

1 Introduction
In 1985, we reported [1] that K[trans-Co(NH3)2(NO2)4] (VI) crystallizes as a conglomerate1 (space group P212121), whose source of chirality is the axial symmetry inherent to the propeller arrangement of the four –NO2 ligands located in the basal plane. This issue was elaborated in more detail in a subsequent publication [2]. Since the earliest study of (VI) was by X-ray diffraction [1], the structure was re-determined by neutron diffraction in order to establish more precisely the positions of the –NH3 hydrogens [3], the results of both studies being essentially identical.
Somewhat later, we found [4] that the salt of composition [cis-Co(en)2(NO2)2][trans-Co(NH3)2(NO2)4] (V) also crystallizes as a conglomerate in space group P21. An attempt to produce useful crystals of [cis-α-Co(trien)(NO2)2][trans-Co(NH3)2(NO2)4] failed [4]; this result has recently been verified by us and other methods of crystal growth (diffusion, etc.) are currently being tried. Interestingly, the isomeric salt [cis-β-Co(trien)(NO2)2][trans-Co(NH3)2(NO2)4]·H2O (VII) readily crystallizes as a racemate in space group P21/c [5]. These observations suggest that the crystallization mode of salts of the anion [trans-Co(NH3)2(NO2)4] may be driven by the nature of the cation–anion interionic bonds which are exclusively hydrogen bonds. Such a suggestion has been formulated and discussed before [2–10]. As a result of that hypothesis, we decided to explore the crystallization behavior of additional Co(III) amine cations with the [trans-Co(NH3)2(NO2)4] anion, in order to focus on the effect of a single variable. The results of these explorations are detailed below.
2 Experimental
2.1 General considerations
[Co(tren)(NO2)2][[trans-Co(NH3)2(NO2)4] and K[trans-Co(NH3)2(NO2)4] were prepared according to procedures given before (Refs. [6] and [1], respectively). Those of [(tren)Co(NO2)2]Cl [11], [(tren)Co(ox)]Cl [12], [(en)2Co(ox)]Cl [12] and [trans-(pn)2Co(NO2)2]Cl [13] were prepared according to their respective literature citations [11–13].
2.2 Syntheses of crystal growth and X-ray diffraction studies of compounds (I) through (IV)
2.2.1 Syntheses of (I) through (IV)
Deionized water solutions of the cations present in compounds (I) through (IV) were prepared from stock material obtained in the procedures described in Refs. [11–13]. They were treated with equimolar solutions of compound (VI) and allowed to evaporate till crystals suitable for X-ray diffraction were obtained. No elemental analyses were made since the starting materials had been prepared and analyzed previously [1,6,11–13].
2.2.2 X-ray diffraction studies
Room temperature (295 K) data were obtained with a Nonius Diffractis 585 instrument using Mo Kα radiation and absorption correction data measured by the Psi-scan technique. A somewhat redundant data set was collected for each of the four substances. Details of data collection and processing of the 295 K data are not presented here; however, they are available in the CCDC compilations (for CIF data on these structures, see Section 5, below for deposition numbers and other relevant information). Data collection at 120 K was carried out with a Nonius CCD instrument using Mo Kα radiation. Half-a-hemisphere of data was collected in each case. Data were corrected with SADABS [14]. In all the cases, the structures were solved, refined and illustrated with the WinGx [15] package of programs. Packing diagrams were generated with DIAMOND [16]. Detailed information concerning molecular parameters can be found in the CIF documents quoted in section `Supporting information available'.
3 Discussion
3.1 Cations and anions
Figs. 1–4 depict the cation–anion pairs present in compounds (I) through (IV). Fig. 1 shows a rather unique array of cations and anions constituting the asymmetric unit present in (I). The array consists of four cations in general positions, three anions in general positions and two additional anions present at inversion centers, or a total of nine ions in the asymmetric unit. This rather unusual arrangement produces a total of 36 cobalt ions in the unit cell of the centrosymmetric space group P21/n.
