) have proven to be remarkably ubiquitous host lattices for a number of SDA run at high concentration, we still are able to report the synthesis of over 20 different lattice hosts. Our studies confirm that low-density products, forming at high concentration reaction conditions, give way to denser silicate lattices (clathrates and sieves of reduced dimensionality) as the reaction conditions experience dilution. Does the behavior of F– in solution have a pivotal role in determining these structure selectivities? Finally in surveying the crystalline products observed vs. the SDA employed we raise a new issue. Is the guest/host packing in the crystal altered relative to conventional (OH– based) zeolite syntheses? Does this provide an avenue for finding new structures, which might not be achievable under conventional reaction conditions?
2 Experimental
Zeolite syntheses and their preparations involving evaporation, H2O/SiO2 adjustment and then HF addition were carried out as described by us in a recent report [30]. In general we strove to use 5 mmol of SDA OH–, 5 mmol HF and 10 mmol of SiO2 whenever we could. In a few instances we had to reduce the overall values while keeping the ratios the same, to minimize the use of our limited supply of SDA. Reactions were monitored by SEM analysis, as it was not always obvious that crystallization had occurred by visual examination of the solution/solid separation [31]. A Hitachi S 370 instrument was used. If crystalline material was detected then a pH was measured and the reactor contents were worked up by washing in a glass filter. pH values were typically close to 6 for many of the reactions carried out using equimolar amounts of HF and SDA OH–. Table 1 contrasts some generalizations for the use of this HF reaction system over the more conventional high pH reaction. While valuable and sometimes unique products emerge through the HF-route, the system has some drawbacks as noted in Table 1. Analysis of crystalline phases was carried out with a Siemens D-500 instrument. Elemental analyses were from Galbraith Labs via ICP methods. Solid state NMR experiments were performed on a Bruker DSX-500 spectrometer operating at 500.2 and 99.4 MHz for 1H and 29Si nuclei, respectively. The standard Bruker 4 mm CPMAS probe was used. Specifics of NMR experiments are given with the data in the text.
Some issues for use of HF reaction and large SDA
2.1 SDAs in the syntheses
A variety of SDA molecules were run in the synthesis screening experiments. The results are presented in Tables 2–9 where we show the structure of the SDA, a bookkeeping number for it, a reference for its synthesis and then finally the X-ray diffraction data of formed zeolites. If the compound has not been described in the literature before, then it is described or discussed in Appendix 1 and coded in the tables as A1 under reference.
Zeolites products from diaza bicyclo octane derivatives
Zeolites products from Diels-Alder derivatives [52]
Zeolite product from imidazole derivatives
Piperidine derivatives [52]
SDA from enamines derivatives
Zeolites products from sparteine derivatives
SDA from Michael-addition and Beckman-rearrangement derivatives [53]
Zeolites from miscellaneous derivatives
2.2 Molecular sieve synthesis and diffraction results
The data in Tables 2–9 show the phase selectivities for the various trial runs. The key variable in most runs was the overall reaction concentration. All the runs were carried out in Teflon liners for Parr 4745 reactors, tumbled at 43 rpm at 423 K on a spit in a Blue M convection-heated oven unless otherwise stated in the text. Samples were examined for crystallinity after filtration and washing to achieve a low conductivity. The determination of whether or not to work up a larger sample was often based upon whether the zeolite crystallization run yielded evidence of product via SEM surveys during the run. Runs were usually terminated after about 50 days if there was still no evidence of crystallinity by SEM. The sample was sent for XRD analysis, nonetheless. The data reported as amorphous in Tables 2–9 typifies this situation.
3 Results
3.1 Survey of products made
Fig. 3 is a summary of the various host silicate structures we encountered in carrying out the SDA survey of reactions, usually at two or more concentrations (these were usually bracketed by H2O/SiO2 ratios as low as 3.5 and then upwards of 15; occasionally a run was made at 25). While more than 20 host structures were obtained, with almost no exceptions they were all materials observed before. On the other hand, some of the products have only newly been reported as a result of experiments carried out in the HF regime in other laboratories. This would be true of such discoveries as ITQ-7 (ISV) and ITQ-12 (ITW); see [24].
A wide number of zeolites can be made as all-SiO2 in the HF-route.
