

1 Introduction
The introduction of fluorenyl ligands in the coordination chemistry of early transition elements has opened up a rich domain of investigations, which has proven especially valuable for the polymerization catalysis field. In particular, group 4 metallocenes incorporating various fluorenyl-based ligands represent a unique class of complexes that afford astonishing catalytic activities in the polymerization of ethylene and α-olefins [1]. The fluorenyl moiety can impart exceptional syndio- or iso-specific control in propylene polymerization mediated by ansa-metallocenes [2], an ability that has initiated a strong interest into sandwich complexes bearing sterically expansive substituted fluorenyl ligands [3]. On the other hand, polymerization catalysis mediated by fluorenyl complexes of rare earth metals has been scarcely explored, although the first lanthanide-fluorenyl complexes have been reported by Beletskaya et al. in the early 1980s [4]. This is rather surprising, considering the great abilities of complexes of lanthanides and related group-3 elements for ethylene (α-olefin) polymerization [5].
This mini-review focuses on important structural aspects of metallocene and half-sandwich fluorenyl complexes of group-3 metals and lanthanides incorporating the [Cp-CMe2-Flu]2− (Cp = cyclopentadienyl; Flu = fluorenyl) and [Me2Si(3,6-tBu2C13H7)(NtBu)]2− ligands developed in our laboratory. It describes the main synthetic approaches that have been used for their preparation and the catalytic applications that have been derived from them, with particular emphasis on the polymerization catalysis field.
2 Ansa-lanthanidocenes bearing carbon-bridged fluorenyl-based ligands
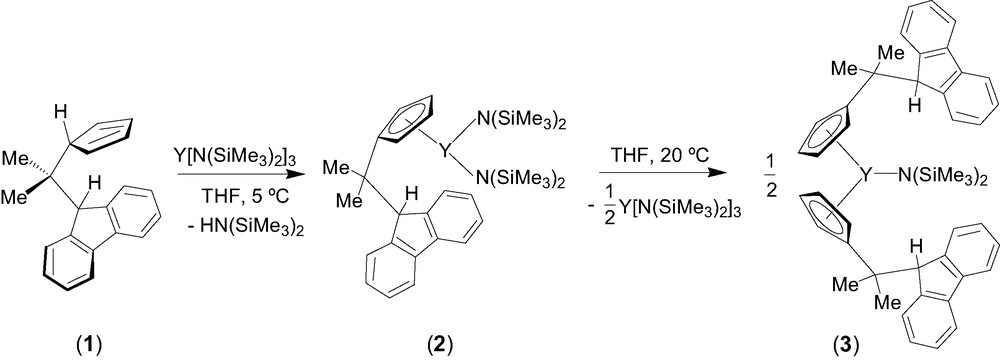
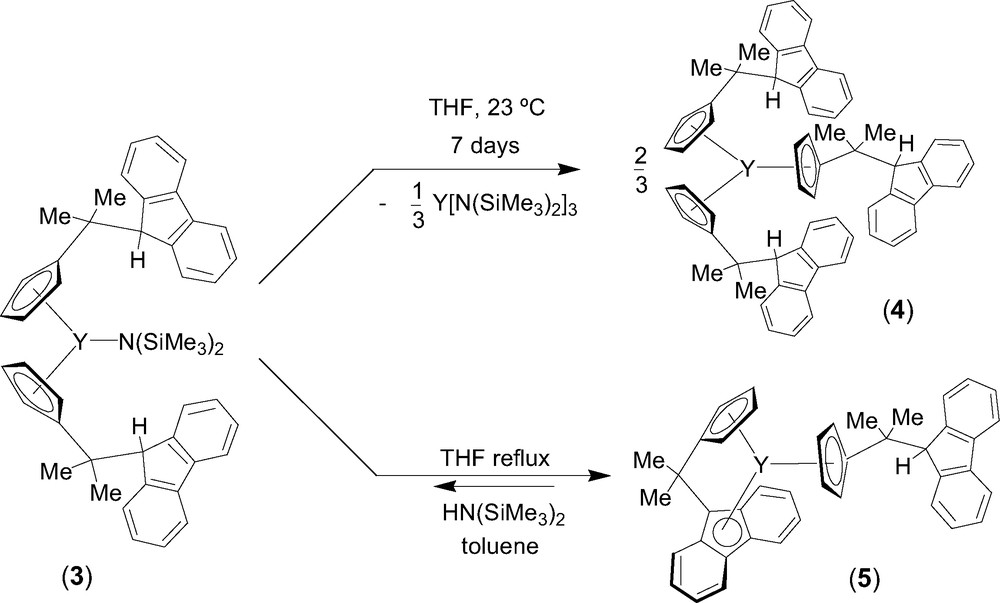
Entry toward fluorenyl-based lanthanidocene amides via amine elimination was investigated in details from the reaction of lanthanide amides Ln[N(SiMe3)2]3 (Ln = Y, La, Nd) with the isopropylidene-bridged ligand CpH–CMe2–FluH (1) [6]. The formation of various cyclopentadienyl derivatives that have free hanging fluorene moieties was observed, presumably because of the much lower acidity of the fluorenyl proton as compared to the cyclopentadienyl proton. Thus, the formation of the mono-substituted intermediate 2 was observed by NMR spectroscopy in the reaction between equimolar amounts of diprotio ligand 1 and Y[N(SiMe3)2]3 in THF at 5 °C (Scheme 1). Subsequent transformation of 2 into the bis(cyclopentadienyl) complex 3 takes place slowly under these conditions and can be accelerated by heating. Unlike the mono-substituted bis(amido) intermediate 2, complex 3 appeared to be kinetically more stable and could be isolated and characterized by NMR spectroscopy in solution and by an X-ray diffraction