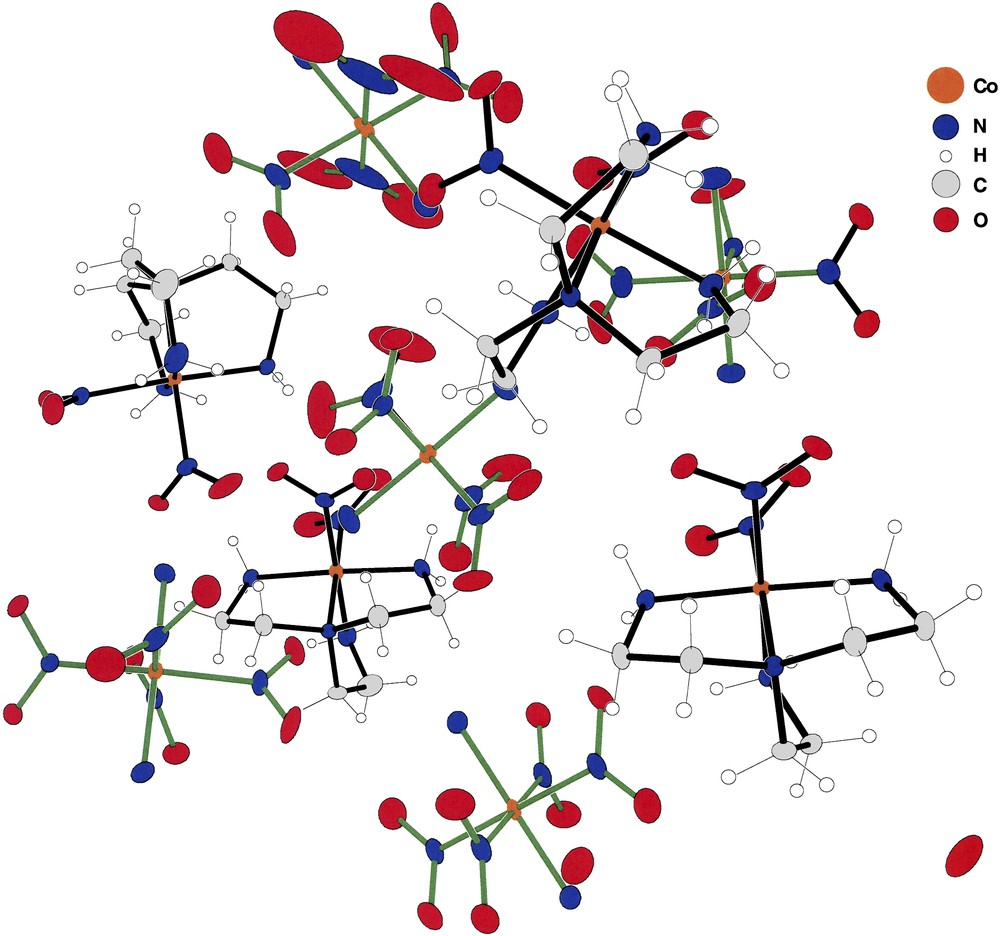
The asymmetric unit of (I). The green bonded species are the [trans-(NH3)2Co(NO2)4] anions while those with blue bonds are the [(tren)Co(NO2)2] cations. Note that the asymmetric unit shown here contains four cations and five anions. This is due to the fact that two of the anions (Co8 and Co9) lie at inversion centers of the space group. Thus, there are two half anions and no violation of charge compensation. For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article. Masquer
The asymmetric unit of (I). The green bonded species are the [trans-(NH3)2Co(NO2)4] anions while those with blue bonds are the [(tren)Co(NO2)2] cations. Note that the asymmetric unit shown here contains four cations and five anions. This is due to ... Lire la suite
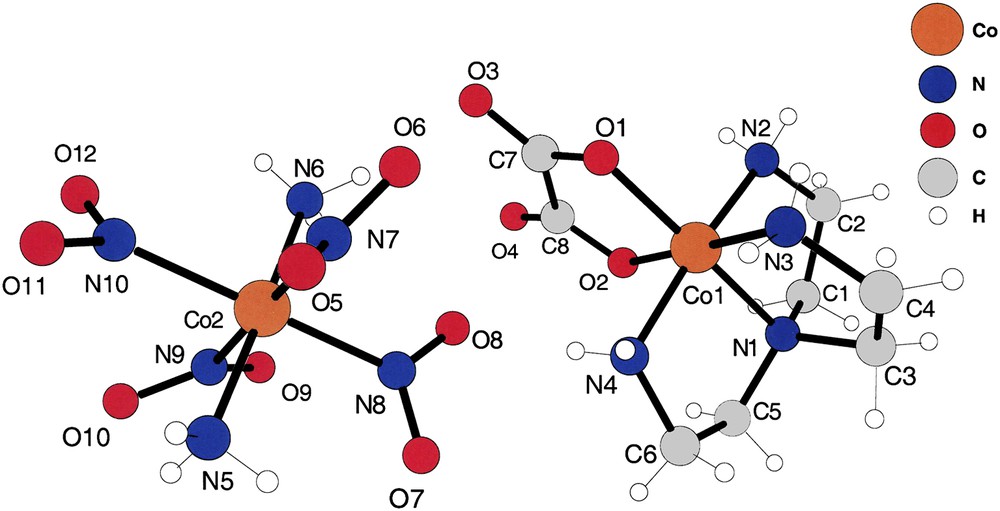
The asymmetric unit of compound (II). Note that the refinement located accurately the positions of the –NH3 hydrogens of the [trans-(NH3)2Co(NO2)4] anions.