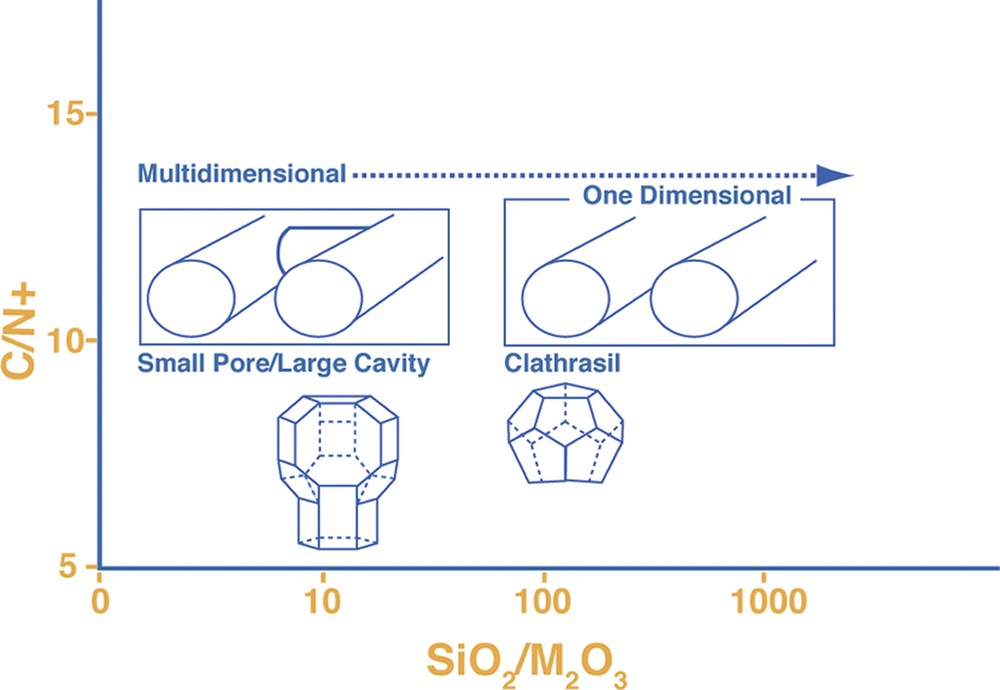
However what is novel is the set of results where some multi-dimensional host frameworks are directly synthesized as all-silica for the first time. One of the first Camblor publications showed that relatively low framework density structures, like chabazite (high micropore volume as commensurate with the low-density; see Fig. 2) could be made for the first time without any aluminum. In this work we also report other relatively open-framework structures prepared in this manner, including SSZ-33 (CON intergrowth), SSZ-37 (NES), EU-1 (EUO) and SSZ-50 (RTH). These are all multi-dimensional zeolite structures. A fifth discovery was an all-silica NU-86 (no structure code yet as this is a disordered material), which crystallized in the presence of an unusual imidazole derivative (see Table 4). Finally an entirely new material, SSZ-61 was found in these reactions (Table 3) and will be discussed further in a later section.
Since our libraries of SDA have been built to some degree on the approach of exploring families of organic reactions, which can lead to variably substituted derivatives of interest, Tables 2–9 are organized accordingly. The categories highlighted in separate tables are:
- • reactions using diaza-bicyclo [2.2.2] derivatives (DABCO), Table 2;
- • SDA built upon Diels-Alder chemistry, Table 3;
- • SDA built upon derivatives of imidazole, Table 4;
- • derivatives of piperidines, Table 5;
- • molecules with a key step involving enamine chemistry, Table 6;
- • derivatives of the polycyclic sparteine diamine system, Table 7;
- • Guest molecules utilizing Michael-addition reactions, Table 8;
- • miscellaneous derivatives, Table 9.
3.2 Multi-dimensional products vs. higher framework density structures
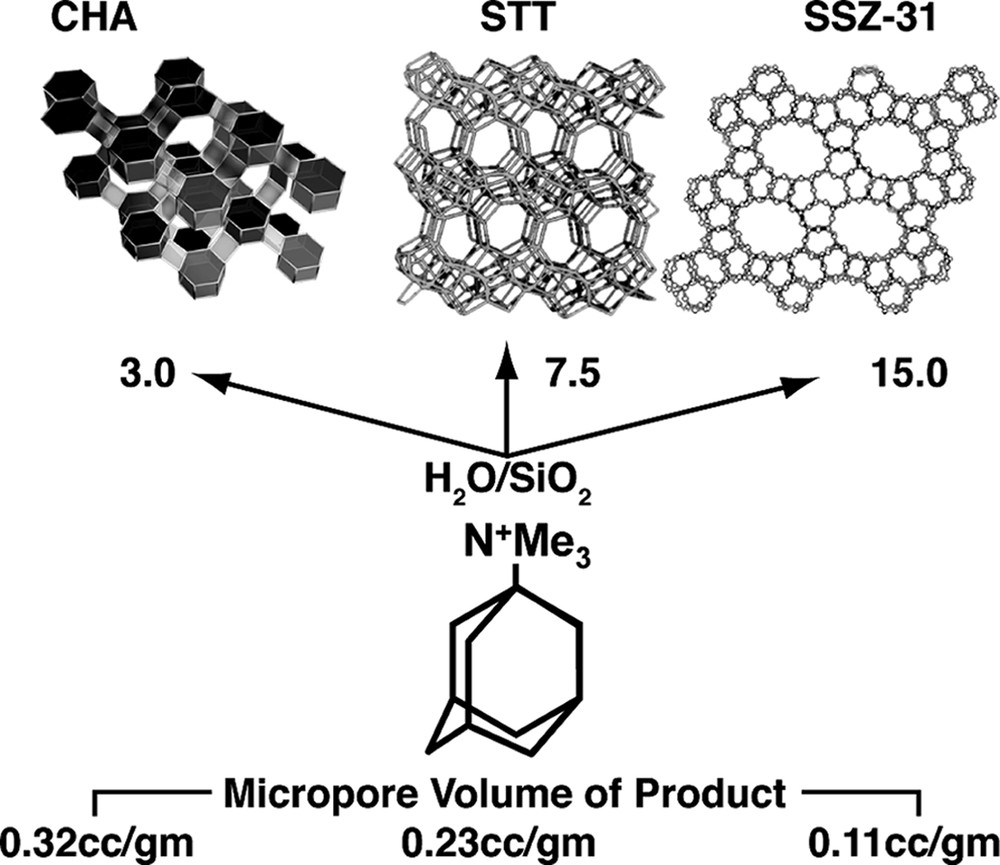
A point was made in the introduction that the discovery of the all-silica but low-density framework silicates was a novel and exciting aspect in using this HF regime at high reactant concentrations. New structures and compositions emerged. The converse then was that as reactions were run under more dilute conditions, two types of frameworks are observed. These are clathrate structures or open-pore products with parallel one-dimensional channel systems. These two products have some correlation to the size of the SDA as we demonstrated in Fig. 1 at the high silica end for reactions carried out in alkaline media. Fig. 2 gives an example of the one-dimensional large pore material, SSZ-31 being formed in the presence of N,N,N-trimethyl-1-adamantammonium cation. Smaller SDA would also lead to clathrates in this chemistry and some examples will be seen in looking at some of the runs in the tables. Interestingly at the high concentration end of the spectrum, a variety of reactions will produce beta zeolite. This is not totally surprising in light of beta zeolite containing a higher population of 4-rings than a number of high silica zeolites. The synthesis relationship of these three materials (in Fig. 2) has already been shown [39] for alkaline reactions, yielding to one-dimensional zeolite products as the trivalent component is reduced in the reaction. ZSM-12 (MTW) is often the ‘default’ product in these reactions. In a similar vein this transformance can be seen in this chemistry. Looking at the DABCO derivatives (Table 2) there are a number of examples of the formation of Beta zeolite. In one series though, one can see that at high enough dilution (H2O/SiO2 = 25) the product eventually becomes MTW. One point to note as well is that the earlier studies in HF-based reactions, many would have been carried out at this H2O/SiO2 ratio or higher, so the tendency was to see one-dimensional or clathrate products crystallize.
