

1 Introduction
The ideal perovskite has the general formula ABX3, where the A-site cations are typically larger than the B-site ones and similar in size to the X-site anions. In this structure, the A cations are surrounded by 12 anions in cubo-octahedral coordination and the B cations are surrounded by 6 anions in octahedral coordination. The ideal structure adopts the cubic space group [1].
One of the most important characteristics of perovskite-related structures is their compositional flexibility. This structure can tolerate anionic non-stoichiometry in the X-site as well as cationic deficiency in the A-site. Cation-deficient perovskite-type oxides A1–xBO3 have been studied for a number of years. Compounds with B = W, Mo or Re are named “bronzes” and A (usually alkali metal) occupies a random position; those with Ti, Nb or Ta, namely “non-stoichiometric perovskites”, exhibit some ordering of A cations. Their structure consists of the framework of BO6 octahedra with partially occupied layers of A-site cations alternating with A-vacant layers along the c-axis resulting in a doubling of the perovskite cell along the c-axis (Fig. 1). Several A1–xBO3 perovskite oxides do exist, such as Ln2/3TiO3 [2,3], Ln1/3BO3 (B = Nb, Ta) [4–9] or Th1/4NbO3 [10]. For this latter compound, an additional long-range ordering between Th atoms and vacancies, giving rise to modulation or localized diffusion, has been detected by electron and X-ray diffraction. This kind of material has also been shown to exhibit interesting properties as electrodes for the Li ion insertion reactions in batteries or for their dielectric behavior and magnetic response [11–14].
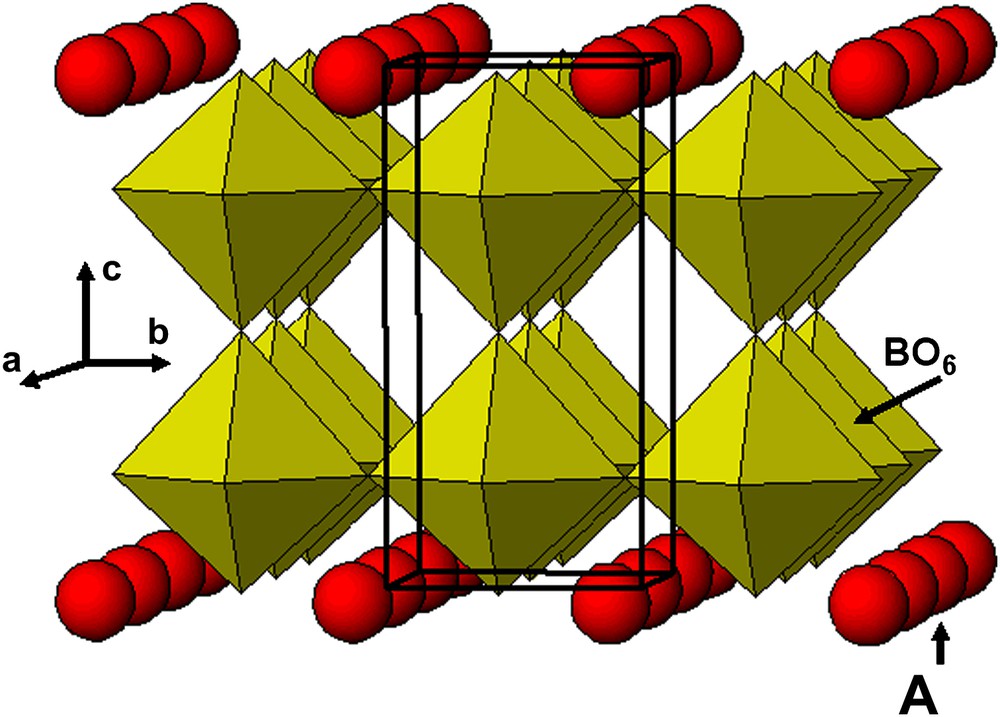
Idealized structure of A1–xBO3: B = Ti (x = 0.33) or Nb (x = 0.66) showing the half (00l) empty planes and half (001) partially filled planes.
