

1 Introduction
Dendrimers with a π-conjugated backbone have generated significant research efforts in the past years [1]. The electronic properties of such compounds can be easily tailored by either introducing various substituents [2], changing the conjugation lengths of the different fragments within the dendritic shell [3], or modulating the substitution pattern of the branching aromatic units [4,5]. The characteristic features of these compounds make them attractive photoactive components for the preparation of new photochemical molecular devices [6]. In particular, their absorption properties have been widely exploited for the design of light harvesting systems [7–9] in which the π-conjugated dendritic chromophore is able to transfer the collected light energy to the central core of the dendrimer [10]. Among the various terminal energy acceptors used in such light harvesting systems, [60] fullerene (C60) has proven to be particularly interesting [8,9]. Effectively, its first singlet and triplet excited-states are relatively low in energy and photo-induced energy transfer events have been evidenced in numerous fullerene-based dyads [11]. In this article, we report the synthesis of fullerene derivatives functionalized with isomeric phenyleneethynylene-based dendrons possessing 1,3,5-triethynylbenzene or 1,2,4-triethynylbenzene branching units (Fig. 1). Whereas the π-conjugation of the dendritic antenna in C60-G1 and C60-G2 is rather limited due to the all-meta-branching scheme, the dendritic scaffold of C60-Y1 and C60-Y2 exhibits an increase of the conjugation length when going from the first to the second generation compound.
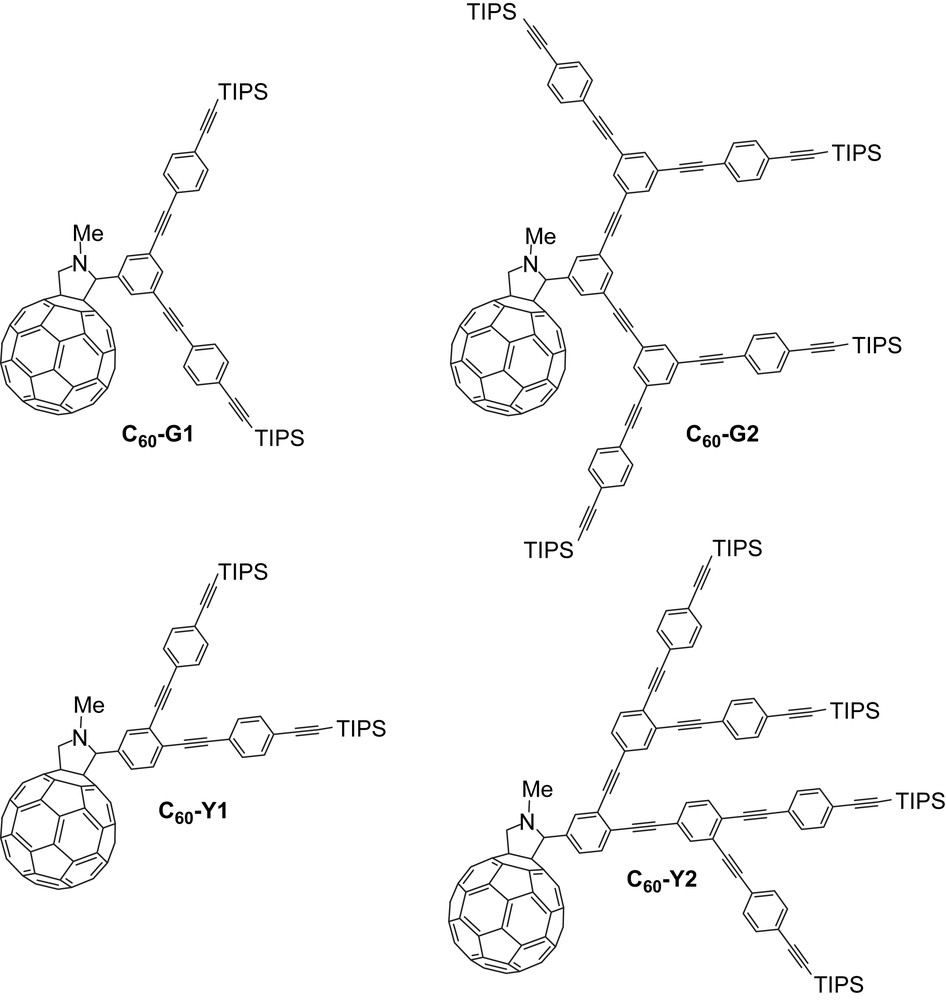
Compounds C60-Gn and C60-Yn (n = 1 or 2; TIPS = triisopropylsilyl).