Another interesting contrast to note is that in the history of the alkaline-based syntheses using SDA, there is a large variety of reactions which produce the MFI (ZSM-5) zeolite. In this chemistry, ZSM-5 is virtually never seen! It can be easily made in the HF-media in the presence of tetrapropyl ammonium compounds but otherwise is rarely seen. This may be attributable in part to the bifurcation in this chemistry, forming multi-dimensional products, which are richer in 4-rings, or clathrasils and one-dimensional products, which may contain few to no 4-rings in their framework. The contrast of 4-ring formation for either trivalent lattice substitution in the alkaline reactions, or as a function of reaction concentration in the HF-based reactions, is an important illustration of the inorganic chemistry contribution to product selectivity. In hindsight, there are probably a number of the MFI-producing reactions in the alkaline media, where the SDA used may act more as a pore-filler than a true SDA, with the inorganic chemistry having a role in making the MFI formation more likely. A good example would be the synthesis of MFI under SiO2/Al2O3 conditions below 50 with smaller amines [40]. But this contribution from the trivalent ions possibly preferring to be situated in 4-rings, and the influence of hydrated alkali cations, are both missing in the HF-based reactions.
Returning to the DABCO-based SDA reactions, another point to be made is that some of the product diversity can be lost in the HF-based reactions when beta zeolite (including related phases like beta, possibly containing some amount of type A or C, and ISV) can be a product using so many SDAs. In the alkaline chemistry phases like SSZ-45 and 47 were discovered but they are missing in these reactions [41,42]. Likewise, some of the product diversity in using Diels–Alder derivatives is lost at the more concentrated reactions with beta zeolite favored. Good examples are the fates of SDA B08 and B71 (Table 3) which normally can be used to make SSZ-33 (CON) in alkaline reactions but make beta here. One derivative, B75 did succeed in making CON as all-silica, for the first time1.
The Michael-addition SDAs, shown in Table 8, represent a transition between multi-dimensional pore products and more clathrate-like host structures. The Michael-addition SDAs we have made are characterized by having bicyclo ring systems and then enough methyl and ethyl branching to afford a molecule with good surface contacts to a host cage structure. This relationship was pointed out in a recent study we carried out in making RTH with great compositional flexibility (SSZ-50) using SDA B168 [38]. There are not a large number of entries as the synthesis of Michael-Addition SDA is usually accomplished in a range of five to eight synthetic steps so the amount of available material will be more limited.
Besides RTH, STF (SSZ-35) and ITE are found in some experiments. The close synthetic relationship of those two materials (STF and ITE) has been described for a range of molecules in the alkali reaction system [44]. STF has a large cage which extends from the pore mouth opening of a 10-ring, one-dimensional system. ITE and RTH both have a multi-dimensional system which contains 8-rings. SSZ-36 is an intergrowth of these two phases as determined by a DIFFaX study [44]. In one instance, in the table, there is an example of CIT-5 formation at the higher dilution. This is consistent with our observation of the higher dilution leading to clathrates or one-dimensional high silica frameworks. CIT-5 (CFI) fits this criteria. The findings concerning CFI will be important for a different discussion to follow.
Table 4 shows several imidazole-based derivatives. They are quite different in their behavior in this chemistry in that the product formed is now relatively insensitive to the H2O/SiO2 factor. While the value may influence the rate of the crystallization, the product is determined by the SDA structure. In this capacity, we can think of the imidazole SDA as providing a circumstance where the SDA nucleation selectivity dominates the inorganic chemistry. This is the exception in this chemistry. Fig. 4 lists a few other SDA, which have very great host selectivity and are unaffected by the H2O/SiO2 parameter.
SDA which are stronger at structure direction than F–.