The crystal structure of Nd1/3NbO3 was first reported by Iyer et al. [9] as isotype of β-La1/3NbO3, in which the lanthanide ions order into alternate (00l) planes, doubling the c-parameter. Nd1/3NbO3 crystallizes in the orthorhombic space group Pmmm with cell parameters a = 3.878 Å, b = 3.907 Å and c = 7.840 Å (Table 1) at room temperature. Later on, Carrillo et al. [15], using electron microscopy, observed a superstructure in β-La1/3NbO3 (2a × 2b × c). Nevertheless, their X-ray diffraction data, even with a careful analysis, did not reveal any features which could be related to the presence of the superstructure observed by electron microscopy. Thus, they interpreted the superstructure as resulting from distorted octahedra on the basis of simulation of the X-ray diffraction patterns.
Structural data of Nd1/3NbO3 from Iyer et al. [9]
| Nd1/3NbO3 | |
| Crystal system | Orthorhombic |
| Space group | Pmmm |
| Z | 8 |
| Lattice parameters (Å) | a = 3.8807 (1) b = 3.9067 (1) |
| c = 7.8365 (1) | |
| α = β = γ = 90 °C | |
| Volume (Å3) | 118.8 |
The aim of the present work is to characterize Nd1/3NbO3 using XRD (Rietveld refinement) and electron microscopy observations in order to establish whether the superstructure observed in β-La1/3NbO3 is present or not in this compound.
2 Experimental
Powder of Nd1/3NbO3 was obtained by solid-state reaction. The starting materials Nd2O3 and Nb2O5 oxides (Aldrich, purity 99.9%) were first dried in air, at 1000 °C, for 24 h. A stoichiometric mixture of these oxides was ground with ethanol in an agate mortar and then heated in an alumina crucible at 1100 °C in air for 10 h, and, after intermediate regrinding, at 1300 °C for 36 h, in air.
The XRD pattern of Nd1/3NbO3 was recorded at room temperature on a Philip X'pert Pro powder diffractometer in the Bragg–Brentano geometry with a back monochromator, using Cu Kα1 radiations (λ = 1.5406 Å). The data collection was performed in the 6–130° 2θ range with a 0.0080° step, using an X'celerator detector (linear PSD covering 2.122 mm).
The diffraction data were analyzed using the Rietveld technique as implemented in the Fullprof program (software winPLOTR, version June 2005 LLB Saclay – LCSIM Rennes).
Electron diffraction experiments were carried out with a JEOL 2000FX electron microscope equipped with a double-tilt specimen stage. The powder was crushed in ethanol and a drop of this suspension was then deposited on a holey carbon-supported film. High-resolution images were obtained using a JEOL 2200FS microscope.
3 Results and discussion
3.1 X-ray powder diffraction
The X-ray diffraction pattern of the as-prepared material, Nd1/3NbO3, is shown in Fig. 2. Most of the reflections can be indexed in the orthorhombic system with the following cell parameters, a = 3.881 Å, b = 3.907 Å, c = 7.836 Å, close to the ones previously reported in the literature for this compound [9]; however, additional reflections can be noted (arrows in the figure). Further heat treatments at 1300 °C did not modify the X-ray diffraction pattern. Annealing at higher temperatures up to fusion (≈1400 °C) led to a decomposition into NdNbO4 and Nb2O5. In order to understand the origin of these additional reflections, electron diffraction was then performed.