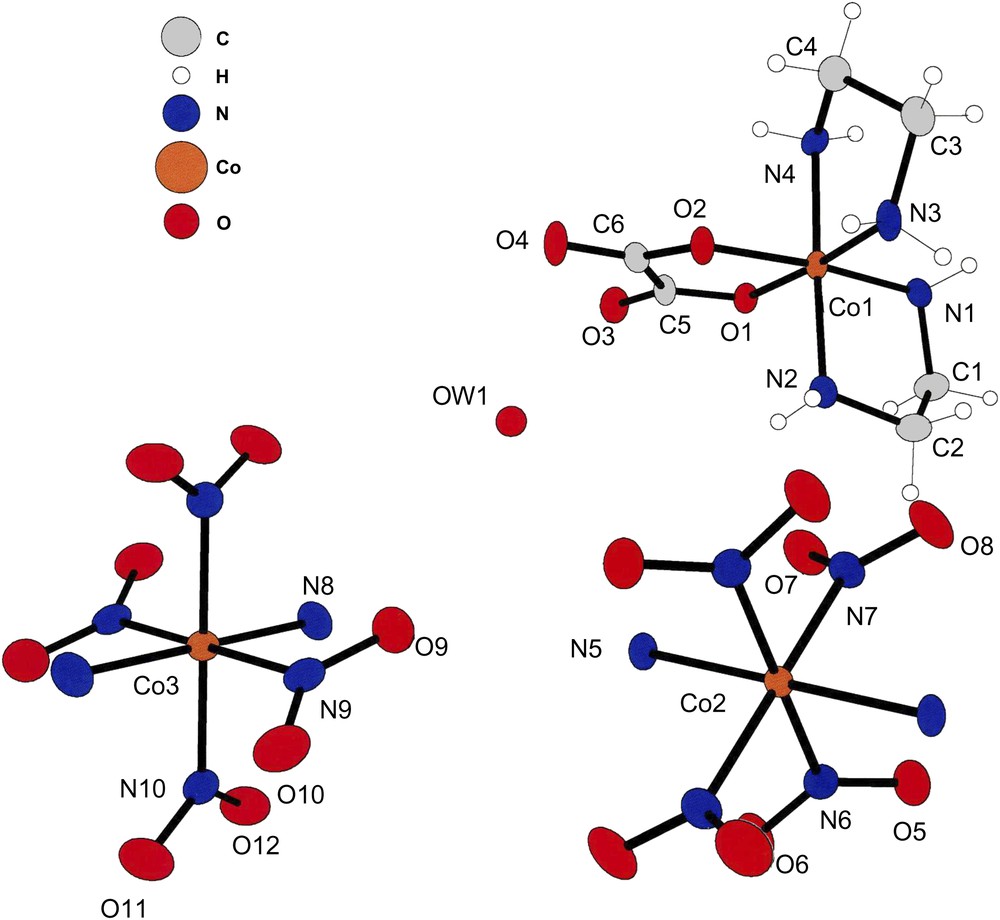
The asymmetric unit of compound (III). The [trans-(NH3)2Co(NO2)4] anions are also at inversion center in this lattice; that is the reason for having labels on only half of the atoms of the anion ligands.
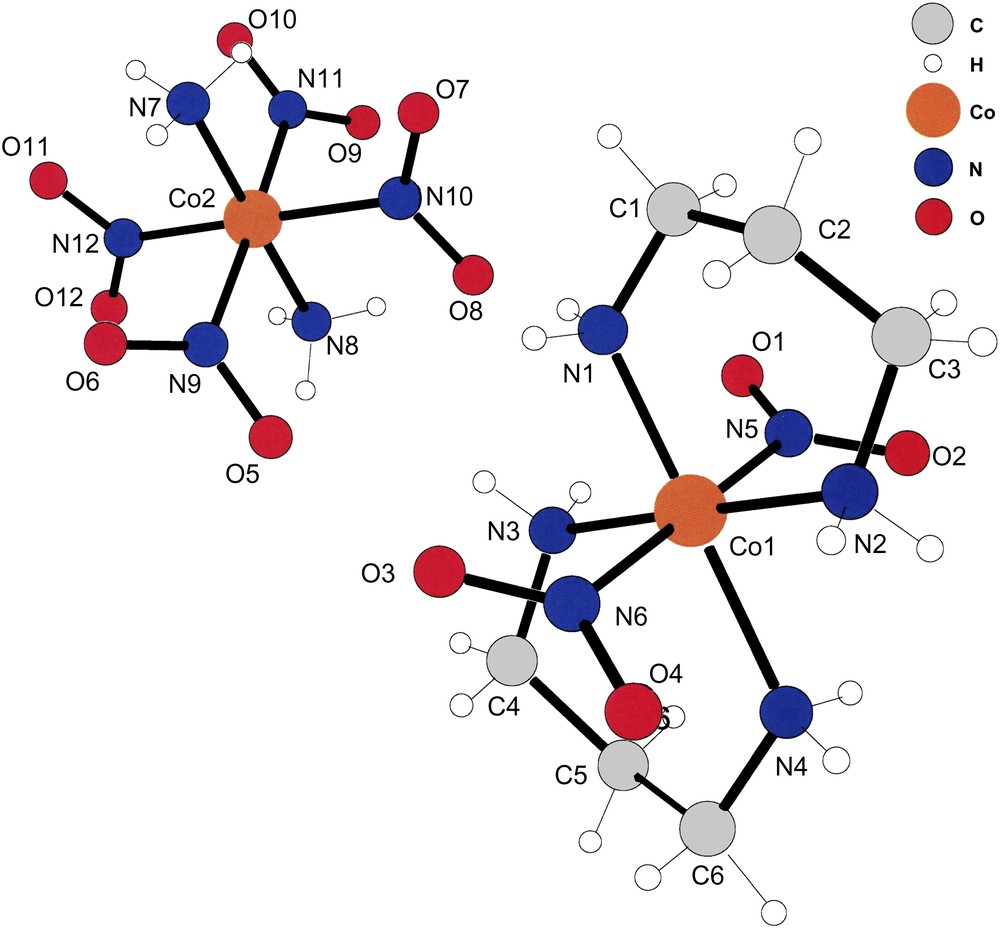
The asymmetric unit of compound (IV). Note that the refinement located accurately the positions of the –NH3 hydrogens of the [trans-(NH3)2Co(NO2)4] anions in this case also. The cations and anions lie at inversion centers; thus, only the asymmetric unit is shown.