study in the solid state. In THF solution, 3 progressively converts into the tris(cyclopentadienyl) product 4 via further disproportionation (Scheme 2). However, upon refluxing in THF and removing of the concomitantly formed amine, complex 3 undergoes an intramolecular amine elimination reaction to give ansa-metallocene 5, which has a pendant fluorenyl arm. This process is reversible as demonstrated by the retroformation of the bis(cyclopentadienyl)amide 3 upon addition of excess of amine HN(SiMe3)2 to a toluene solution of 5 (Scheme 2). Similar reactivity trends were observed for lanthanum and neodymium amides.
Effective direct ansa-chelation of 1 onto lanthanides was achieved afterwards by using a salt metathesis route (Scheme 3) [7]. Salt metathesis reactions between LnCl3(THF)n (Ln = Y, La) and one equivalent of the dilithium salt of the isopropylidene-bridged ligand, [Flu-CMe2-Cp]Li2, in diethylether solution led to the isolation of new ionic metallocene complexes, [(Cp-CMe2-Flu)2Ln]–[Li(ether)n]+ (ether = Et2O, THF) (Ln = Y, n = 4, 6; Ln = La, n = 2, 7), which contain two chelating ligand units per metal center. The ionic complex 6 presumably originates from ligand redistribution in the primary formed heteroleptic ate-complex [(Cp-CMe2-Flu)YCl2]–[Li(ether)4]+, which exists in solution (vide infra), but may be unstable upon crystallization; ligand redistribution reactions are in fact quite common in lanthanide chemistry. Complex 6 was selectively prepared on using 2 equiv. of [Cp-CMe2-Flu]Li2 vs. YCl3(THF)3.5. The solid-state structures of 6 and 7 were established by X-ray diffraction studies. Interestingly, three polymorphic varieties of 6 were identified and shown to correspond all to a fully dissociated ion-pair with the formula [(η3,η5-Flu–CMe2–Cp)(η1,η5-Flu–CMe2–Cp)Y]–[Li(Et2O)(THF)3]+ (6). The fluorenyl ligands in 6 show an unprecedented η1- and a rare η3-bonding modes involving, respectively, a carbon atom of a phenyl ring, and the bridgehead carbon atom of the central ring and the two adjacent carbon atoms of one six-membered ring (Scheme 3). DFT computations carried out on the anionic fragment of 6 corroborated the nature of these bonding modes [4,7]. Only the exocyclic η3-bonding mode is observed for the fluorenyl moieties in complex 7, which features an associated ion-pair structure with the formula [(η3,η5-Flu–CMe2–Cp)2-La]–[Li(OEt2)2]+. The bonding modes of the fluorenyl ligands in this series of ionic complexes may thus appear dependent on the nature, i.e. ionic radius, of the metal center. Moreover, the flat energy surfaces revealed by DFT computations indicate easy switching between different coordination modes. Ionic complexes 6 and 7 constitute the first structurally characterized examples of bis(ansa)lanthanidocenes.
