

1 Introduction
Lanthanide complexes possessing a high degree of structural organization associated with tunable electronic and magnetic properties are promising precursors for molecular and supramolecular devices [1–3]. As a result of the poor stereochemical preferences and variable coordination numbers of Ln(III) ions, the structural control of the final architecture essentially arises from weak, but crucial interactions between the wrapped ligand strands according to the induced fit concept [1–3]. In this context, semirigid symmetrical tridentate units containing a central pyridine ring L1–4 (Scheme 1) have been shown to produce nine-coordinate pseudo-tricapped trigonal prismatic sites [4–9] but an improved control of the electronic properties requires the development of unsymmetrical tridentate units (such as L5,6, scheme 1) possessing two different side arms connected to the 2- and 6-positions of the central pyridine ring [7,10–13]. However, the controlled facial arrangement of three unsymmetrical tridentate binding units around Ln(III) implies the use of non-covalent d-block based tripods (L6) or their connection to a tripod [14–17] which prevents facial/meridional isomerization [18].
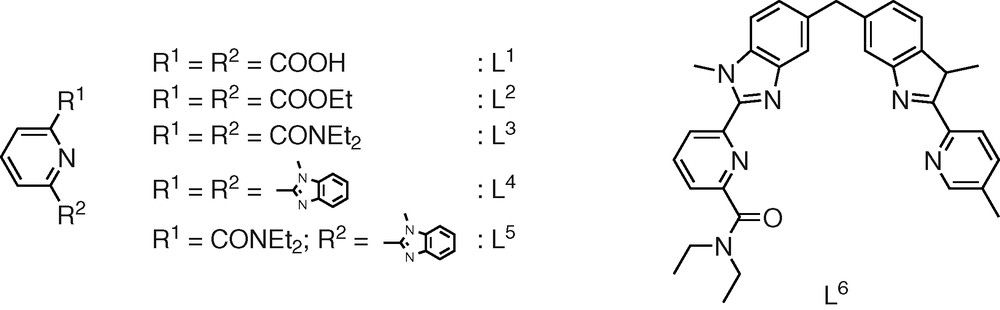
2 Synthesis of the ligands
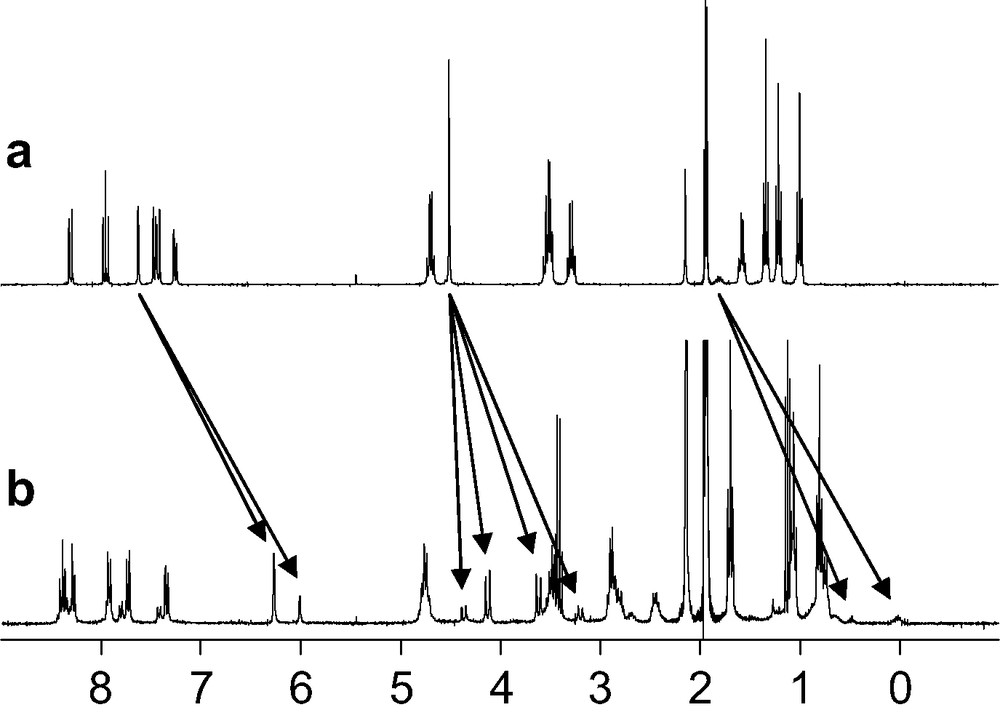
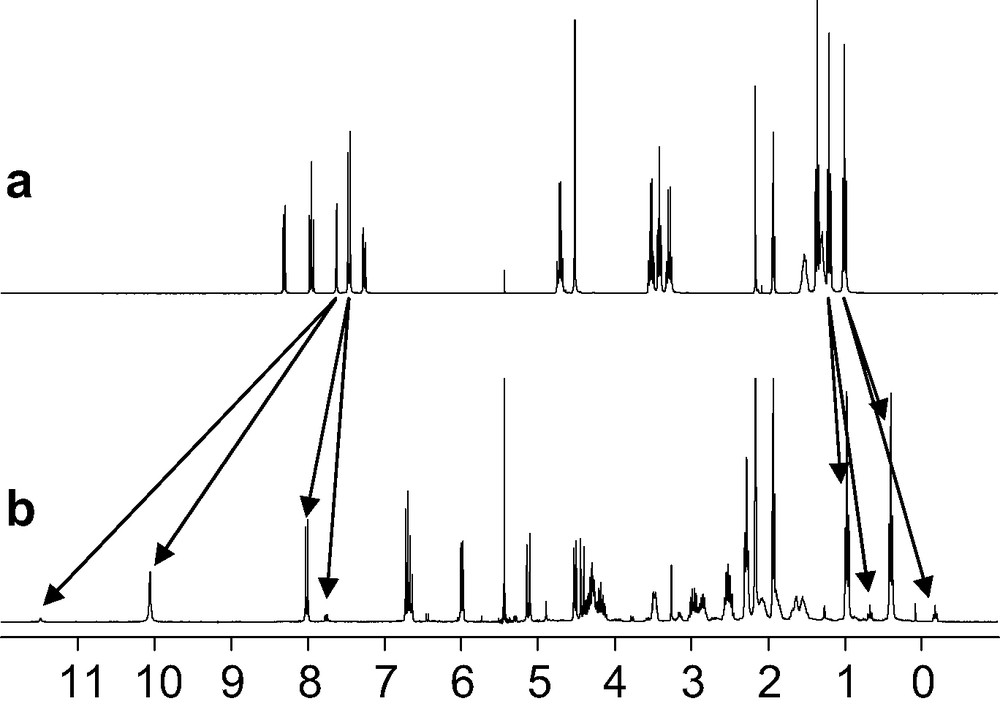
The ligands tris-{2-[2-(6-diethylcarbamoyl-pyridin-2-yl)-1-ethyl-1H-benzoimidazol-5-ylmethoxy]-ethyl}-methane (L7) [19], 1,1,1-tris-{2-[2-(6-diethylcarbamoyl-pyridin-2-yl)-1-ethyl-1H-benzoimidazol-5-ylmethoxy]-ethyl}-ethane (L8) [20] and tris-{3-[2-(6-diethylcarbamoyl-pyridin-2-yl)-1-ethyl-1H-benzoimidazol-5-ylmethoxy]-propyl}-methane (L9) were obtained according to a convergent multi-step strategy (Scheme 2). The 13C-NMR spectra of Lx (x = 7–9) in acetonitrile display the signals expected for trigonal symmetry (C3 or C3v point groups). The 1H-NMR signals confirm threefold symmetry and the systematic observation of enantiotopic methylene protons points to flexible side arms providing a dynamically average C3v for Lx (x = 7–9) in solution (Figs. 1a, 2a and 3a).