A search of the CSD2 for compounds crystallizing in P21/c (and equivalents) and having z = 36, produced only one hit, ZZZKEY [17], for which no coordinates are available. It is the [(lithium butoxide)-(t-butyl lithium)] complex. Therefore, the only documented example of a monoclinic crystal with 36 molecules in the unit cell is compound (I).
The cation and anion of compound (II) are shown in Fig. 2. They are obviously hydrogen-bonded by oxalato oxygens of the cation to the end –NH3 hydrogens of the anion and from –NO2 oxygens of the anion to –NH2 hydrogens of the cation. That pair of ions is the asymmetric unit of this substance, crystallizing in the centrosymmetric space group I2/a. Note that the –NH2 and –NH3 hydrogens were found experimentally at chemically reasonable positions, allowing us to calculate intermolecular hydrogen bonds of those ligands.
Compound (III) crystallizes in the centrosymmetric space group P21/n with a cation at a general position and two anions at special positions. Such an arrangement is shown in Fig. 3.
Fig. 4 displays a cation–anion pair in positions leading to the shortest hydrogen bonds between the two ions present in (IV). It is obvious that the oxygens of N9 and N10 form hydrogen bonds with the amine hydrogens of N1 and N3. Although not depicted in Fig. 1, the hydrogens of the –NH3 ligands of the anions also form inter-molecular hydrogen bonds with the oxygens of the cationic –NO2 ligands.
3.2 Crystallization pathway selected and crystal architecture
Previously, we observed that whereas K[trans-Co(NH3)2(NO2)4] (VI) [1] and [cis-Co(en)2(NO2)2][trans-Co(NH3)2(NO2)4] (V) [4] crystallize as conglomerates, [cis-β-Co(trien)(NO2)2][trans-Co(NH3)2(NO2)4]·H2O (VII) [5] crystallizes as a racemate. In those days, we did not have the graphics facilities we have now; consequently, we have remedied this by depicting the packing diagrams of (V), (VI) and (VII) in Figs. 5–7, drawn with DIAMOND [16]. The addition of these three packing diagrams will illustrate the packing modes of all the compounds of the type C[trans-Co(NH3)2(NO2)4] (with C = a cation).