2 Results and discussion
The synthetic approach to prepare compounds C60-Gn and C60-Yn (n = 1 or 2) relies upon the 1,3-dipolar cycloaddition of an azomethine ylide generated in situ from the corresponding aldehydes and N-methylglycine. This methodology has proven to be a powerful procedure for the functionalization of C60 due to its versatility and the ready availability of the starting materials [12]. The synthesis of dendrons 3 and 6, the key building blocks for the synthesis of C60-G1 and C60-G2, respectively, is shown in Scheme 1. Reaction of 3,5-dibromobenzaldehyde (2) with mono-protected bisalkyne 1 [5] under Sonogashira conditions gave the first generation dendron 3 in 85% yield. Dibromoolefination according to Corey–Fuchs [13] then provided 4, which, after treatment with an excess of LDA in THF at −78 °C and quenching with NH4Cl, afforded terminal alkyne 5. Compound 5 was subjected to a Pd-catalyzed cross-coupling reaction with 2 to yield the second generation dendron 6.

Reagents and conditions: (i) PdCl2(PPh3)2, CuI, PPh3, Et3N, THF (85%); (ii) CBr4, PPh3, Zn dust, CH2Cl2 (68%); (iii) LDA, THF then NH4Cl, H2O (71%); (iv) 2, PdCl2(PPh3)2, CuI, PPh3, Et3N, THF (30%).
Compounds 8 and 11 were obtained from 3,4-dibromobenzaldehyde (7) by following a similar synthetic approach as that described above for the preparation of the corresponding isomers 3 and 6 (Scheme 2). Pd-catalyzed cross-coupling reaction of terminal alkyne 1 with 3,4-dibromobenzaldehyde (7), Corey–Fuchs dibromoolefination and treatment with an excess of LDA gave 10 in an overall 87% yield. Subsequent Sonogashira coupling with 7 yielded the second generation dendron 11. It is worth mentioning here that the methodology based on successive Sonogashira coupling reactions of a terminal alkyne with a halobenzaldehyde, Corey–Fuchs dibromoolefination and treatment with LDA was also successfully applied to the synthesis of linear oligophenyleneethynylene derivatives [14]. It is an interesting alternative to the approaches reported by the groups of Tour [15] and Godt [16]. On the one hand, compared to the strategy based on the trimethylsilyl and the 3,3-diethyltriazene functions as complementary protecting groups for terminal alkyne and aryl iodine, respectively, it avoids the use of large amounts of rather volatile carcinogenic methyl iodide [15]. On the other hand, compared to the strategy based on the bromine–iodine selectivity of the Pd-catalyzed alkyne–aryl coupling which is not always completely iodo-selective, it prevents the formation of undesirable symmetric by-products [16].

Reagents and conditions: (i) 1, PdCl2(PPh3)2, CuI, PPh3, Et3N, THF (75%); (ii) CBr4, PPh3, Zn dust, CH2Cl2 (95%); (iii) LDA, THF then NH4Cl, H2O (92%); (iv) 7, PdCl2(PPh3)2, CuI, PPh3, Et3N, THF (78%).
Owing to the presence of the triisopropylsilyl (TIPS) substituents, dendrons 3–6 and 8–11 are highly soluble in common organic solvents (CH2Cl2, CHCl3, THF), and 1H- and 13C NMR spectroscopic characterization was easily achieved. The 1H NMR spectra of compounds 3 and 6 recorded in CDCl3 are shown in Fig. 2. For both compounds, the resonance arising from the aldehydic proton is observed at ca. 10 ppm. In the aromatic region, the spectrum of 3 shows two sets of signals in a typical pattern for a 3,5-disubstituted benzaldehyde moiety as well as a singlet at 7.50 ppm for the protons of the two para-disubstituted phenyl groups. The 1H NMR of 6 is also in full agreement with the proposed structure. In addition to the signals corresponding to the protons of the central 3,5-diethynylbenzaldehyde moiety, the resonances arising from the two equivalent 1,3,5-triethynylbenzene units are observed at 7.68 ppm. The 1H NMR spectra of 8 and 11 recorded in CDCl3 are depicted in Fig. 3. When compared to the spectra of 3 and 6 shown in Fig. 2, the spectra of 8 and 11 are more complicated due to their reduced symmetry resulting from the 1,3,4-branching motif. In the aromatic region, the 1H NMR spectrum of 8 is characterized by three sets of signals for the aromatic protons of the central 1,3,4-trisubstituted phenyl ring (Ha, Hb and Hc) as well as a pseudo-singlet for the protons of the two para-disubstituted phenyl groups. The 1H NMR spectrum of 11 is also consistent with the proposed structure. In addition to the signals corresponding to the protons of the central 3,4-diethynylbenzaldehyde moiety, the resonances arising from the two 1,3,4-triethynylbenzene units appear as a single set of three signals. These two aromatic rings are in principle non-equivalent, however both benzene rings are substituted by three alkyne groups and must be in a similar chemical environment; they are therefore pseudo-equivalent. Finally, the peaks of the peripheral para-diethynyl phenyl moieties appear between 7.44 and 7.47 ppm. Compounds 3–6 and 8–11 were further characterized by IR spectroscopy. For all the derivatives, the characteristic CC stretching band is observed at 2151–2152 cm−1. In the IR spectrum of 3, 6, 8 and 11 the diagnostic aldehyde band is observed at ca. 1700 cm−1. In the case of 5 and 10, the C–H stretching band characteristic of the terminal alkyne function is seen at ca. 3300 cm−1.