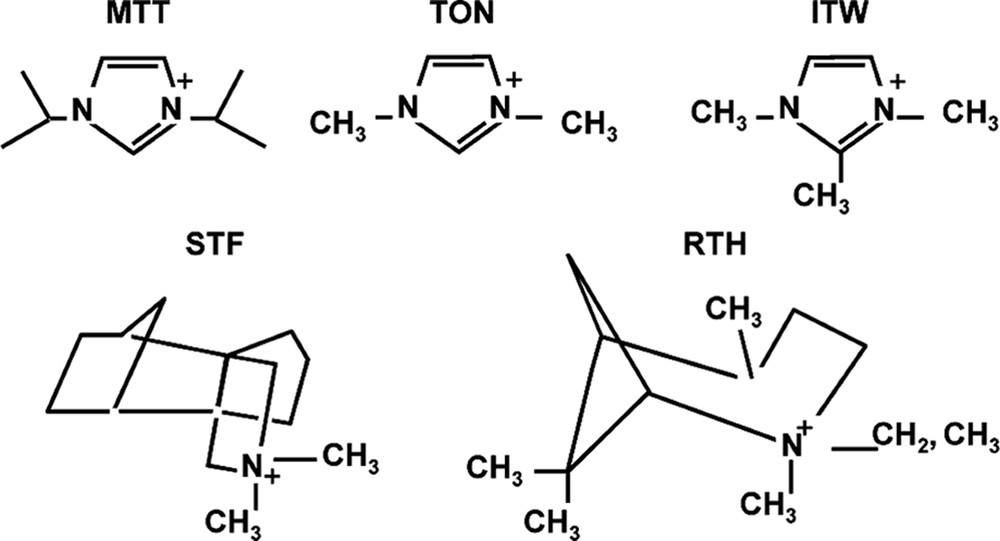
Returning to the concept of the rate, but not the product being affected by H2O/SiO2 values, a comparative study was carried out for rates of formation of MTT versus ITW as the H2O/SiO2 ratio was changed. The ITW has double-4 rings and should be favored by the more concentrated reaction conditions. Indeed it was found that it forms faster under more concentrated conditions and conversely MTT forms more rapidly under dilute conditions [45]. MTT has no 4-rings in the structure. The two frameworks are shown in Fig. 5. The rate data are in Fig. 6. So the results may indicate that the use of silicate or fluorosilicate species in host nucleation may be different in the two concentration regimes and nucleation of different types of structures (ITW vs. MTT) will be favored in some conditions over others.
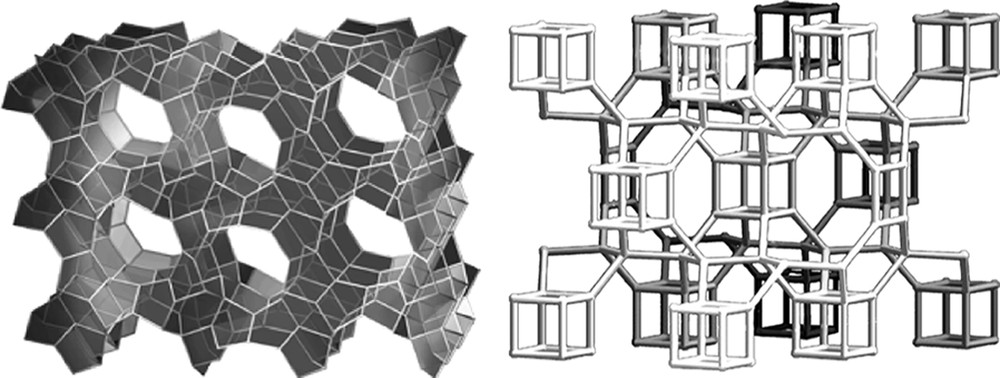
Structures of MTT and ITW.
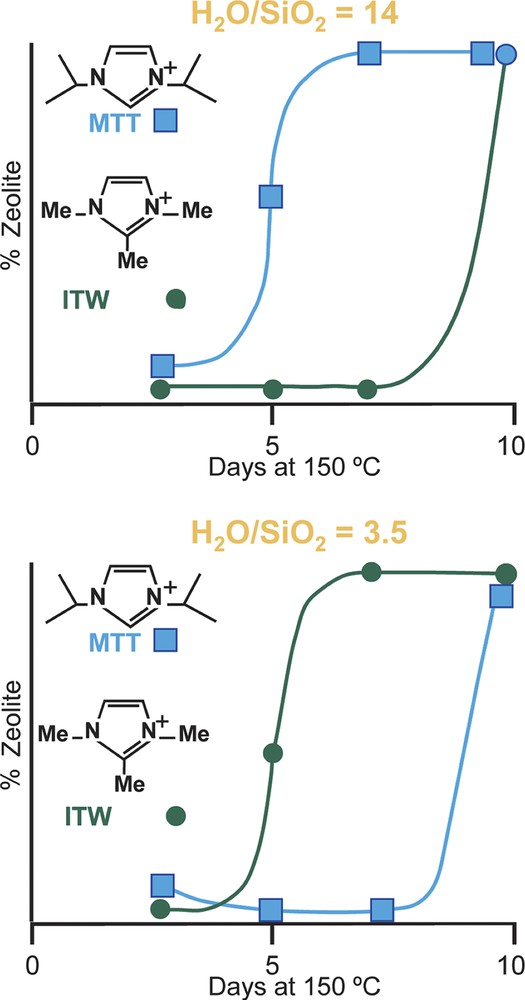
Rate of formation of MTT and ITW.
Given that we have assembled in this collection a few hundred runs at 423 K, it is also noteworthy that no quartz product was observed. We did see some cristobalite in just one series. The absence of quartz is all the more surprising when one considers that a number of these runs were heated out to 7 weeks. The intriguing possibility is that there is a key element in the solution chemistry of these SiFx systems which precludes the successful nucleation of quartz. By contrast, it is all too often a product in the hydroxide-based zeolite syntheses under high silica conditions.
A final point from the survey studies concerns the prospect of forming larger single crystals. Two examples of this result with products large enough for single crystal work are shown in Fig. 7. The products from making SSZ-50 (Table 8) and SSZ-55 (Table 9) proved large enough for single crystal work. Studies on locating the SDA and F anion attachment sites are underway for these two materials.

SEM of SSZ-50 (left) and SSZ-55 (right).