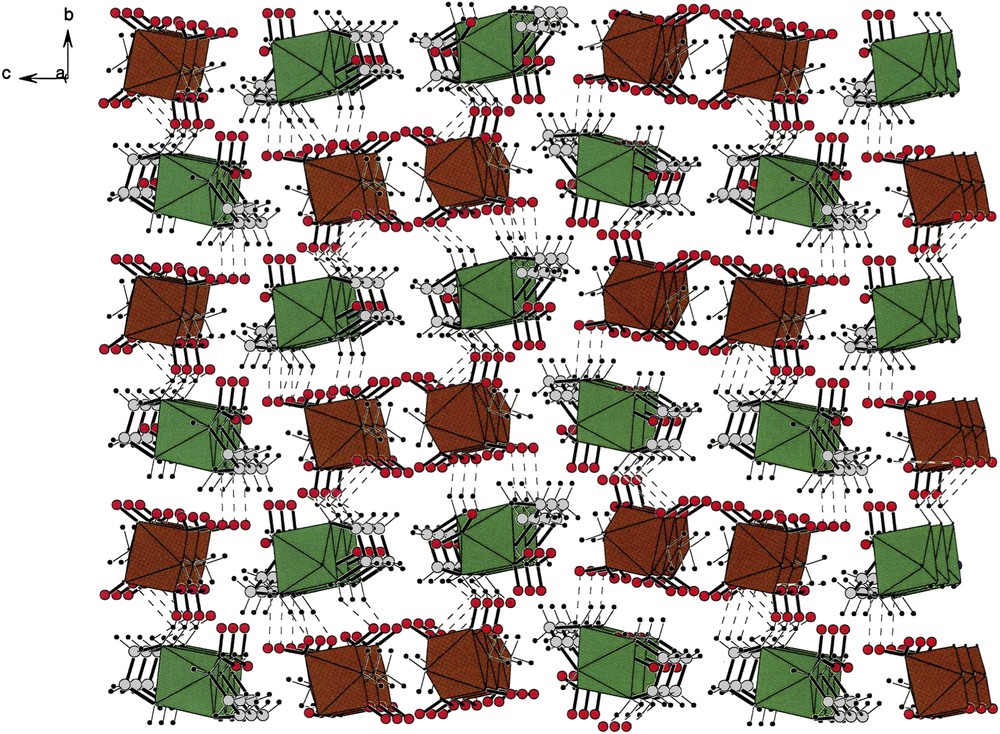
The unit cell of the [cis-(en)2Co(NO2)2][trans-(NH3)2Co(NO2)4] (V). This unit cell was plotted in order to make a uniform comparison of the unit cells of all the seven compounds containing [trans-(NH3)2Co(NO2)4] anions. The quality of the graphics available before did not permit this comparison – a neglect that we remedy now. The same comment applies for the packing diagrams of compounds (VI) and (VII). See below. The octahedral polyhedron in green is that of the cations, while those of the anions are in brown. Note the helical chains formed by cations as well as anions and that they interpenetrate one another. This is an a-projection. There are strings running along the b-axis composed of ….cation–anion–cation–anion…. While the strings observed along the c-direction are made up of ….cation–cation–anion–anion…. For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article. Masquer
The unit cell of the [cis-(en)2Co(NO2)2][trans-(NH3)2Co(NO2)4] (V). This unit cell was plotted in order to make a uniform comparison of the unit cells of all the seven compounds containing [trans-(NH3)2Co(NO2)4] anions. The quality of the graphics available ... Lire la suite
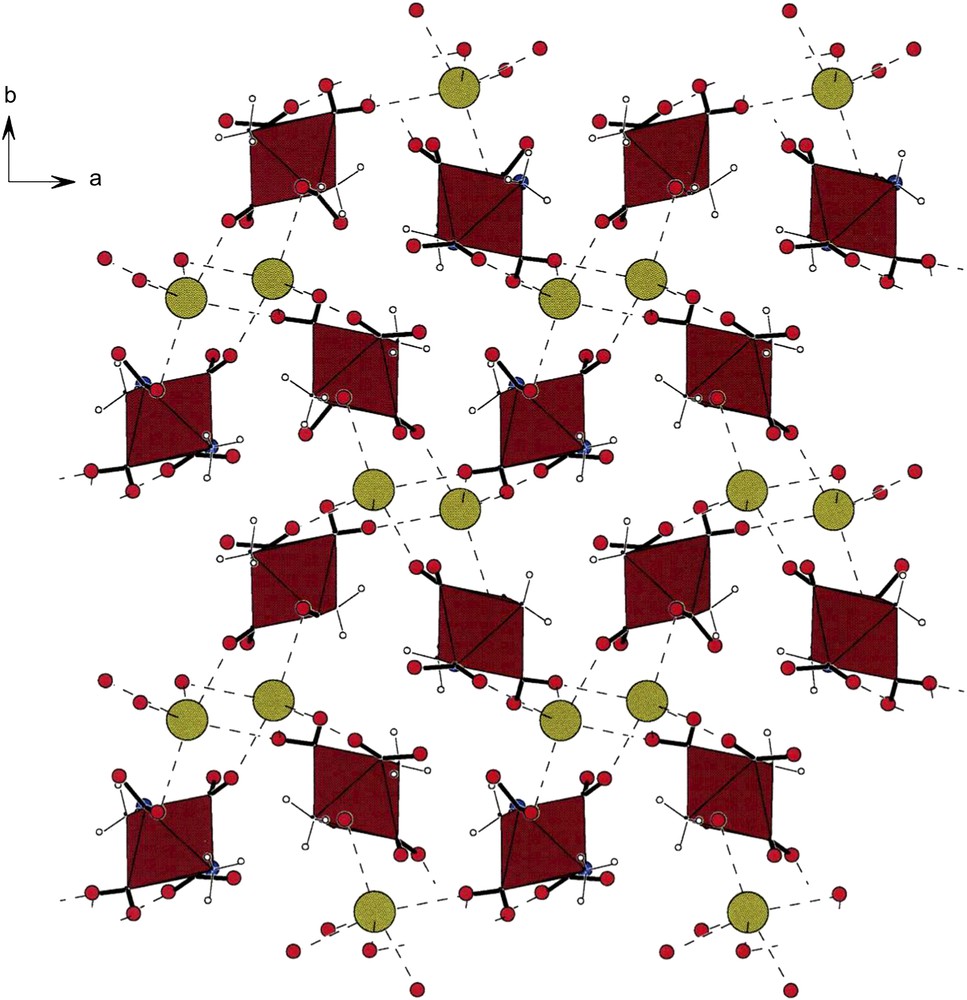
A c-projection of the asymmetric unit of compound K[trans-(NH3)2Co(NO2)4] (VI). Note the helical arrays of anions running along the a- and b-direction. The potassium cations are located in holes formed by groups of four anions. The dotted lines are the bonds formed by the oxygens of –NO2 ligands and the potassium cations. For comments on the lengths of these contacts see the text.