1H NMR spectra (300 MHz, CDCl3) of compounds 3 (bottom) and 6 (top).
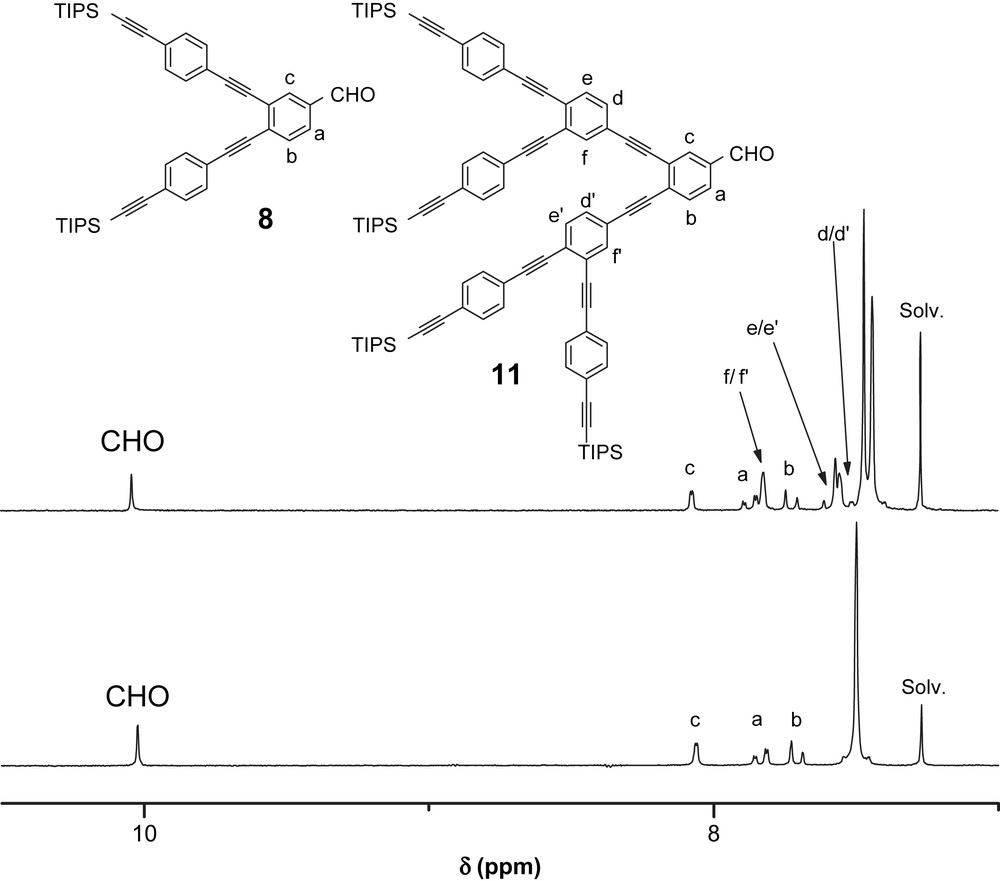
1H NMR spectra (300 MHz, CDCl3) of compounds 8 (bottom) and 11 (top).
Reaction of aldehydes 3, 6, 8 and 11 with N-methylglycine and C60 in refluxing toluene gave the corresponding pyrrolidinofullerene C60-Gn and C60-Yn (n = 1 or 2) in 27–52% isolated yield (Scheme 3). The structure of fullerodendrimers C60-Gn and C60-Yn (n = 1 or 2) was confirmed by analytical and spectroscopic data. The 1H NMR spectra of C60-Gn and C60-Yn (n = 1 or 2) recorded in CDCl3 exhibit the expected features with the signals arising from the dendritic branches, two doublets and a singlet for the pyrrolidine protons as well as a singlet for the N–CH3 group. It is also important to note that the signals corresponding to the protons of the phenyl group directly attached to the pyrrolidine ring are broad at room temperature. As previously described for phenyl-pyrrolidinofullerene derivatives [17], this indicates restricted rotation of the phenyl substituent on the pyrrolidine ring. This was confirmed by variable-temperature NMR studies showing a clear coalescence and a reversible narrowing of all the peaks for all the compounds. Indeed, the 1H NMR spectra of C60-Gn and C60-Yn (n = 1 or 2) recorded at high temperature (90–120 °C) are all well resolved, with sharp signals for the protons of the aromatic substituent attached to the pyrrolidine ring. The 13C NMR spectra of C60-Gn and C60-Yn (n = 1 or 2) were also in full agreement with their C1 symmetry resulting from the presence of the asymmetric C atom in the pyrrolidine ring. Finally, the structure of fullerodendrimers C60-Gn and C60-Yn (n = 1 or 2) was confirmed by mass spectrometry. As a typical example, the FAB mass spectrum of compound C60-G2 is shown in Fig. 4. The expected molecular ion peak is observed at 2176.4. Two characteristic fragments are also observed at 1455.1 and 720.0 corresponding to [M − C60]+ and [C60]+, respectively. However, no signals corresponding to defected dendrimers could be detected, thus showing the monodispersity of C60-G2.