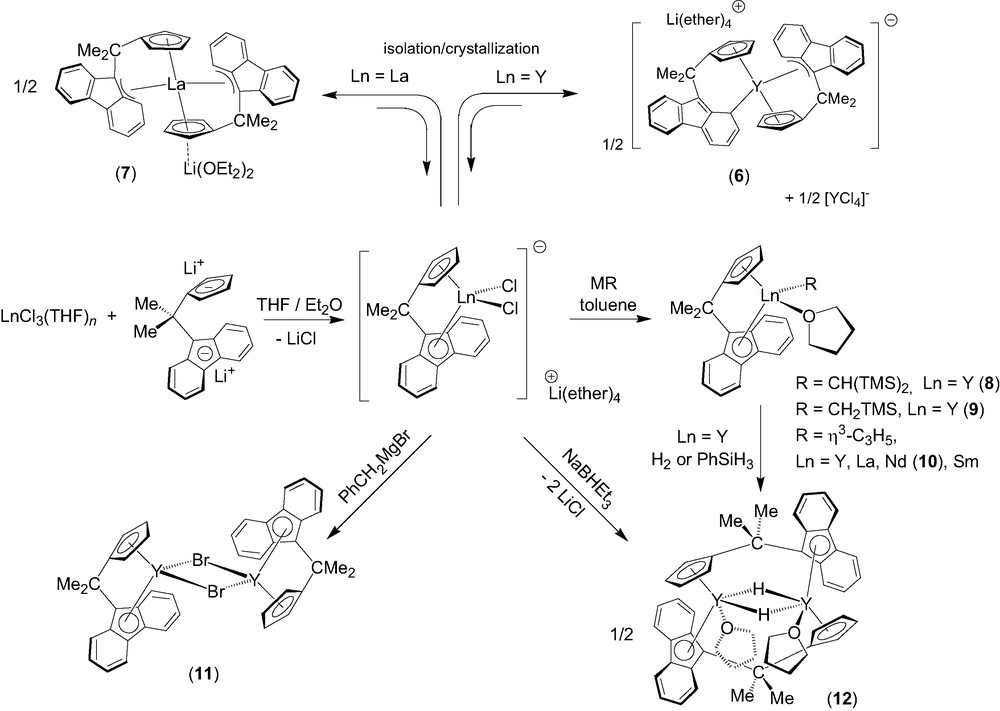
The dichloro ate-complex [(Cp–CMe2–Flu)YCl2]–[Li(ether)4]+ (prepared in solution by the aforementioned salt metathesis) was successfully applied to the preparation of neutral yttrocene alkyl derivatives [(Cp–CMe2–Flu)]Y(R)(THF) (R = CH(SiMe3)2, 8; CH2SiMe3, 9) in high yields (Scheme 3) [8]. These complexes hold both a coordinated THF molecule. The solid-state structure of 8 was established by X-ray diffraction, showing the fluorenyl moiety symmetrically coordinated to yttrium in an intermediary η3–η5 mode. Similarly, the reaction of the anionic complex [(Cp–CMe2–Flu)YCl2]–[Li(ether)4]+ (ether = Et2O, THF) with the Grignard reagent ClMgC3H5 (1 equiv. vs. Ln, as a THF solution) in toluene at 20 °C cleanly gives the neutral allyl complex [Flu–CMe2–Cp]Y(C3H5) (THF) as a yellow microcrystalline powder [9]. The parent La, Nd (10) and Sm complexes were prepared analogously (Scheme 2). Those allyl compounds were characterized by elemental analysis, NMR spectroscopy (for diamagnetic species) and a single crystal X-ray diffraction study for 10. The reaction of [(Cp–CMe2–Flu)YCl2]–[Li(ether)4]+ with PhCH2MgBr gives, instead of a benzyl derivative, the neutral base-free bromo complex {[(η5,η5-Cp–CMe2–Flu)]Y(μ-Br)}2 (11) [8], which shows a dimeric structure in the solid-state with chelating [Cp–CMe2–Flu]2− and bridging bromide ligands. Hydrogenolysis of complexes 8 and 9 with H2 or PhSiH3 gives the hydride {[(μ:η5,η5-Cp–CMe2–Flu)]Y(μ-H)(THF)}2 (12). Alternatively, 12 can be prepared in a straightforward procedure via reduction of the dichloro ate-complex [(Cp–CMe2–Flu)YCl2]–[Li(ether)4]+ with 1 equiv. of NaBHEt3. The solid-state structure of complex 12 was determined by X-ray diffraction, revealing a dimeric structure with both bridging Cp–CMe2–Flu and hydride ligands (Y−H = 1.99(4)−2.01(4) Å). Complex 12 constitutes the first structurally characterized example of a group-3 metal/lanthanide hydride stabilized by a fluorenyl ligand.