3.3 Use and location of fluorine anion in formation of guest/host structures
A recent computational study by Catlow, Attfield and Sokol dealt with the prospect of F in three different environments within a zeolite host structure [46]. In one scenario, the F– was part of an ion-pair in a host like MFI, in another instance the ion was present within a cubic cage and interacted ionically with several centers. This configuration was exemplified by the cage-based structure AST. The third situation was a nest region within the framework where the fluorine was bonded directly to a distinct five coordinate silicon center. This relationship was tested for IFR.
We decided to study these possibilities from an experimental approach. We chose AST and IFR as targets but decided to enhance the chance of an ion-pair formation in a channel by studying MTT, which has no 4-rings in the structure (just units of 5 and 6). These three structures and the candidate SDA are shown in Fig. 8. We also show zeolite SGT as it formed as the AST system was diluted.
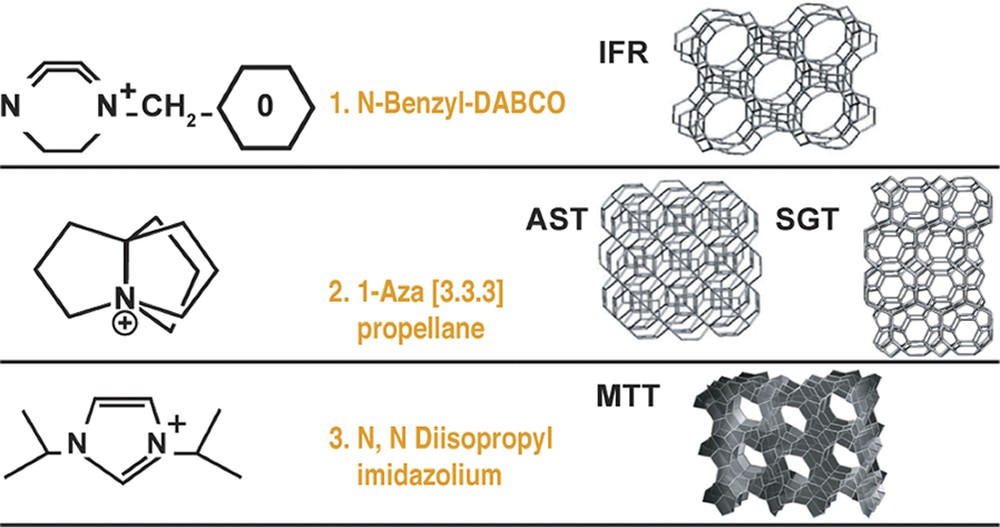
Organo-cations (SDAs) used in the zeolite synthesis study.
Table 10 is a compendium of the run conditions used for the study where we attempted to look at both the effect of dilution and reaction time. The runs were fixed at 150 °C and 43 rpm. In the case of the benzylDABCO SDA, while IFR was specified at more concentrated reaction conditions, with dilution out to H2O/SiO2 = 14, the one-dimensional 12-ring MTW became the product. This is consistent with the view stated above of more open-framework products giving way to those more condensed. Likewise the AST product, selectivity was supplanted by more dense SGT. In the case of MTT there was no change as we had noted above for this surprising SDA selectivity exhibited by imidazole derivatives. SEM images in Fig. 9 show how unique and well formed the product crystals can be in the synthesis under HF conditions.
Reaction conditions for synthesis of fluoride-based all-silica molecular sieves

SEM images of IFR, MTT SGT and AST.
Some corresponding relationships can be seen in terms of the guest components. Table 11 shows the products as a function of the conditions for H2O/SiO2. Table 12 shows the same reaction relationships but now shows the wt.% C found in the guest/host complex. It can be seen that the amount of guest molecule is at its greatest for the most concentrated reaction conditions. The MTT data are invariant. Table 13 shows what happens for the fluoride found in the product complexes. Again the most open structures have the higher amounts of F associated with their formation. But interestingly, the MTT case is not invariant and in fact shows an inverse relationship for reaction concentration and the net amount of fluorine found in the product. Given that the formation of MTT is also slowed in a more concentrated system, the question raised in combining these two results is whether there is a fluorosilicate solution equilibrium which favors the formation of some species at each end of the dilution spectrum. This theory is shown schematically in Fig. 10 where we attempt to describe two possible states for the fluorine–silica interaction. We recognize that the most likely case will be a range of interchanging species in solution (as is seen in studies of alkali silicate systems), but it is also possible that certain key species may be favored in one medium over another.
Products in the study with three unique SDAs
Weight % fluorine in products
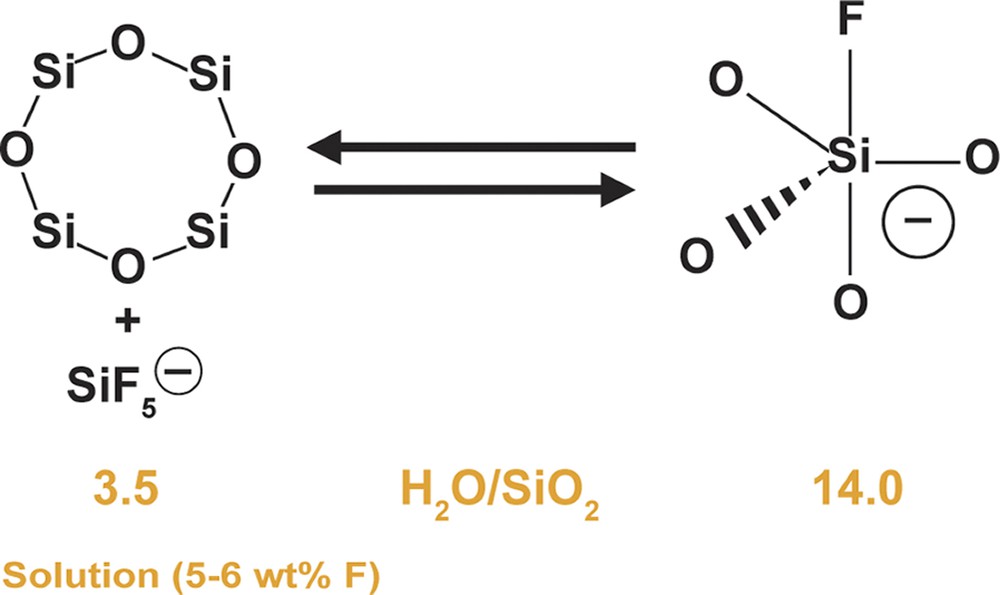
Some theoretical concepts concerning the role of F– at high concentration.