Reagents and conditions: (i) C60, N-methylglycine, toluene, Δ.
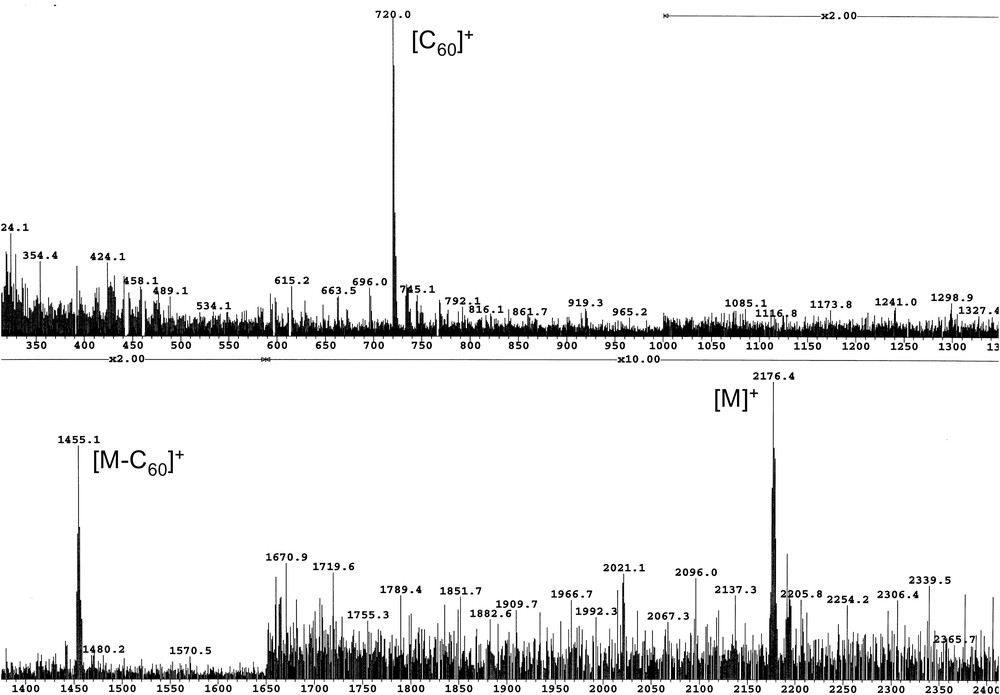
FAB mass spectrum of compound C60-G2.
3 Conclusion
Two series of isomeric dyads with differently branched phenyleneethynylene-based moieties and a pyrrolidinofullerene core have been prepared. Preliminary luminescence measurements reveal no emission from the π-conjugated dendritic branches in C60-Gn and C60-Yn (n = 1 or 2) thus indicating a strong quenching of the phenyleneethynylene fluorescence by the fullerene moiety in all compounds. Detailed photophysical studies are currently under investigation to elucidate the nature of the intramolecular photo-induced processes in C60-Gn and C60-Yn (n = 1 or 2).
4 Experimental section
4.1 General
Reagents and solvents were purchased as reagent grade and used without further purification. THF was distilled over sodium benzophenone ketyl. Compounds 1 and 8–11 [5] were prepared according to previously reported procedures. All reactions were performed in standard glassware under an inert Ar atmosphere. Evaporation and concentration were done at water aspirator pressure and drying in vacuo at 10−2 Torr. Column chromatography: silica gel 60 (230–400 mesh, 0.040–0.063 mm) was purchased from E. Merck. Thin Layer Chromatography (TLC) was performed on glass sheets coated with silica gel 60 F254 purchased from E. Merck, visualization by UV light. IR spectra (cm−1) were measured on an ATI Mattson Genesis Series FTIR instrument. NMR spectra were recorded on a Bruker AC 200 or AC 400 with solvent peaks as reference. FAB mass spectra (m/z; % relative intensity) were taken on a ZA HF instrument with 4-nitrobenzyl alcohol as matrix. Elemental analyses were performed by the analytical service at the Institut Charles Sadron, Strasbourg.