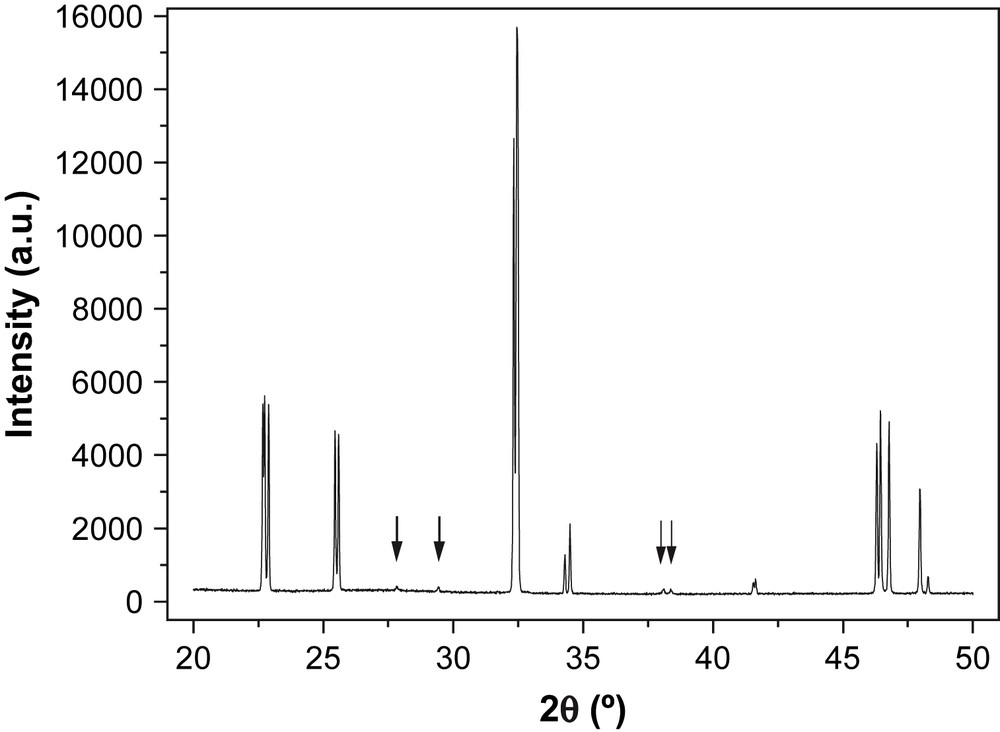
XRD pattern of Nd1/3NbO3 at room temperature; the arrows indicate the reflection not indexed in the a = 3.881 Å, b = 3.907 Å, c = 7.836 Å cell.
3.2 Electron diffraction
Typical electron diffraction patterns of Nd1/3NbO3 powder are shown in Fig. 3. Using the c* axis which can be easily identified (≈7.8 Å), Fig. 3a characterizes the [100] zone axis; tilting of about 45° around the c* axis leads to pattern (3b). To be fully interpreted, this pattern needs to double the a and b initial parameters. Obviously for instance, the reflection labeled (110) cannot be indexed without doubling a and b parameters. Thus, this pattern corresponds to zone axis. Starting from the pattern (3a) ([100] zone axis) and tilting of about 45° around the b* axis leads to pattern (3c) corresponding to the zone axis. This pattern further confirms the superstructure 2a × 2b. Starting from the zone axis, any rotation of 45° around the c* axis gives patterns similar to Fig. 3d or a. Note that the a* and b* axes cannot be distinguished as they are metrically almost identical. The reciprocal lattice (Fig. 4) reconstructed from these patterns implies as for the X-ray data, a superstructure with the following cell parameters: aC ≈ 7.76 Å, bC ≈ 7.81 Å, cC ≈ 7.84 Å. From the systematic condition of reflections h + k = 2n, which appears in the patterns, a C Bravais lattice can be deduced. Since we did not observe any additional conditions in the (0kl)* and (h0l)* reciprocal planes, the absence of glide planes orthogonal to a and b directions can be asserted.

Electron diffraction patterns for Nd1/3NbO3: zone axes (a) [100], (b) , (c) and (d) [010].