The fluorine was found associated with silicate centers in all three systems we set out to study and the location of fluorine in the MTT structure was verified by both MAS NMR and single crystal studies [30]. Why there is a greater amount of fluorine associated with the MTT formed under more dilute conditions seems not to be necessarily so much a function of the directing role of fluoride in solution as perhaps an ability to either form the structure from units containing perhaps only one fluorine, or from forming a structure which actually provides more defect sites as it grows, which can be filled by fluoride anions as the crystal grows.
3.4 Differences in guest packing for frameworks from alkali and HF-based reactions; some critical reaction possibilities
It has been pointed out before that the products made with near neutral pH reactions and using fluoride anions, have many fewer framework defects than the corresponding products made under alkaline pH conditions [47]. In turn this can mean that there is less disruption to the internal wall surface in the F-products. These disruptions would occur because there are incomplete or dangling bonds with negative charge which might also have water molecules associated with them. In a study by Koller et al. [48], he pointed out that each defect site would have three SiOH and one Si–O–, as determined by NMR work. Unless the extra Si–OH and O– sites can be directed away from the Nonasil cages (as an example) a special intrusion into the cage will be created. This can affect the packing of the SDA occluded and might provide a difference for successful SDA incorporation in Nonasil crystallized in OH– vs. HF medium. One should be able to see these differences by NMR. If there is less surface wall disruption within the structure is there a possibility that there can be a tighter framework fit for the SDA and the silica host? Assuming that the bonding details for all-silica hosts created in either the presence of OH– or F– might be the same as far as angles and lengths related to Si–O (obviously there will be changes in local order for the minority of silicate centers also bonded to fluoride) then we might have the same void region but a contrast in how well it can be filled.
The prospect of this phenomenon, and the basis for this section of our study, came about as a result of two anomalous results we observed in the Tables 2–9. In the Table for Diels–Alder data we had noted that B71 made Nonasil under the more dilute conditions. The one carbon smaller B09 (also the same table) had been the biggest molecule we had found to make Nonasil under alkaline conditions. This was shown in both synthesis work and molecular modeling [49]. At the same time, B71 had been found to make only 12-ring zeolite products; multi-dimensional SSZ-33 at higher SiO2 lattice substitution and SSZ-31 (higher framework density) under less substitution. This relationship had been generalized in Fig. 1. Now we were finding that B71 could make a clathrate as the reaction became more dilute in the HF system. Consistent with Fig. 2 though, multi-dimensional product beta was made under more concentrated reaction conditions. These relationships are summarized in Table 14. Fig. 11 shows how the 29Si CPMAS NMR change from beta (BEA*) to NON, and the spectra indicates that there is not too much Q3 in the spectra for NON. We then returned to looking at the Nonasil sieves made by B09 and studied under both OH and F media (see Table 15). Fig. 12 shows cross-polarization (CP) MAS NMR dynamic curves with the hope of seeing differences in CP time constants for the comparative Nonasil samples. A comparison with the Nonasil made from B71 is also given.
Some anomalies for SDA in HF versus OH– medium
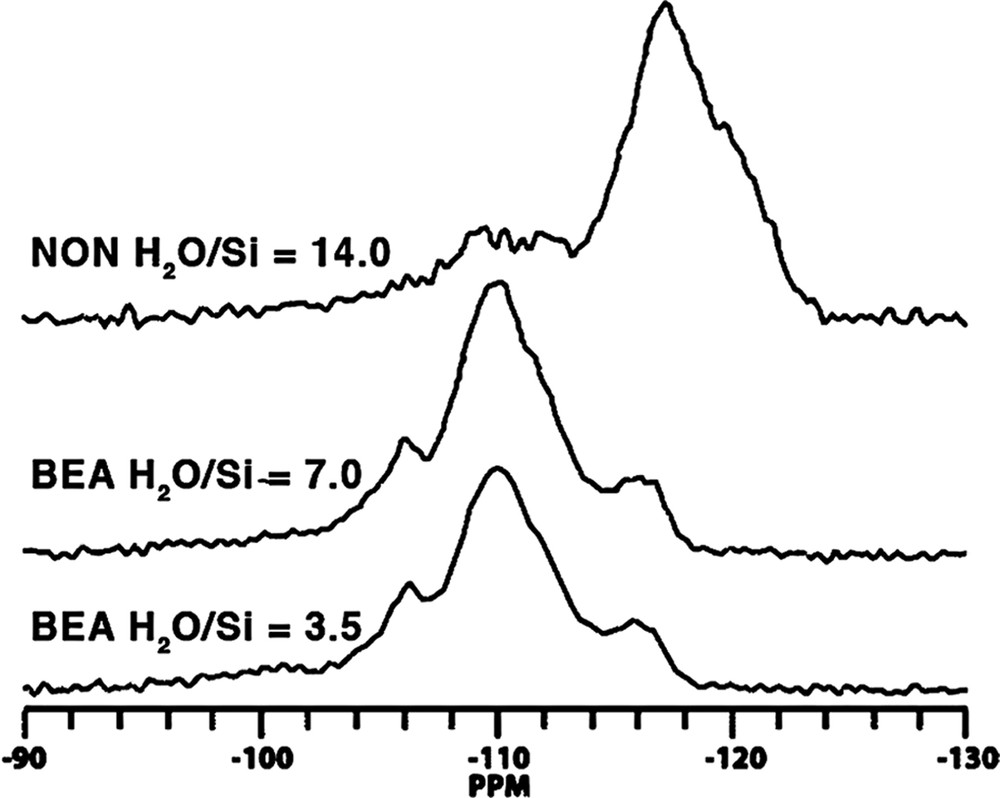
29Si CP MAS NMR of NON versus Beta, where each is made from SDA B71. Samples were spun at 6 kHz and 2 ms of cross-polarization contact time was used.