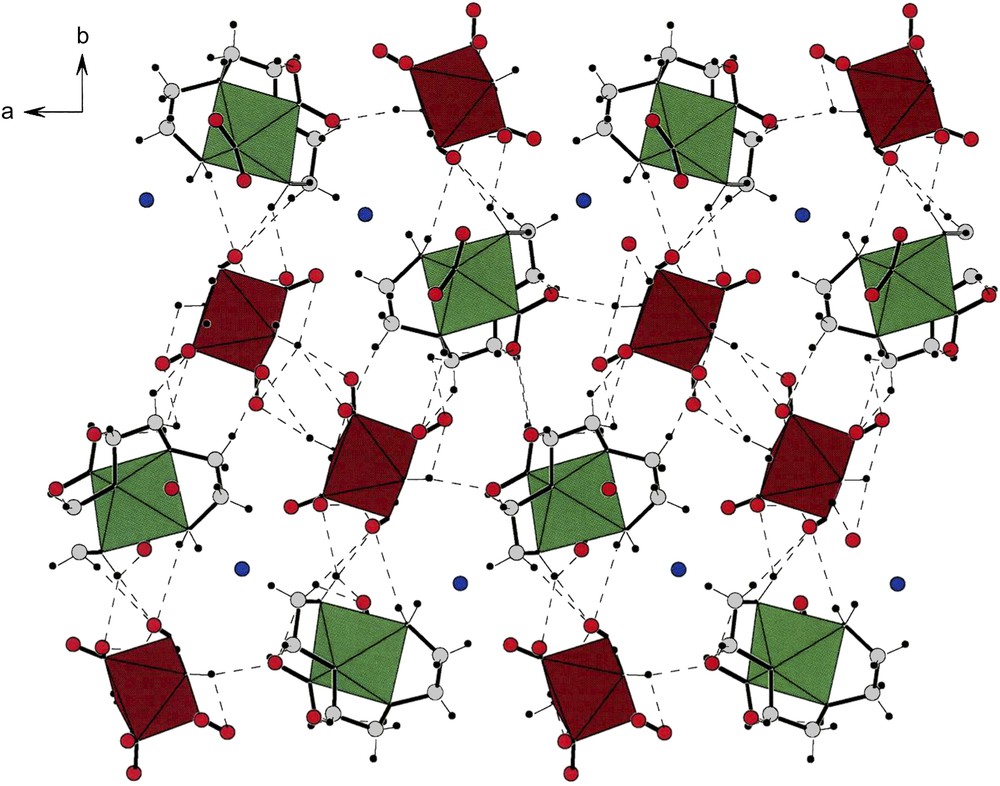
An a-projection of the unit cell present in the centrosymmetric compound [cis-b-(trien)2Co(NO2)2][trans-(NH3)2Co(NO2)4] (V). Note the inversion center at the center of the projection. All pairs of adjacent cations and anions are related by an inversion center. Therefore the helical arrays of cations and anions, easily noted along the b- and c-directions, are made up of internally racemic strings of the type …R–S–R–S–R–S–R–S… Such an array differs from other ones we have observed in other centrosymmetric packings in which there are strings of homochiral molecules or ions; but adjacent strings of such entities are of opposite chirality. That is, they are not internally compensated as is the case in this compound. Masquer
An a-projection of the unit cell present in the centrosymmetric compound [cis-b-(trien)2Co(NO2)2][trans-(NH3)2Co(NO2)4] (V). Note the inversion center at the center of the projection. All pairs of adjacent cations and anions are related by an inversion center. ... Lire la suite
Fig. 5 shows the packing of the cations and anions present in [cis-Co(en)2(NO2)2][trans-Co(NH3)2(NO2)4] (V) [4] where it is evident that cations and anions form helical strings running along the b-axis. The cationic and anionic helical strings interpenetrate one another such that strings can also be viewed as alternations of cations and anions that are hydrogen bonded to one another. A list of such hydrogen bonds can be found in the original report [4].
Fig. 6 shows the packing in crystals of K[trans-Co(NH3)2(NO2)4] (VI) [1]. As in the case of [cis-Co(en)2(NO2)2][trans-Co(NH3)2(NO2)4] (V) [4], the anions also form helical arrays readily noted along the a- and b-direction of the lattice. The homochiral strings consist of anionic moieties and are stitched together by K⋯O bonds, as is evident in this figure. Each potassium forms six such oxygen bonds with distances under 3.000 Å and three longer ones ranging from 3.016(4) Å to 3.153(4) Å. If all nine contacts are accepted as bonded interactions, each potassium makes contacts with seven of the eight oxygens of the anions [1].
Fig. 7 displays the architecture of the crystalline lattice of [cis-β-Co(trien)(NO2)2][trans-Co(NH3)2(NO2)4]·H2O [5] (VII). Along the b-axis one can see the helical strings of cations and anions. However, unlike (V) and (VI), compound (VII) crystallizes as a racemate. This is clearly illustrated by the fact that there is an inversion center in the middle of the cell. Therefore, the cell is divided into two enantiomeric halves containing cations and anions of opposite chirality.
Fig. 8 shows the packing of compound (I). Along the b-axis, one can readily see green cationic strings as well as brown anionic strings that wind about the two-fold screw axis. In the center of the cell is an inversion center, as was the case of the packing found in (VII). However, the similarities end there since the lattice of compound (I) is far more complex than that found in compound (VII). In particular, note the complex channels along the c-direction that enclose columns of anions.