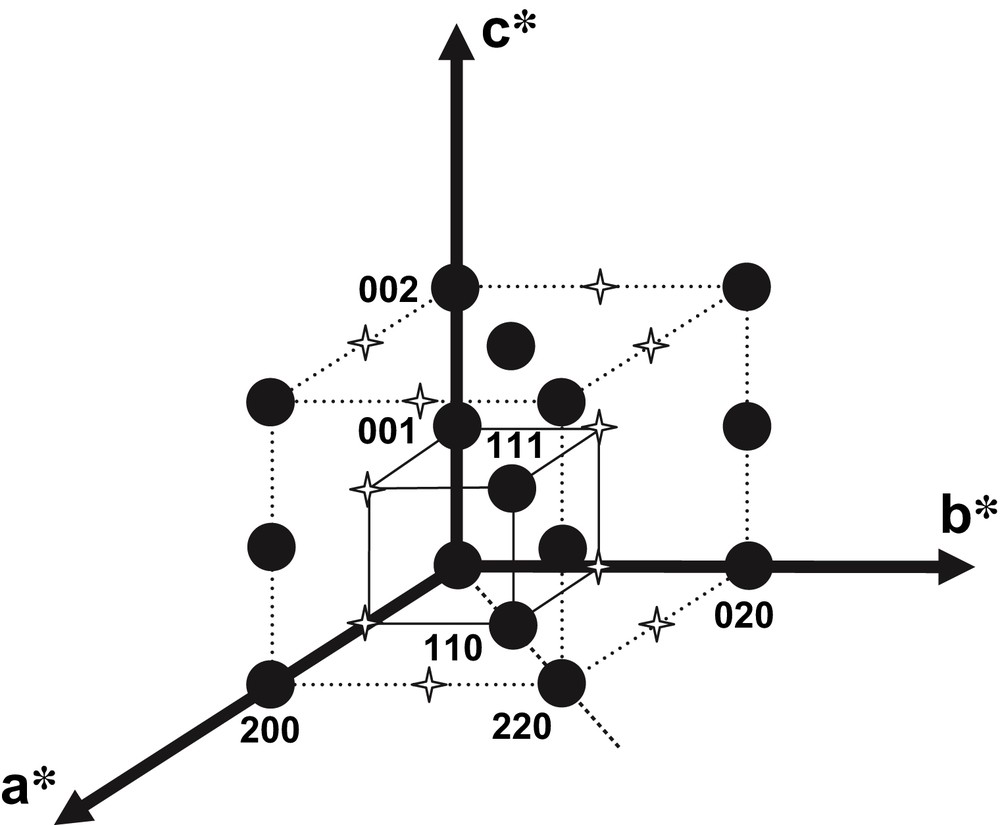
Reciprocal lattice for Nd1/3NbO3 characterizing the C-type Bravais lattice.
Unfortunately it has not been possible to get information on a possible presence of a glide plane orthogonal to the c direction, as the (hk0)* reciprocal plane has not been observed.
In addition, one should stress that in most patterns, after long exposure, additional spots of weak or very weak intensity, more or less diffuse, can be evidenced, as shown in Fig. 5. The previous cell describes the main reflections but not the very weak additional ones which are observed around the main spots. These additional reflections are reproducible and present in various reciprocal planes. Due to their very weak intensity, it has not been possible to obtain additional information such as the exact location of these diffuse scattering or even their nature (spots, surface, cylinder, sphere…).

Electron diffraction patterns of the (a) and (b) zone axes for Nd1/3NbO3 showing weak additional spots.
3.3 Rietveld structural refinements
On the basis of the electron diffraction study, the X-ray diffraction pattern of Nd1/3NbO3 was then refined using the Fullprof program. With respect to the work of Carillo et al. [15], the most symmetrical space group, i.e. Cmmm, was chosen with the initial cell parameters obtained from the previous electron diffraction study. In a first refinement, the initial atomic positions of Nd1/3NbO3 were deduced from those of La1/3NbO3 [15]. In the course of this work, Zhang et al. published the crystal structure of Nd1/3NbO3 based on neutron diffraction data [16]. A second refinement was therefore performed using the atomic positions (Cmmm space group). We also included a second phase, NdNbO4, which could be identified in small amount. This resulted in a slightly improved refinement.
The peak shape was described by a pseudo-Voigt function, and the background level was fitted with linear interpolation between a set of 28 given points with refinable heights. The refined parameters include the scale factor, zero point correction, cell parameters, isotropic thermal and positional parameters for all atoms and three coefficients to describe the angular dependence of line width and asymmetry factors.
Data and details of the refinements are given in Table 2. The various reliability factors show that the refinement # 2, based on the atomic positions of Zhang et al., is improved, the final values being Rexp = 11.69; Rwp = 13.2; RB = 4.72; χ2 = 1.28.