SDAs used comparative NMR study for NONASIL
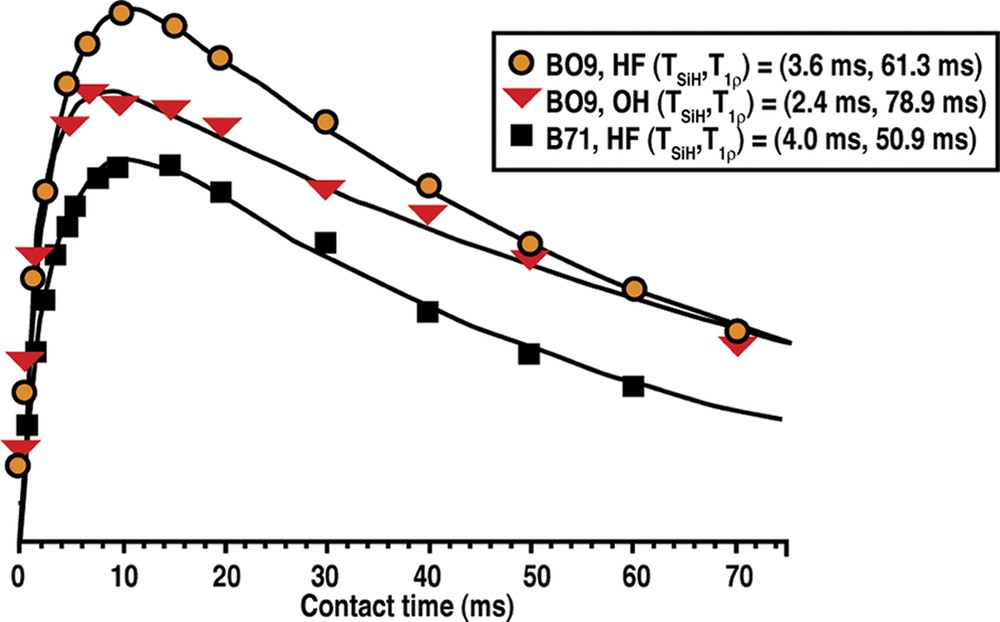
The 1H–29Si cross-polarization dynamic curves from as synthesized NON zeolites prepared with different SDA (BO9 and B71) and in different media (OH) and HF).
With these samples in hand we then proceeded to see if the measurement of cross-polarization rate constants could provide an indication that there is tighter packing for the Nonasil made under HF conditions than under OH–. A key parameter in the cross-polarization experiment is the rate constant (1/TSiH) that can be directly correlated with the distance between SDA and framework Si atoms. Rates can be measured for the cross-polarization transfer from abundant spin (like H) to less abundant nuclei (like C or Si) under a spin-locking condition (or contact period). From Fig. 12 the solid lines are iterative fits using the standard CP dynamics equation,
So in this attempt we are, at present, unable to show a closer packing (using NMR) for B71 over BO9. It should also be noted (from Table 3) that a variety of N-substituted [5.2.1.0] tricyclodecanes were made with an emphasis on larger R-groups at N+. None of these made NON. So the ring enlargement in going from BO9 to B71 may be the only case that does this in HF-mediated reactions.
Another example of this unexpected reaction selectivity in the HF system as compared to alkaline media, is the example in Table 8 where Michael-addition SDA B140 makes CIT-5 (after several weeks of reaction). This SDA makes no products in the various screening reactions we have tried in alkaline conditions. In Table 7 there are some examples of the synthesis of CIT-5 using sparteine derivatives. This 14-ring silicate was discovered by Wagner et al. [51] during study of these SDAs. Note that for the sparteines, the product can be made in both fluoride and alkaline reactions. In the case of B140, we also see that the CIT-5, the only product it makes, is made under the more dilute reaction conditions, consistent with the speculation that one-dimensional zeolites (even 14-rings) would be favored at this end of the spectrum of concentrations.
Again, we decided to ask whether we could see differences in packing for the guest molecules within these various CIT-5 materials by using cross-polarization MAS NMR experiments. As above the idea was to look at whether the CP dynamic behavior and the rate constants (TSiH, T1p) can be correlated to the distance for the SDA to the silicate host wall. Response times in milliseconds were recorded and an argument could be made that the cross-polarization is rapid in looking at the behavior for B140 within the CFI host structure. But so far, for a series of sparteine derivatives, when used as either ethyl or methyl quaternary ammonium compounds, and then studied in either OH or HF medium results have been less clear in terms of polarization rate constants related to closeness of fit. More work needs to be done here and we cannot directly compare B140 in both systems as it makes CIT-5 in only the HF reaction. Molecular modeling may also help to provide answers to this question, as not all orientations may be capable of giving favorable packing.