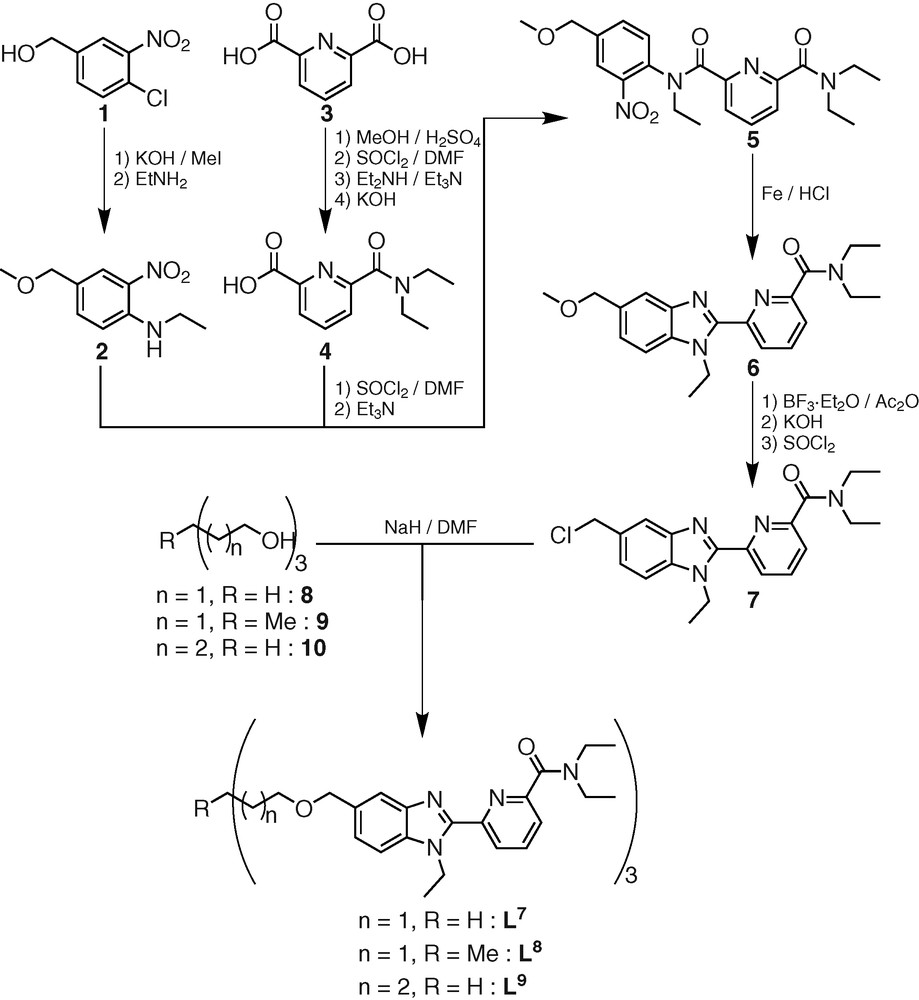
1H-NMR spectra of a) L7 and b) [La(L7)]3+ in CD3CN (298 K).
1H-NMR spectra of a) L9 and b) [Eu(L9)]3+ in CD3CN (298 K).

1H-NMR spectra of a) L8 and b) [Eu(L8)]3+ in CD3CN (298 K).
3 Solution structure of the complexes [Ln(Lx)](ClO4)3 (x = 7, Ln = La; x = 8, Ln = Eu; x = 9, Ln = Eu)
ESI-MS titrations of Lx (x = 7–9, 10–4 mol.dm–3, acetonitrile) with Ln(ClO4)3·n H2O (Ln = La, Eu, Lu; n = 1–4) for Ln:Lx (x = 7–9) ratios in the range 0.5–2.0 show the exclusive formation of the complex [Ln(Lx)]3+ (x = 7–9) together with their adduct ions [Ln(Lx)(CF3SO3)i](3–i)+ (i = 1–2). On the other hand, the 1H-NMR spectra of [La(L7)]3+ and [Eu(L9)]3+ systematically display two different sets of signals corresponding to two different C3-symmetrical species in approximately 7:3 and 13:1 ratios, respectively (Figs. 1b and 3b).