Structural data and X-ray Rietveld refinement parameters at room temperature of Nd1/3NbO3
| Nd1/3NbO3 | Positions from Carillo [15] | Positions from Zhang [16] |
| Crystal system | Orthorhombic | Orthorhombic |
| Space group | Cmmm | Cmmm |
| Z | 8 | 8 |
| a (Å) | 7.7614 (1) | 7.7614 (1) |
| b (Å) | 7.8134 (1) | 7.8134 (1) |
| c (Å) | 7.8364 (1) | 7.8365 (1) |
| Volume (Å3) | 475.22 | 475.23 |
| Theoretical density (g cm−3) | 5.28 | 5.28 |
| Experimental density (g cm−3) | 5.27 | 5.27 |
| Wavelength (Å) | λKα1 = 0.15406 | λKα1 = 0.15406 |
| Step scan increment (deg 2θ) | 0.008 | 0.008 |
| 2θ range (deg) | 7–130 | 7–130 |
| Program | Fullprof | Fullprof |
| Scale factor | 0.1882 × 10−3 | 0.2221 × 10−3 |
| Zero point (deg 2θ) | −0.0083 | −0.0078 |
| Pseudo-Voigt function | η = 0.2729 (4) | η = 0.2801 (4) |
| [PV = ηL + (1 − η))G] | ||
| Caglioti law parameters | U = 0.01137 | U = 0.01109 |
| V = −0.00184 | V = −0.00160 | |
| W = 0.00290 | W = 0.00285 | |
| No. of reflections | 264 | 264 |
| No. of refined parameters | 31 | 30 |
| RBragg | 6.35 | 4.72 |
| cRwp (%) | 14.2 | 13.2 |
| cRexp (%) | 11.69 | 11.69 |
| χ2 | 1.47 | 1.28 |
Fig. 6 shows the excellent agreement between the calculated and observed diffraction profiles for Nd1/3NbO3. The atomic positions, isotropic thermal displacement and reliability factors (RB and Rwp) of Nd1/3NbO3 are given in Table 3 and the interatomic distances in Table 4, only for the refinement based on Zhang atomic positions.
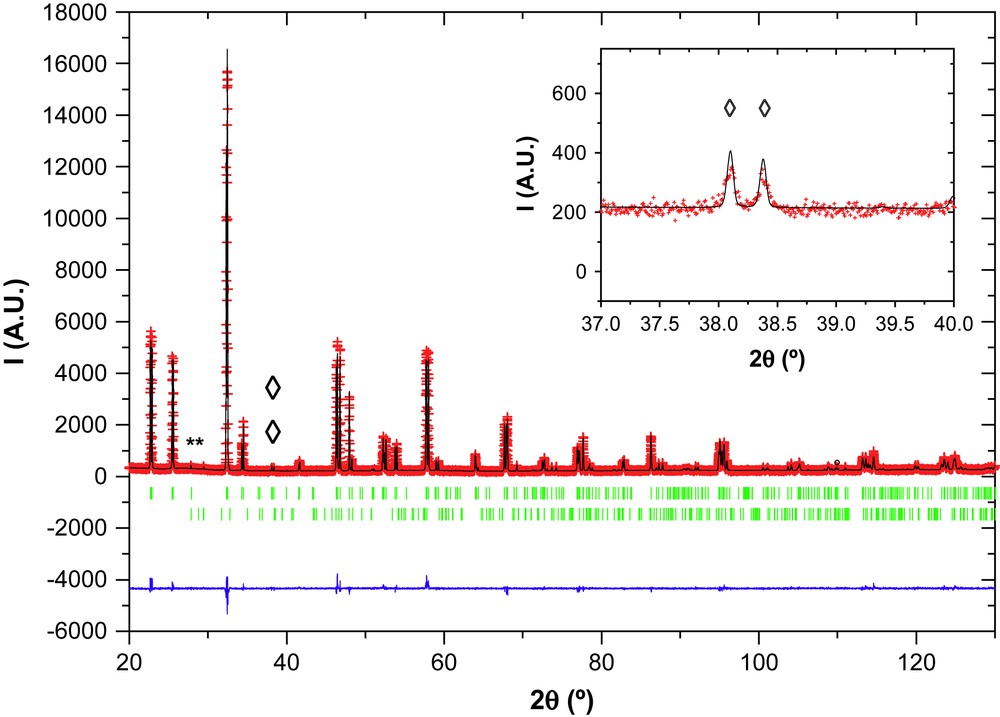
Rietveld refinement of the powder XRD pattern of Nd1/3NbO3. The observed (+), calculated (—), and difference (bottom line) profiles, as well as Bragg positions (vertical bars) are shown. (*) Indicates peak positions of secondary phase NdNbO4 (JPDS 85–1110). (♢) Indicates positions of characteristic superstructure peaks.