4.2 General procedure for the Sonogashira cross-coupling reactions
To an oven dried glass screw capped tube were added all solids including the aryl bromide, alkyne, CuI, PPh3 and palladium catalyst. The atmosphere was removed via vacuum and replaced with dry argon (3×). THF and triethylamine were added by syringe and the reaction was conducted at 65 °C in an oil bath while stirring. Upon cooling the reaction mixture was filtered via gravity filtration to remove solids and diluted with dichloromethane. The reaction mixture was extracted with an aqueous NH4Cl solution. The organic layer was dried with MgSO4 and filtered through a plug of SiO2 (CH2Cl2). The solvent was evaporated and the product purified as outlined in the following text.
4.2.1 Compound 3
This compound was prepared from 1 (4.0 g, 14.2 mmol), 2 (1.25 g, 4.73 mmol), Pd(PPh3)2Cl2 (99.5 mg, 0.14 mmol), CuI (30.7 mg, 2 mmol) and PPh3 (92 mg, 0.36 mmol) in a 4/1 THF/Et3N mixture (40 mL). Column chromatography (SiO2, Hexane/CH2Cl2 9:1) yielded 3 (2.68 g, 85%). Colorless oil. IR (KBr): 1705 (CO), 2152 (CC). 1H NMR (CDCl3, 200 MHz): 10.01 (s, 1H), 7.97 (d, J = 2 Hz, 2H), 7.91 (t, J = 2 Hz, 1H), 7.48 (s, 8H), 1.09 (s, 42H). 13C NMR (CDCl3, 75 MHz): 190.6, 139.4, 136.7, 132.0, 131.9, 131.5, 124.7, 124.0, 122.2, 106.4, 93.3, 91.2, 88.8, 18.6, 11.3. Anal. Calcd for C45H54OSi2, 0.5 H2O: C 80.90, H 8.30; found: C 80.44, H 8.29.
4.2.2 Compound 6
This compound was prepared from 5 (1.2 g, 1.81 mmol), 2 (0.19 g, 0.73 mmol), Pd(PPh3)2Cl2 (16 mg, 0.02 mmol), CuI (5 mg, 0.04 mmol) and PPh3 (23 mg, 0.09 mmol) in a 4/1 THF/Et3N mixture (10 mL). Column chromatography (SiO2, Hexane/CH2Cl2 8:2) yielded 6 (0.31 g, 30%). Yellow glassy solid. IR (neat): 1700 (CO), 2150 (CC). 1H NMR (CDCl3, 300 MHz): 10.02 (s, 1H), 7.99 (d, J = 2 Hz, 2H), 7.90 (t, J = 2 Hz, 1H), 7.67 (m, 6H), 7.48 (s, 16H), 1.10 (s, 84H). 13C NMR (CDCl3, 75 MHz): 190.5, 139.6, 136.8, 134.6, 134.2, 132.3, 132.0, 131.5, 124.5, 124.1, 123.9, 123.3, 122.5, 106.5, 93.2, 90.6, 89.9, 89.3, 88.2, 18.7, 11.3. Anal. Calcd for C99H110OSi4, 0.5 H2O: C 82.73, H 7.78; found: C 82.56, H 8.10.
4.2.3 Compound 8
This compound was prepared from 1 (4.40 g, 15.57 mmol), 7 (1.53 g, 5.80 mmol), Pd(PPh3)2Cl2 (0.49 g, 0.70 mmol), CuI (57 mg, 0.29 mmol) and PPh3 (0.23 g, 0.88 mmol) in THF/Et3N 4:1 (60 mL). Column chromatography (SiO2, Hexane/CH2Cl2 4:1) yielded 8 (2.90 g, 75%). Yellow solid (mp. 178 °C). IR (KBr): 2152 (CC), 1702 (CO). 1H NMR (200 MHz, CDCl3): 10.02 (s, 1H), 8.06 (d, J = 2 Hz, 1H), 7.83 (dd, J = 7 and 2 Hz, 1 H), 7.70 (d, J = 7 Hz), 7.50 (s, 4H), 1.15 (s, 42 H). 13C NMR (50 MHz, CDCl3): 189.9, 135.2, 132.8, 132.0, 131.9 (2C), 131.4, 131.3, 130.8, 128.0, 126.3, 124.1, 123.8, 122.4, 122.2, 106.4, 106.3, 97.0, 94.5, 93.2, 93.0, 89.4, 88.7, 18.6, 11.2. Anal. Calc. for C45H54Si2O: C 81.02, H 8.16; found: C 80.74, H 8.19.