A c-projection of the cell describing the packing of compound [(tren)Co(NO2)2][trans-(NH3)2Co(NO2)4] (I). Cations are green and anions brown as in all packings shown in this report. Note that both, cations and anions form cavities containing their counter-ions. The lattice has an inversion center at the middle of the ab-plane. For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.
Fig. 9 displays the packing found in compound (II). Along the a-direction, there are alternating strings of cations (green) and anions (brown) and at the center of the cell is an inversion center. A pair of anions is very obviously related by an inversion operation; therefore, the members of the anionic strings alternate in chirality. Finally, the two cationic (green) strings on the left are enantiomorphically related to the two strings on the right of the figure.
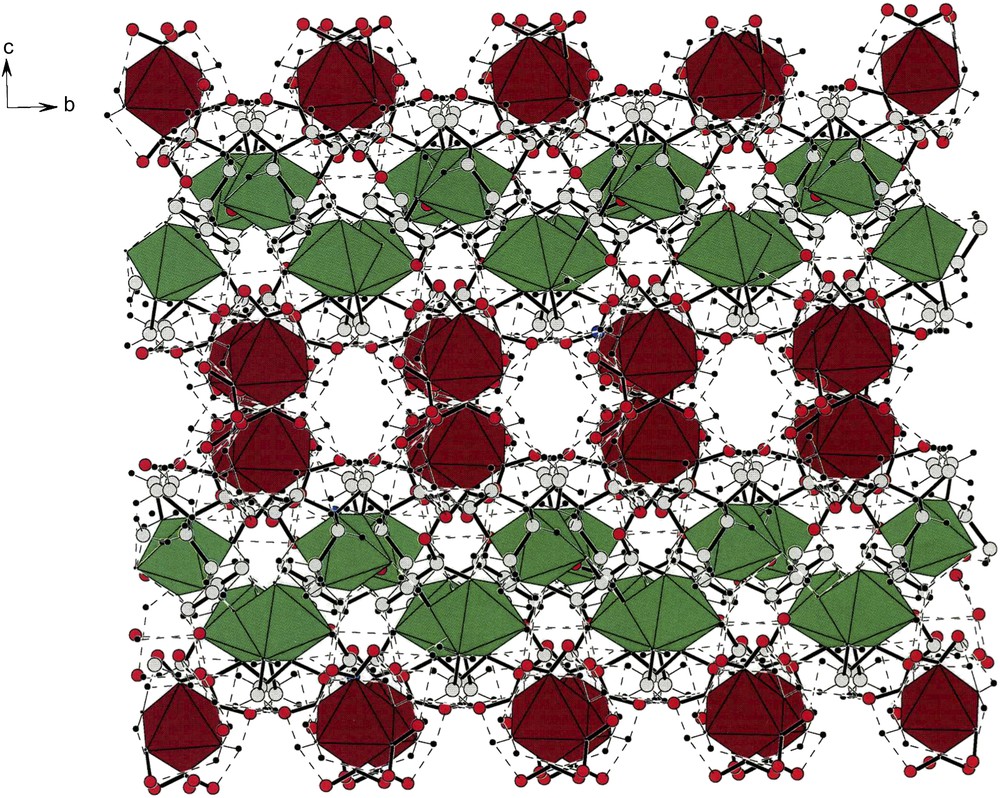
An a-projection of the unit cell contents of compound [(tren)Co(ox)][trans-(NH3)2Co(NO2)4] (II). Note the inversion center that creates two halves. The brown strings (anions) in the middle of the cell clearly show the division. The contents of the right side are enantiomorphically related to those of the left. For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.
Fig. 10 shows brown strings of anions and green strings of cations that pack in alternating fashion, held together by hydrogen bonds. Given the space group (P21/n) of compound (III), there is an inversion center which relates, enantiomorphically, the upper and the lower halves of the figure.
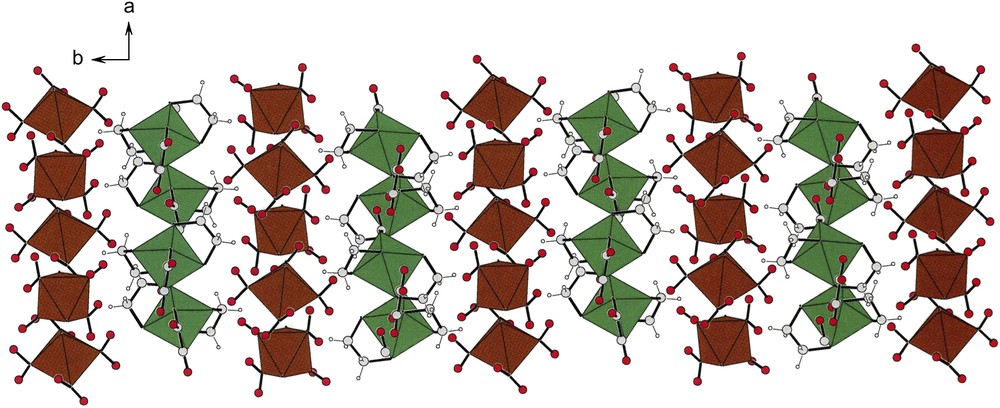
Alternating rows of cations (green) and anions (brown) in the unit cell of compound [(en)2Co(ox)][trans-(NH3)2Co(NO2)4] (III) in c-projection. Note that while rows of cations and anions alternate, there seem to be many more anions than cations. In fact there are twice as many due to the fact that all of the anions sit at inversion centers of the lattice. See the asymmetric unit in Fig. 3. For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article. Masquer
Alternating rows of cations (green) and anions (brown) in the unit cell of compound [(en)2Co(ox)][trans-(NH3)2Co(NO2)4] (III) in c-projection. Note that while rows of cations and anions alternate, there seem to be many more anions than cations. In fact ... Lire la suite
Finally, Fig. 11 shows the packing observed for compound (IV). As shown in that diagram, the X-ray results were of very high quality since we found, experimentally, the six hydrogens of the trans –NH3 ligands. The packing shows that the cations (green) and the anions (brown) still form well defined, alternating, strings. These are easily observed along the c-direction. A very rich pattern of hydrogen bonds links cationic and anionic strings. It is also true that there are strings in the diagonal between the b and c axes in which there is an alternation of the cations and anions along such strings. An interesting feature of the cations and anions of this pair is that they are both trans derivatives.