The minor variation of the chemical shifts of the same nucleus in the two isomers points to similar chemical environments and closely related arrangements of the strands. We have shown that these two isomers did not result from an endo/exo isomerization of the apical proton, but from a blocked conformation (on the NMR time scale) of the ethyleneoxy spacers of the capping moiety [19]. The 13:1 ratio observed for the two isomers in the 1H NMR spectrum of [Ln(L9)]3+ – versus the 7:3 ratio observed for [Ln(L7)]3+ – reflects the gain of flexibility induced by the longer spacer in L9. The high activation energy ([Ln(L7)]3+, ΔG≠ > 71 kJ mol–1; [Ln(L9)]3+, ΔG≠ > 67 kJ mol–1) estimated for this interconversion suggests that it requires the partial decomplexation of the ligand. On the other hand, the 1H-NMR spectrum of [Eu(L8)]3+ displays a single set of 22 signals which confirms the formation of a single C3-symmetrical species (Fig. 3b), in contrast with the systematic detection of two different inert conformers for [Ln(Lx)]3+ (x = 7, 9). The formation constants log(ßLn11) = 7.2–8.2, observed for the complexes [Ln(L8)]3+ (Ln = La–Lu) do not vary significantly along the lanthanide series within experimental errors and point to negligible size-discriminating effects (Fig. 4). These constants are slightly larger than log(ßLn11) = 6.5–7.6 obtained in the same conditions for the analogous nine-coordinate podates [Ln(L7)]3+ (Ln = La–Lu), which suggests that the methylation of the remote capping carbon atom influences the binding of the tridentate side arms and slightly increases their affinity for Ln(III) [19,20].
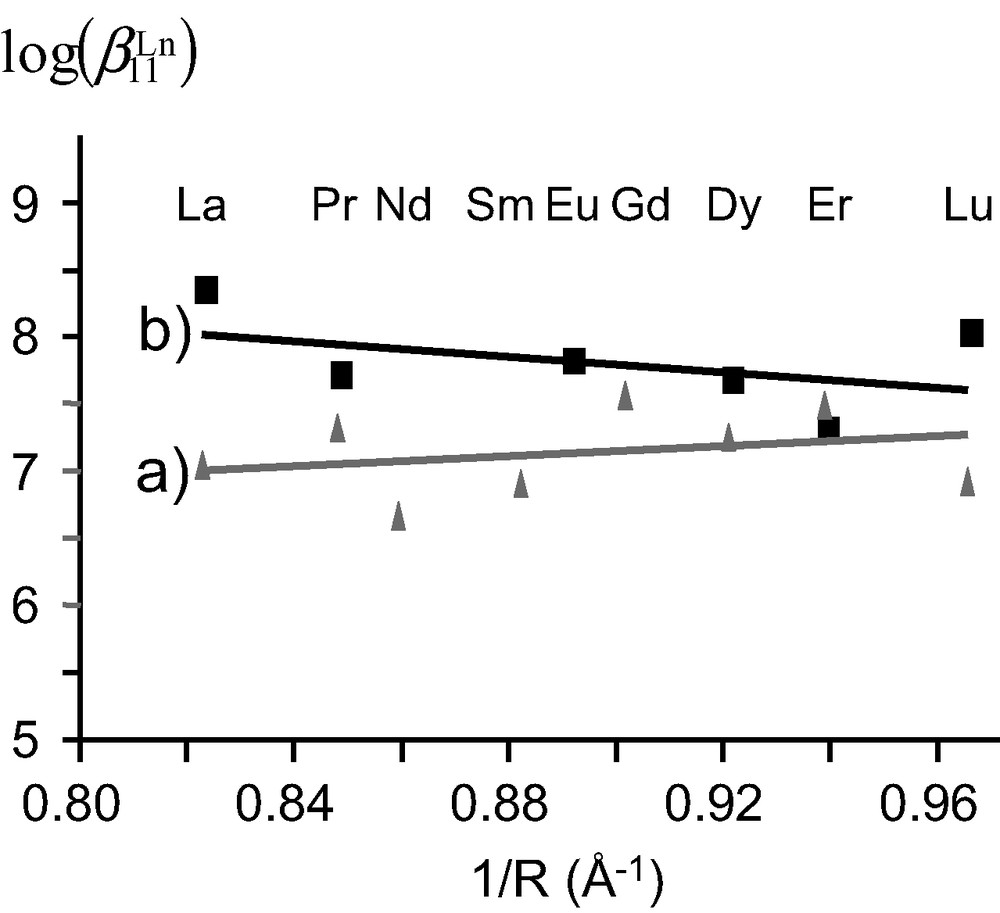
Formation constants log(ßLn11) for the complexes a) [Ln(L7)]3+ and b) [Ln(L8)]3+ in acetonitrile (10–2 mol·dm–3 [N(nBu)4]ClO4, 293 K) versus the inverse of nine-coordinate ionic radii (Å–1).