3 Constrained-geometry fluorenyl-based systems
The σ-bond metathesis approach was intensively investigated as a convenient entry toward half-sandwich lanthanide complexes incorporating constrained geometry fluorenyl-based ligands [10]. Quantitative coordination of the diprotio ligand Me2Si(3,6-tBu2C13H7)(NHtBu) (13) onto yttrium to form the half-sandwich complex 14 was readily achieved (Scheme 4). The crystal structure of 14 revealed that, in the solid state, the fluorenyl moiety is bonded onto yttrium in an unusual exocyclic η3-fashion. The presence of two coordinated THF molecules in 14, as compared to ‘constrained geometry’ cyclopentadienyl-based yttrium analogues that contain a single THF ligand [11], constitutes a specific feature of this 12-electron complex. It is difficult to ascertain whether the coordination of a second THF molecule in 14 results from the reduced hapticity of the fluorenyl ligand or vice-versa. Interestingly, this reduced η3 hapticity of the fluorenyl unit seems to be preserved in solution, as judged by variable temperature NMR spectroscopy. One of the two coordinated THF molecules is labile on the NMR time scale and involved into a dissociative process between the mono- and di-solvated forms of complex 14.
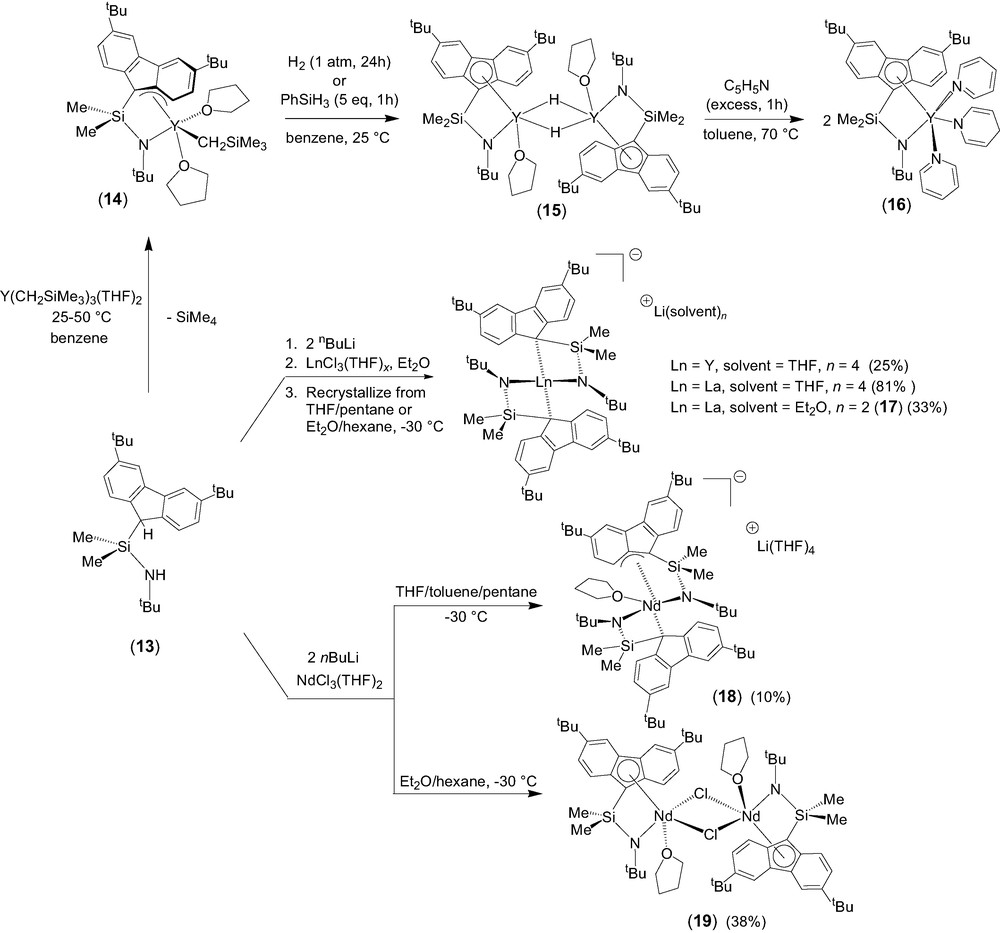