The triclinic unit cell of the conglomerate crystals of the compound of composition [trans-(pn)2Co(ox)][trans-(NH3)2Co(NO2)4] (IV). In this b-projection one notes that the green cations alternate with the brown anions along the diagonal between the b- and c-axis. Cations and anions form four-sided boxes enclosing their counter-ions. Hydrogen bonds are profuse and are shown as dotted lines. For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article. Masquer
The triclinic unit cell of the conglomerate crystals of the compound of composition [trans-(pn)2Co(ox)][trans-(NH3)2Co(NO2)4] (IV). In this b-projection one notes that the green cations alternate with the brown anions along the diagonal between the b- and ... Lire la suite
4 Summary
Our experience with all the compounds we have studied in order to establish their crystallization mode is that they crystallize as conglomerates (a) if the optically active species form helical strings which themselves are chiral by virtue of the left- or right-handedness inherent in a helical object; (b) for neutral compounds, each string is surrounded by additional strings which may be of the same handedness as the first; if so, the crystal is a conglomerate. If there is an equal number of left- and right-handed helices, the crystal is a racemate. If there is a different number of left- and right-handed strings, the crystal is said to be unbalanced. Thus the nature of the inter-string interactions decides the outcome; (c) if the crystal contains ions, the one that is chiral normally forms infinite helical strings also, and those strings are now stitched together by their counter-ions. Thus, the counter-ion decides the outcome since it may stitch strings of the same or of opposite chirality. This is our 11th paper on the subject of counter-ion influence on the crystallization pathway [2–10] and, thus far, we have found no exceptions to the statements within conclusion (c); (d) if all the ions present are chiral, helical strings of cations and anions may form which can produce chiral or racemic crystals in the same manner as described, see (b) and (c); (e) if there are waters of crystallization and counter-ions, they control the crystallization pathway as detailed, also, in (b) and (c); (f) the common thread in all the cases of conglomerate crystallization we have studied is that helical arrays are formed. In this fashion it makes no difference whether the constituent molecules are organic, inorganic or organometallic. Moreover, it makes no difference whether the constituent molecules are inherently chiral or not since a helix is a chiral entity in itself. That is the reason why inherently achiral molecules, such as simple hydroxyphenols, often crystallize in enantiomorphic space groups.
5 Supporting information available
X-ray crystallographic files in CIF format have been deposited with the Cambridge Structural Database as files CCDC 203150, 203151, 203148, and 203149, respectively, for crystals I, II, III and IV. The material can be obtained, free of charge, on application to CCDC, 12 Union Road, Cambridge CB2 1EZ, UK (fax: +44 1223 336033 or e-mail: deposit@ccdc.cam.ac.uk).
Acknowledgements
We thank the Robert A. Welch Foundation for support of these studies (Grant 592 to IB) and for postdoctoral support to Dr. Manas K. Saha. We also thank the Institute for Mexican American Studies (UH) for their support. We also thank Dr. Frank R. Fronczek (LSU) for collecting the low-temperature data.
1 Notes: (a) conglomerate crystallization is the phenomenon whereby a racemic solution produces a mechanical mixture of pure optically active crystals; for an authoritative, and thorough, review of the subject, the reader is recommended the monograph by J. Jacques, A. Collet and Sam H. Wilen “Enantiomers, Racemates and Resolutions.” New York: Wiley Interscience, 1981. We also recommend reading the material presented in the first 13 references. (b) Tren = tris(2-aminoethyl)N, en = 1,2-diaminoethane, pn = 1,3-diaminopropane, ox = oxalato dianion.
2 Cambridge Structural Database, Cambridge Crystallographic Data Centre, 12, Union Road, Cambridge CB2 1EZ, UK. Tel.: +44 1223 336408; fax +44 1223 336033; WWW: http://www.ccdc.cam.ac.uk. Released by Wavefunction, Inc., 18401 Von Karman Ave., Suite 370, Irvine, CA 92612, (949) 955 2120, (949) 955 2118 (fax), http://www.wavefun.com. Release of April, 2001.