Atomic positions, isotropic thermal displacement and reliability factors of Nd1/3NbO3
| Atom | Site | x | y | z | B | Occ. |
| # 1 – Nd1/3NbO3 (from Carillo) | ||||||
| a = 7.7614 Å, b = 7.8134 Å, c = 7.8364 Å, RB = 6.35, Rwp = 14.2 | ||||||
| Nd (1) | 2a | 0 | 0 | 0 | 0.41 (52) | 0.080 |
| Nd (2) | 2b | 0.5 | 0 | 0 | 0.34 (46) | 0.088 |
| Nb | 8m | 0.25 | 0.25 | 0.260 (1) | 0.46 (4) | 0.5 |
| O (1) | 8n | 0 | 0.248 (9) | 0.230 (2) | 1.88 (32) | 0.5 |
| O (2) | 8o | 0.228 (4) | 0 | 0.226 (2) | 0.73 (40) | 0.5 |
| O (3) | 4e | 0.25 | 0.25 | 0 | 3.31 (60) | 0.25 |
| O (4) | 4f | 0.25 | 0.25 | 0.5 | 2.95 (57) | 0.25 |
| # 2 – Nd1/3NbO3 (from Zhang) | ||||||
| a = 7.7614 Å, b = 7.8134 Å, c = 7.8365 Å, RB = 4.72, Rwp = 13.2 | ||||||
| Nd | 4i | 0 | 0.251 (1) | 0 | 0.56 (4) | 0.1667 |
| Nb | 8o | 0.250 (2) | 0 | 0.261 (1) | 0.70 (3) | 0.5 |
| O (1) | 4g | 0.285 (1) | 0 | 0 | 1.82 (47) | 0.25 |
| O (2) | 4h | 0.218 (1) | 0 | 0.5 | 1.95 (47) | 0.25 |
| O (3) | 4k | 0 | 0 | 0.196 (1) | 0.67 (42) | 0.25 |
| O (4) | 4l | 0 | 0.5 | 0.271 (1) | 0.29 (40) | 0.25 |
| O (5) | 8m | 0.25 | 0.25 | 0.229 (1) | 1.42 (22) | 0.5 |
Bond lengths and angles (refinement # 2)
| Bonds lengths (Å) | Bond angles (°) | ||
| Nb–O1 | 2.061 | O1–Nb–O2 | 179.8 |
| Nb–O2 | 1.892 | O3–Nb–O4 | 167.7 |
| Nb–O3 | 2.006 | Nb–O5–Nb | 165.6 |
| Nb–O4 | 1.941 | Nb–O2–Nb | 165.1 |
| Nb–O5 | 1.969 | ||
| Mean Nb–O | 1.972 |
Additional efforts for positioning some amount of Nd in the empty plane and fitting with other space groups of lower symmetry were carried out without success. In all cases, the goodness of the fit did not improve. Finally, the amount of the impurity, NdNbO4, was estimated to be around 1–2%.
3.4 Discussion: structural features
The structure of Nd1/3NbO3 is known to be of the deficient A-site perovskite type, with Nb5+ and Nd3+ ions located in octahedral and in dodecahedral sites, respectively. The refinement of the XRD pattern confirms the presence of empty Nd3+ sites in every second layer. The high-resolution image (Fig. 7) is consistent with this structure and evidences the cation ordering; the inset shows a simulated image calculated with the structural data obtained from the X-ray refinement that nicely confirms these results. This makes a framework of NbO6 octahedra and partially occupied layers of Nd3+ cations at z = 0, alternating with Nd-vacant layers (z = 1/2), which causes the c-axis to be doubled (Fig. 8). At z = 0, Nd3+ ions occupy partially and randomly the dodecahedral sites in agreement with both refinements, the average composition being Nd1/3NbO3. In addition, Nb5+ atoms are slightly shifted from the octahedra center towards the empty Nd layer, which results in alternating long and short Nb–O bonds along the c-axis (cf. Nb–O1 and Nb–O2 in Table 4). Similarly, O3, O4 and O5 atoms are slightly moved away from their ideal positions in the z = 1/4 plane due to electrostatic repulsions. Finally, the NbO6 octahedra are somewhat distorted (see bond lengths and bond angles in Table 4), but the average Nb–O distance, 1.973 Å, is typical of Nb (V) oxides. The unit cell dimensions of a ≈ b ≈ 2ap result from the tilting of NbO6 octahedra around the b-axis. The superlattice reflections arising from this small rotation are very weak in the X-ray diffraction pattern, due to the much stronger scattering factors of Nb and Nd.