Generation of the related hydrido derivative from the yttrium carbyl 14 was achieved by reaction with H2 or PhSiH3 (Scheme 4). The resulting yellow product (15) is insoluble in common organic solvents (aromatic hydrocarbons, chlorinated solvents, THF), despite the presence of two tert-butyl substituents. The reaction of this putative hydride with an excess of pyridine at 70 °C resulted in the selective formation of the 1,4-addition product 16, the identity of which was established by NMR spectroscopy and X-ray analysis [12]. Complex 16 displays a pseudo-five-coordinate geometry very similar to its alkyl parent 14, with the THF molecules and the alkyl group in 14 being replaced by pyridine molecules and a dihydropyridinyl group in 16, respectively. However, in contrast with the unusual non-symmetric, exocyclic η3 coordination mode of the fluorenyl moiety observed in the solid-state structure of 14, that of 16 features a symmetric η5 coordination, resulting in a higher electron-count.
Salt elimination reactions between the dilithium salt [Me2Si(3,6-tBu2C13H6)(NtBu)]Li2 and lanthanide trichloride precursors were explored and resulted in most cases in the formation of the anionic bis(ligand) disproportionation products of general formula [{Me2Si(3,6-tBu2C13H6)(NtBu)}2Ln]−[Li(ether)n]+ (Ln = Y, ether = THF, n = 4; Ln = La, ether = Et2O, n = 2; 17) [10]. These diamagnetic complexes were isolated in good yields and authenticated by NMR spectroscopy and an X-ray diffraction study for 17. When the same salt metathesis approach was applied to neodymium chemistry, the disproportionation product [η3,η1-{(3,6-tBu2C13H6)SiMe2NtBu}2Nd(THF)]–[Li(THF)4]+ (18) was isolated in poor yield (Scheme 4). Crystallization of the crude product from Et2O/hexane solution led to the isolation of the neutral chloro complex [η5,η1-{(3,6-tBu2C13H6)SiMe2NtBu}Nd(μ-Cl)(THF)]2 (19), which is the major product formed in this process. The identity of both complexes was unambiguously established by single crystal X-ray diffraction studies.