One other note can be made about the close-packing aspect of guest/host formation in this chemistry. In the Diels–Alder Table, there is a large molecule, B139, which makes a novel material SSZ-61. This product forms at high dilution and its TEM images (Fig. 13) suggest that it has parallel, one-dimensional pores. This would be consistent with the type of material and extent of dilution we might expect in the HF reaction. Also, as in our discussion above concerning B140 and CIT-5 zeolite, we also get no products at all for using B139 in alkaline reaction systems. Work is underway to investigate whether we can show a strong cross-polarization correlation related to a ‘closeness-of-packing’ in this novel material.

TEM of SSZ-61. The end-on view of sizable pores can be seen in the main field view and the parallel lattice fringes in the lower right corner segment. Parallel pores would be a characteristic we had expect for one-dimensional channels, which is what would be consistent with SSZ-61 crystallizing at higher dilution.
4 Conclusions
A survey was made of the crystallization of all-silica molecular sieves in the HF-based synthesis reaction, attempting to study the results with two or more dilution experiments. A wide number of products were found and generally conforming to the previous notion that multi-dimensional host silicates crystallize best from highly concentrated reaction systems. By comparison, more dilute reactions generate higher framework density silicate products, being uni-dimensional or clathrate in structure. Some exceptions were found where the same SDA can make the same host structure over a variety of concentration values. In these instances we have come to view the SDA as having a stronger structure-determining role than the inorganic SiFx chemistry in solution. Five new all-silica compositions are reported in this survey and one new crystalline phase. A few other results are marked as unknowns in the Tables and they are currently under study to determine if they are layered or fully connected silicate structures. Most of these are found at the most concentrated reaction conditions.
Within the context of the survey results, a few SDA were studied in greater detail as providing an opportunity to follow how the system selects a crystallization product and the eventual fate of the fluoride ions. Following a computational study from Catlow's group we attempted to find three different host environments for three different structures, which were specified by selective SDA (structures IFR, AST and MTT). We looked at how the structures rates were affected by dilution, and then at the resulting pore contents of SDA and fluoride. Both parameters increased for more concentrated conditions for synthesis of AST and IFR. MTT showed its pore filling to be invariant and an oddly inverse relationship was shown for the F– content in the product vs. reactant concentration. AST and IFR can be considered to be lower density more open-framework structures as contrasted with a high framework density for MTT. We thought it was possible that the MTT system might give the best prospect of creating an ion-pair of SDA and F– within the pores of the molecular sieve (MTT has no 4-rings). But even here crystallographic and NMR studies showed a fluorine bonded to a framework Si site [30]. Further studies are underway to better understand how fluoride is used in these contrasting syntheses.
Finally an attempt was made to understand the possibility that larger molecules may be able to fit into a host lattice when the medium is HF rather than OH–. Results that supported this observation came from two SDA in the Diels–Alder group of SDA and in a molecule from the Michael-addition group. Then, these anomalies were studied with NMR, the aim being to establish whether the SDA was closer to the host walls in HF type reactions. The results of these cross-polarization experiments do not unequivocally support this theory so further work in synthesis, NMR, and modeling is needed concerning this possibility. Nonetheless the empirical observation is that phases can be obtained under these conditions with large SDA which do not function in the conventional high pH zeolite synthesis reactions.
Acknowledgements
At ChevronTexaco, we thank Dr. Ignatius Chan for microscopy images of SSZ-61, Thomas Rea for Scanning Electron Microscope images, LunTeh Yuen for help in some of the synthesis runs, Greg Lee for the invention and synthesis of the Michael-Addition SDA. Additionally we thank ChevronTexaco for support of research at California Institute of Technology. The NMR Facility at Caltech was supported under Grant #9724240 from the National Science Foundation.
Appendix A1
List of syntheses not referenced in the Tables 2–9:
L36
Synthesized from the commercially available free amine. It was exhaustively methylated with CH3I in methanol with 2 equiv. of KHCO3. After several days, the solution was stripped off (CH3I and CH3OH). The dry solids were extracted twice with CHCl3. The latter was dried with anhydrous Na2SO4, filtered and then stripped to produce the crude product separate from K salts. The crude product was recrystallized from a minimum of hot methanol.
E45
Starting with the commercially available di-anhydride Diels Alder compound, the material is treated with an excess of ethylamine (70% in water) and a small amount of 4-dimethylaminopyridine (caution; toxic) in a closed Parr Reactor, heated to 85C for 3 days. This procedure is presented in [32]. The reaction is cooled to room temperature, poured into extra water and the crystals which form are filtered off. After drying, the di- N-ethyl derivative is reduced by lithium aluminum hydride in THF [39] at room temp and then under reflux for a few days. The lithium aluminum hydride is carefully decomposed with 15% NaOH with good mechanical stirring. The amine is extracted into ether, dried with MgSO4, recovered from the dried solution, and then alkylated with ethyl bromide or iodide to produce the correct diquaternary compound.
C36
This SDA was prepared by the alkylation of the 1,2 dimethylimidazole with 2-propyl bromide in chloroform solvent. The product crystallizes from solution and can be recrystallized from a minimum of hot methanol.
C64, 67
These diquaternary compounds were prepared by reacting two equivalents of the imidazole base with an equivalent of the alpha/omega dihalide (1,6 dibromo in the case of C64 and 1,9 for the C67). The C64 product is easily isolated and then recrystallized. The C67 requires a partitioning of the reaction system (after removing solvent) between a minimum of a brine solution and THF for removal of unreacted materials. The brine solution is then evaporated to dryness, the desired product extracted into CHCl3, dried over MgSO4 and then stripped down. The product, at this stage, is an oil giving a good fit for the desired product, by NMR.
1 The first report of an all-silica CON with few defects came from a novel post-synthesis treatment discovery from researchers Jones and Davis as reported by [43].