Experimental image of Nd1/3NbO3 along the [100] projection. Inset in the image: simulated image obtained using the structural data determined by X-ray diffraction. Inset in the top-right angle shows the Fourier transform of the experimental image.
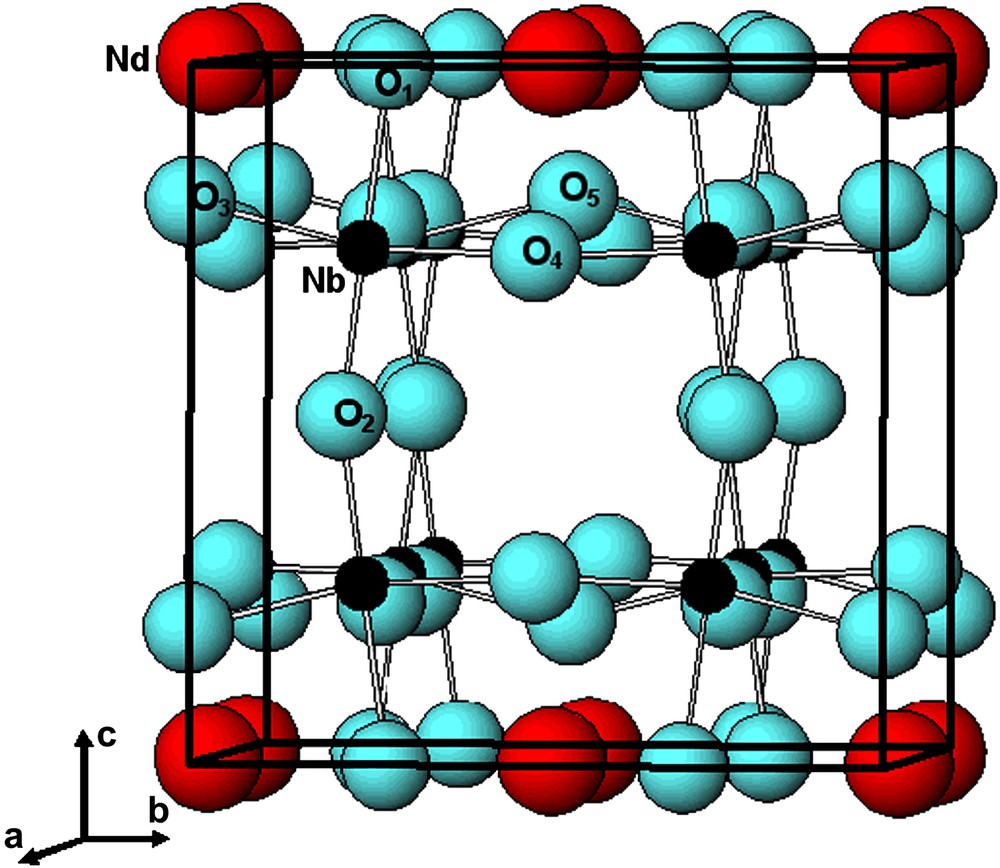
Atomic arrangement in Nd1/3NbO3.
The very weak and more or less diffuse scattering observed in the electron diffraction patterns probably indicates some long-range ordering of neodymium atoms and vacancies. Such additional reflections have already been observed by Alario-Franco et al. [10] for the A-deficient perovskite Th1/4NbO3. These authors interpreted their observation by considering a modulation of the occupation of the Th crystallographic sites associated with a displacive modulation of these atoms and also of surrounding atoms. Preliminary observations indicate that the additional reflections observed in Nd1/3NbO3 by electron diffraction are not located at the same position as those observed for Th1/4NbO3, which likely results from different stoichiometry of the compounds, i.e. the cationic occupation in the central site is different and thus the modulation should be different. One should also note that, due to the weakness of these additional diffuse reflections appearing in the electron diffraction patterns, their presence in X-ray diffraction patterns is doubtful. A single crystal study should now be performed to provide a more complete description of the structure.
Acknowledgements
The “region Aquitaine” is acknowledged for its financial support. In addition, we are indebted to Éric Labraud for his technical assistance.