Reviewing the structural features of metallocene complexes of group-3 metals and lanthanides, in the light of the peculiar examples featured in this review, five limiting coordination modes of fluorenyl ligands can be unequivocally distinguished (Scheme 5). The bonding in A is reminiscent of that of ‘classic’ metallocene architectures. Within the framework of a covalent bonding scheme, C13H9− behaves in A as a six-electron donor. In B and D, C13H9− is a four-electron donor. It is a two-electron donor in C, while the situation in E cannot be discriminated between C13H9− being a two- or a four-electron ligand. In contrast to group 4 metals, the fluorenyl ligand offers a surprisingly large variety of coordination modes in group-3 metal/lanthanide complexes. Type A (η5) and type B (η3) are almost the only ones for group 4 metals and, of course, also by far the most common ones for group-3 metal and lanthanide complexes. The reasons for this preference are essentially electronic in origin. Also, the energy profile of the pathway connecting types A and B is particularly flat, so that the definitive choice between these modes might be made by small perturbations such as steric interactions or crystal packing forces (in the solid state), and solvent and temperature (in solution), for instance. Reasons for more exotic coordination modes, in particular types D and E, remain unclear today. We assume that steric constraints may drive metal slippage towards the adjacent phenyl rings of the fluorenyl ligands; however, electronic factors can not be ruled out. Detailed computations are currently underway on model systems to investigate the influence of these parameters and explore possible pathways connecting coordination modes A–E.
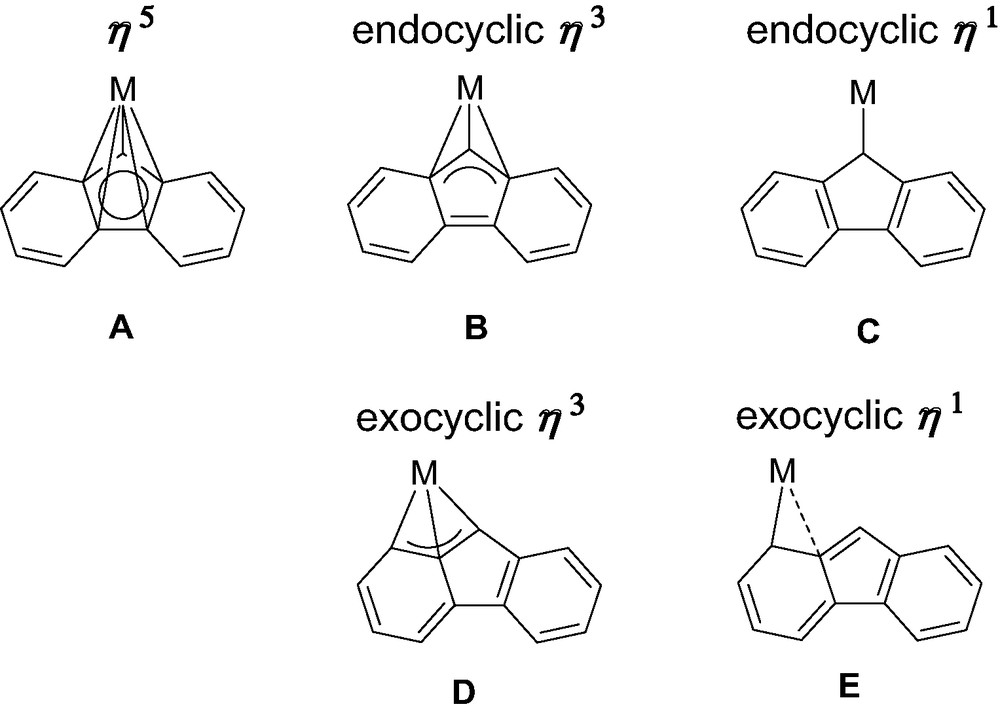
Coordination modes of fluorenyl ligands observed with group 3 metals and lanthanides.
4 Applications in catalysis
When we initiated this project, catalytic applications of group-3 metal/lanthanide fluorenyl complexes were poorly surveyed and limited to a few examples of methyl methacrylate (MMA) polymerization reactions [4]. This was in striking contrast with the extremely rich and still flourishing group 4 metallocene polymerization chemistry and fine chemicals catalysis.