4.2.4 Compound 11
This compound was prepared from 10 (2.1 g, 3.17 mmol), 7 (0.31 g, 1.17 mmol), Pd(PPh3)2Cl2 (99 mg, 0.14 mmol), CuI (12 mg, 0.059 mmol) and PPh3 (46 mg, 0.18 mmol) in THF/Et3N 4:1 (20 mL). Column chromatography (SiO2, Hexane/CH2Cl2 4:1) yielded 11 (1.32 g, 78%). Yellow solid (mp. 150 °C). IR (KBr): 2151 (CC), 1704 (CO). 1H NMR (200 MHz, CDCl3): 10.04 (s, 1 H), 8.07 (d, J = 2 Hz, 1H), 7.87 (dd, J = 7 and 2 Hz, 1H), 7.82 (broad s, 2H), 7.72 (d, J = 7 Hz, 1H), 7.58 (d, J = 7 Hz, 2H), 7.53 (dd, J = 7 and 2 Hz, 2H), 7.47 (s, 8H), 7.44 (s, 8H), 1.15 (s, 42 H), 1.13 (s, 42 H). 13C NMR (50 MHz, CDCl3): 190.1, 135.3, 135.0, 134.9, 132.7, 132.0, 131.9, 131.4, 131.3, 130.9, 126.4, 126.0, 125.9, 125.7, 123.8, 123.6, 122.6, 122.3, 106.6, 96.6, 95.5, 95.4, 94.2, 94.15, 94.1, 93.1, 93.0, 92.95, 92.9, 90.2, 89.8, 89.6, 89.1, 89.0, 18.6, 11.3, 11.2. Anal. Calcd for C99H110Si4O: C 83.25, H 7.76; found: C 83.34, H 7.71.
4.3 General procedure for the dibromoolefination reactions
A mixture of CBr4, PPh3 and Zn dust in dry CH2Cl2 was stirred at room temperature for 24 h. The suspension was then cooled to 0 °C and the appropriate aldehyde dissolved in CH2Cl2 was added at once. The resulting mixture was slowly warmed to room temperature and stirred overnight. The resulting thick suspension was filtered and evaporated. The residue was dissolved in a minimum of CH2Cl2, then hexane was added to precipitate the remaining P-containing by-products. The resulting mixture was filtered and evaporated. The product was then purified as outlined in the following text.
4.4 Compound 4
This compound was prepared from 3 (1.5 g, 2.25 mmol), CBr4 (3.73 g, 11.3 mmol), PPh3 (2.95 g, 11.3 mmol) and Zn dust (0.73 g, 11.3 mmol) in CH2Cl2 (50 mL) and column chromatography (SiO2, Hexane/CH2Cl2 9:1) yielded 4 (1.26 g, 68%). Yellow oil. IR (KBr): 2152 (CC). 1H NMR (200 MHz, CDCl3): 7.66 (t, J = 2 Hz, 1 H), 7.62 (d, J = 2 Hz, 2H), 7.47 (s, 8H), 7.43 (s, 1H), 1.15 (s, 42 H). Anal. Calcd for C46H54Si2Br2: C 67.14, H 6.61; found: C 67.11, H 6.69.
4.4.1 Compound 9
This compound was prepared from 8 (2.80 g, 4.20 mmol), CBr4 (8.36 g, 25.20 mmol), PPh3 (6.61 g, 25.20 mmol) and Zn dust (1.65 g, 25.20 mmol) in CH2Cl2 (200 mL) and column chromatography (SiO2, hexane) yielded 9 (3.30 g, 95%). Pale yellow solid (mp. 140 °C). IR (KBr): 2151 (CC). 1H NMR (200 MHz, CDCl3): 7.75 (d, J = 2 Hz, 1H), 7.57 (d, J = 7 Hz, 1H), 7.51(dd, J = 7 and 2 Hz, 1H), 7.48 (m, 8 H), 7.47 (s, 1H), 1.15 (s, 42 H). 13C NMR (50 MHz, CDCl3): 135.2, 134.8, 131.9, 131.6, 131.3, 127.8, 125.7, 125.5, 123.6, 122.88, 122.8, 106.6, 94.5, 93.7, 92.95, 92.9, 91.2, 90.0, 89.6, 18.6, 11.3. Anal. Calcd for C46H54Si2Br2: C 67.14, H 6.61; found: C 67.37, H 6.67.
4.5 General procedure for the preparation of alkynes from dibromoolefines
A solution of LDA in THF was slowly added to a solution of the appropriate dibromoolefine in THF at −78 °C. After 3 h, a saturated aqueous NH4Cl solution was added. The reaction mixture was diluted with hexane, washed with water, dried with MgSO4 and evaporated. The product was then purified as outlined in the following text.