4 Crystal and molecular structure of the podates [Ln(Lx)](ClO4)3 (x = 7, Ln = La [19]; x = 8, Ln = Eu [20])
In the solid state, the complexes [La(L7)]3+ and [Eu(L8)]3+ display similar metallic environments in which the metal atom is nine-co-ordinate in a distorted tricapped trigonal prismatic site with the three oxygen atoms of the carboxamide groups and the three nitrogen atoms of the benzimidazole rings occupying the vertex of the prism, and the three nitrogen atoms of the pyridine rings capping the rectangular faces (Fig. 5). The striking difference between the crystal structure of [Eu(L8)]3+ and that of the analogous podate [La(L7)]3+ concerns the exo conformation of the apical carbon resulting from its methylation in [Eu(L8)]3+ [20].
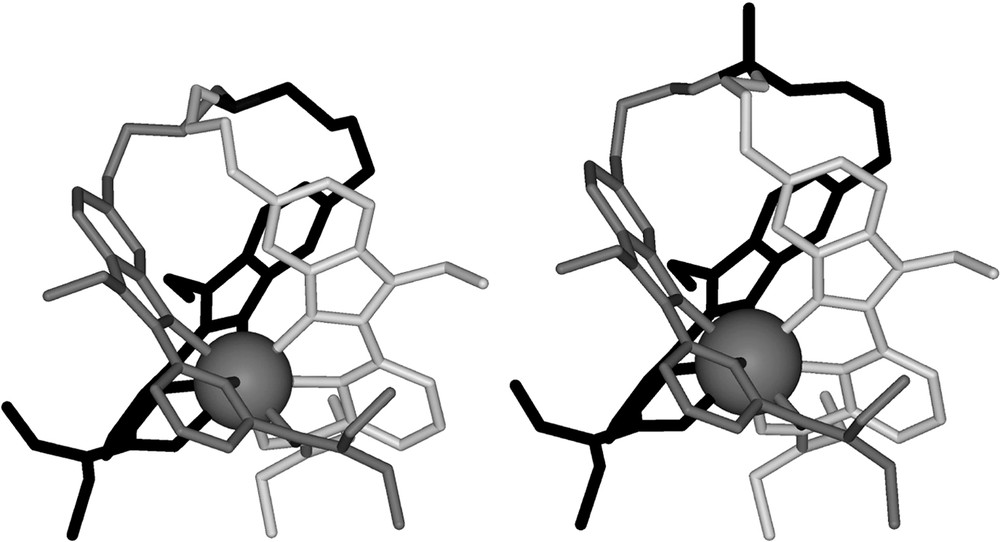
Perspective views of the crystal structure of a) [La(L7)]3+ (major conformer) and b) [Eu(L8)]3+ perpendicular to the C3 axis.
5 Conclusion
To the best of our knowledge, the methylated covalent tripod L8 produces the first unambiguous nine-coordinate podates [Ln(L8)]3+ in which the three helically wrapped aromatic tridentate binding units are connected to a single capping atom displaying no acid-base property. This covalent tripod can be considered as a valuable alternative to the delicate self-assembly processes with post-modification required for assembling identical nine-co-ordinate lanthanide sites in non-covalent podates, and in which the capping atom is an inert d-block ion (CrIII, CoIII) [12,21].
6 Supporting information
Detailed experimental procedures for the preparation of the tripod L9 and its precursors (including references for each known compounds), and relevant spectroscopic characterizations (1H and 13C-NMR spectra).
Acknowledgements
This work is supported through grants from the Swiss National Science Foundation.