The yttrium hydrocarbyls 8 and 9 derived from the [Cp–CMe2–Flu]2− ligand were found to be quite active in the polymerization of MMA under mild conditions (−15 to 20 °C), yielding slightly syndiotactic-enriched PMMA (rr 60–66%) with high molecular weight (Mn = 271,000–376,000) and moderate molecular weight distributions (Mw/Mn = 1.43–1.87) [8]. Contrastingly, the ‘constrained geometry’ yttrium alkyl 14 and ‘hydride’ 15 complexes were found almost inactive towards MMA, whereas the anionic complex of lanthanum 17 smoothly reacts with MMA in the temperature range 20–50 °C to give atactic PMMA with high molecular weight (Mw = 216 000–250 000) and rather large dispersity (Mw/Mn = 2.8–3.6) [10]; the latter data reflect poor initiation efficiency and overall poor control of the polymerization.
Much more interestingly, upon exploring the catalytic activity of the neutral allyl complexes [(Cp–CMe2–Flu)Ln(allyl)(THF)], highly syndiospecific polymerization of styrene was achieved [9]. Polymerization of bulk styrene led to high monomer conversions (70–85%) under smooth conditions (60 °C), while introduction of toluene in polymerization media was found detrimental for catalyst activity. The polymerization activity of the allyl complexes was in the order Nd (10) > Sm > La > Y, featuring a remarkable activity of 250–1710 kg PS mol−1 h−1 for complex 10. The molecular weights were in the range Mn = 10 000–135 000, with narrow to usual molecular weight distributions Mw/Mn of 1.25–2.10. Nearly perfect syndiotacticity of polystyrene was illustrated by the rrrr pentad abundance > 99%, a value which had been achieved solely with complexes of Ti and V so far [13]. A very relevant example from Hou and co-workers [14] appeared at the same time as our contribution, also dealing with highly syndiotactic polymerization of styrene (rrrr > 99%) initiated by the two-component cationic system (Me3SiCp)Ln(CH2SiMe3)2(THF)/[Ph3C][B(C6F5)4] (Ln = Sc, Y, Gd, Lu).
The intramolecular hydroamination/cyclization of α,ω-aminoalkenes catalyzed by the constrained-geometry alkyl complex 14 was briefly investigated (Scheme 6) [15]. High selectivity but moderate activity was observed for this process, in contrast to highly active cyclopentadienyl-based lanthanidocenes, most likely due to competitive coordination between molecules of THF in 14 and the amino substrate.
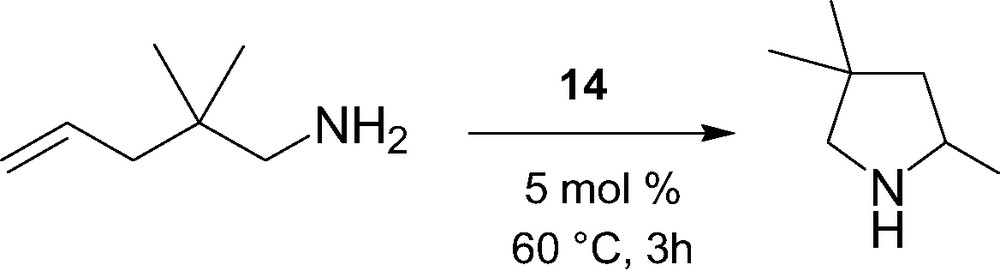
5 Conclusions
The large variety of coordination modes of fluorenyl complexes of group-3 metals and lanthanides provides organometallic and catalysis chemists with many opportunities to control the coordination sphere and saturation of the metal center, and the overall complex geometry. Many efforts are still necessary to understand the factors that govern bonding of fluorenyl and parent ligands onto lanthanides. This shall open up rational design of new ligand architectures to obtain in turn complexes with original structural and reactivity features, making them interesting candidates for a variety of catalytic applications.