4.5.1 Compound 5
This compound was prepared from 4 (1.26 g, 1.53 mmol) and LDA (6.11 mmol) in THF (40 mL) and column chromatography (SiO2, Hexane/CH2Cl2 8:1) yielded 5 (720 mg, 71%). Yellow glassy solid. IR (KBr): 2152 (CC), 3304 (CC–H). 1H NMR (CDCl3, 300 MHz): 7.66 (t, J = 2 Hz, 1H), 7.60 (d, J = 2 Hz, 2H), 5.30 (s, 8H); 3.12 (s, 1H), 1.14 (s, 42H). 13C NMR (CDCl3, 75 MHz): 134.7, 134.6, 132.4, 132.3, 132.0, 131.5, 131.1, 123.9, 123.8, 123.0, 122.5, 106.5, 93.1, 90.4, 89.3, 81.9, 78.5, 18.7, 11.3. Anal. Calcd for C46H54Si2, H2O: C 81.13, H 8.30; found: C 81.15, H 8.28.
4.6 Compound 10
This compound was prepared from 9 (3.28 g, 3.98 mmol) and LDA (20 mmol) in THF (60 mL) and column chromatography (SiO2, hexane/CH2Cl2 20:1) yielded 10 (2.40 g, 92%). Colorless solid (mp. 155 °C). IR (KBr): 3314 (C–H), 2151 (CC). 1H NMR (200 MHz, CDCl3): 7.63 (d, J = 2 Hz, 1H), 7.46 (s, 8H), 7.42 (d, J = 7 Hz, 1 H), 7.34 (dd, J = 2 and 7 Hz), 3.16 (s, 1H), 1.15 (s, 42H). 13C NMR (50 MHz, CDCl3): 135.1, 132.0, 131.6, 131.4, 125.85, 125.8, 123.75, 123.7, 122.8, 122.1, 106.6, 95.1, 93.9, 93.0, 92.9, 89.7, 89.1, 82.4, 79.5, 18.6, 11.3. Anal. Calcd for C46H54Si2: C 83.32, H 8.21; found: C 82.99, H 8.24.
4.7 General procedure for the preparation of pyrrolidinofullerene
N-Methylglycine and the appropriate aldehyde were added to a solution of C60 in toluene under argon. The mixture was heated at 115 °C. After 24 h the mixture was allowed to cool to room temperature and the solvent was removed under reduced pressure. The product was then purified as outlined in the following text.
4.7.1 Compound C60-G1
This compound was prepared from N-methylglycine (542 mg, 6.08 mmol), 3 (500 mg, 0.75 mmol), C60 (595 mg, 0.83 mmol) in toluene (670 mL) and column chromatography (SiO2, Hexane/CH2Cl2 8:2) yielded C60-G1 (287 mg, 27%). Brown glassy solid. IR (KBr): 2149 (CC). 1H NMR (C2D2Cl2, 400 MHz, 100 °C): 8.03 (d, J = 2 Hz, 2H), 7.75 (t, J = 2 Hz, 1H), 7.50 (m, 8H), 5.08 (d, J = 9 Hz, 1H), 5.01 (s, 1H), 4.36 (d, J = 9 Hz, 1H), 2.91 (s, 3H), 1.23 (s, 42H). 13C NMR (CDCl3, 75 MHz): 145.54, 145.49, 145.39, 145.35, 145.26, 145.24, 145.18, 144.70, 144.57, 144.39, 144.36, 143.13, 143.00, 142.69, 142.60, 142.57, 142.25, 142.22, 142.13, 142.10, 142.04, 142.00, 141.94, 141.90, 141.68, 141.63, 140.20, 140.12, 139.69, 138.10, 137.11, 136.52, 135.95, 135.66, 134.92, 132.24, 132.17, 132.01, 131.48, 123.68, 122.65, 106.57, 93.07, 90.32, 90.24, 82.85, 69.98, 69.01, 40.13, 31.91, 29.70, 29.65, 29.35, 18.66, 11.30. Anal. Calcd for C107H59NSi2, 2H2O: C 88.58, H 4.38, N 0.97; found: C 88.23, H 4.73, N 1.15. FAB MS: 1415.6 [M + H]+ calculated for C107H60NSi2 (1415.8).
4.7.2 Compound C60-G2
This compound was prepared from N-methylglycine (76 mg, 0.85 mmol), 6 (150 mg, 0.105 mmol), C60 (84 mg, 0.116 mmol) in toluene (95 mL) and column chromatography (SiO2, hexane/CH2Cl2 8:2) yielded C60-G2 (120 mg, 52%). Brown glassy solid. IR (KBr): 2152 (CC). 1H NMR (C2D2Cl2, 400 MHz, 110 °C): 8.09 (d, J = 2 Hz, 2H), 7.79 (t, J = 2 Hz, 1H), 7.74 (d, J = 2 Hz, 4H), 7.72 (t, J = 2 Hz, 2H), 7.51 (m, 16H), 5.09 (d, J = 10 Hz, 1H), 5.04 (s, 1H), 4.38 (d, J = 10 Hz, 1H), 2.93 (s, 3H), 1.24 (s, 84H). 13C NMR (CDCl3, 75 MHz): 144.70, 144.56, 144.36, 143.13, 142.56, 142.27, 142.14, 142.01, 141.91, 141.67, 140.20, 138.30, 136.54, 135.98, 135.66, 134.29, 132.02, 131.47, 123.95, 123.79, 123.69, 122.53, 106.55, 93.14, 90.48, 89.47, 89.02, 69.01, 40.10, 29.70, 18.66. Anal. Calcd for C161H115NSi4, CH2Cl2: C 86.06, H 5.22, N 0.62; found: C 86.39, H 5.55, N 0.86. FAB MS: 2176.4 [M]+ calculated for C161H115NSi4 (2176.0).
4.7.3 Compound C60-Y1
This compound was prepared from N-methylglycine (210 mg, 2.2 mmol), 8 (150 mg, 0.22 mmol), C60 (210 mg, 0.29 mmol) in toluene (230 mL) and column chromatography (SiO2, hexane/toluene 4:1) yielded C60-Y1 (137 mg, 45%). Brown glassy solid. 1H NMR (CDCl3, 200 MHz): 7.50–7.46 (m, 11H), 5.03 (d, J = 10 Hz, 1H), 4.95 (s, 1H), 4.31 (d, J = 10 Hz, 1H), 2.83 (s, 3H), 1.14 (s, 42H). 13C NMR (CDCl3, 50 MHz): 152.85, 147.27, 146.44, 146.24, 146.18, 146.12, 146.07, 145.93, 145.89, 145.82, 145.69, 145.54, 145.51, 145.45, 145.30, 145.22, 145.19, 144.66, 144.54, 144.36, 144.33, 143.10, 142.96, 142.65, 142.56, 142.18, 142.11, 142.06, 142.00, 141.95, 141.84, 141.65, 141.57, 140.15, 139.99, 139.64, 136.99, 136.43, 135.94, 135.66, 133.65, 132.98, 130.48, 129.71, 125.88, 106.54, 93.09, 69.05, 17.39, 17.20, 12.44, 10.07. Anal. Calcd for C107H59NSi2: C 90.84, H 4.20, N 0.99; found: C 90.45, H 4.27, N 0.91. FAB MS: 1415.2 [M]+ calculated for C107H59NSi2 (1414.8).
4.7.4 Compound C60-Y2
This compound was prepared from N-methylglycine (63 mg, 0.7 mmol), 11 (100 mg, 0.07 mmol), C60 (66 mg, 0.09 mmol) in toluene (130 mL) and column chromatography (SiO2, hexane/toluene 4:1) yielded C60-Y2 (60 mg, 43%). Brown glassy solid. 1H NMR (CDCl3, 200 MHz): 7.82–7.43 (m, 25H), 5.00 (d, J = 10 Hz, 1H), 4.96 (s, 1H), 4.30 (d, J = 10 Hz, 1H), 2.85 (s, 3H), 1.14 (s, 42H), 1.12 (s, 42H). 13C NMR (CDCl3, 50 MHz): 155.91, 153.69, 152.86, 152.46, 147.29, 146.42, 146.30, 146.28, 146.19, 146.17, 146.14, 146.09, 145.95, 145.92, 145.79, 145.70, 145.57, 145.52, 145.47, 145.37, 145.33, 145.24, 145.21, 145.17, 144.68, 144.55, 144.38, 144.33, 143.13, 142.97, 142.68, 142.58, 142.55, 142.53, 142.23, 142.20, 142.13, 142.09, 142.08, 142.01, 141.98, 141.88, 141.66, 141.59, 140.18, 139.68, 138.01, 137.04, 136.43, 135.98, 135.64, 135.04, 134.97, 132.09, 132.03, 131.94, 131.45, 131.36, 130.86, 126.12, 125.77, 125.54, 123.81, 123.71, 123.05, 122.70, 122.67,95.39, 94.13, 93.52, 93.41, 93.12, 93.02, 90.66, 89.84, 89.16, 89.13, 69.01, 40.05, 29.69, 18.66, 11.30. Anal. Calcd for C161H115NSi4, CH2Cl2: C 86.06, H 5.22, N 0.62; found: C 86.28, H 5.42, N 0.56. FAB MS: 2176.8 [M]+ calculated for C161H115NSi4 (2176.0).
Acknowledgements
This work was supported by the CNRS. S. Z. and M. U. thank the French Ministry of Research, and A. G. the ADEME – Région Alsace for their fellowships.