

1 Introduction
Although extensively studied in the last decade, the fullerenes continue to be a promising challenge in current chemical research. After a first phase characterized by theoretical, physical and physicochemical studies, a well-established ‘organic chemistry of the fullerenes’ developed in a very rapid progression to the point that, currently, a wide variety of functionalized fullerenes are available through simple and accessible synthetic routes [1]. Among the several derivatives, those containing covalently linked TEMPO moieties (TEMPO = 2,2,6,6-tetramethylpiperidine-1-oxyl) have been studied by means of EPR spectroscopy (standard and time-resolved) to elucidate and understand the peculiar characteristics of important species such as fullerene anions or triplet excited states. These species are connected with relevant fullerene features, such as optical limiting properties, ferromagnetism, singlet oxygen sensitization. Here we review our most significant achievements in the field of nitroxide-labeled fullerenes that were synthesized by means of the addition to [60]fullerene of azomethine ylides.
2 Synthesis
Among the many reactions that were successfully developed, the 1,3-dipolar cycloaddition of azomethine ylides, introduced in 1993 [2], provides a valuable synthetic procedure that is used by many for the preparation of functionalized fullerenes for applications in materials science and for the study of fundamental physicochemical properties of the fullerene spheroid. The azomethine ylide cycloaddition was employed to prepare the spin-labeled fulleropyrrolidines discussed in this work whose structure is presented in Fig. 1 and Figs. 7 and 8 (vide infra). Derivatives 1–3 have the TEMPO moiety spiro-linked (1 and 2) or linked (3) to position 2 of the pyrrolidine ring. Derivative 1 was prepared by condensing N-methylglycine, 2,2,6,6-tetramethyl-4-piperidone-1-oxyl and [60]fullerene [3], whereas for derivative 2, 4-amino-4-carboxy-2,2,6,6-tetramethylpiperidine-1-oxyl (TOAC) and paraformaldehyde were used along with [60]fullerene [3]. Fulleropyrrolidine 3 was synthesized starting from 4-formyl-TEMPO, N-methylglycine and [60]fullerene [3]. N-acylated derivative 4 was obtained by allowing N–H fulleropyrrolidine to react with 4-carboxy-TEMPO in the presence of 1-(3-(dimethylamino)propyl)-3-ethylcarbodiimide (EDC) and N-hydroxybenzotriazole (HOBt) as coupling agents [3]. Derivatives 5 and 6 are donor–acceptor dyads in which a nitroxide spin-labeling has been introduced to study the photoinduced electron transfer from the ferrocene to the fullerene. They were synthesized by condensing the corresponding N-alkylferrocenyl glycine with 2,2,6,6-tetramethyl-4-piperidone and [60]fullerene, followed by oxidation with 4-chloroperbenzoic acid [4]. Bis-fulleropyrrolidines 7–11 and binitroxide 12 are interesting examples of biradicals. A standard bis-addition protocol to [60]fullerene was used to prepare biradicals 7–11 in which two 2,2,5,5-tetramethylpyrrolidine-1-oxyl moieties were placed at precise distance and mutual orientation on the fullerene sphere [5,6]. TOAC and 4-formyl-TEMPO were employed to synthesize biradical 12 [7].
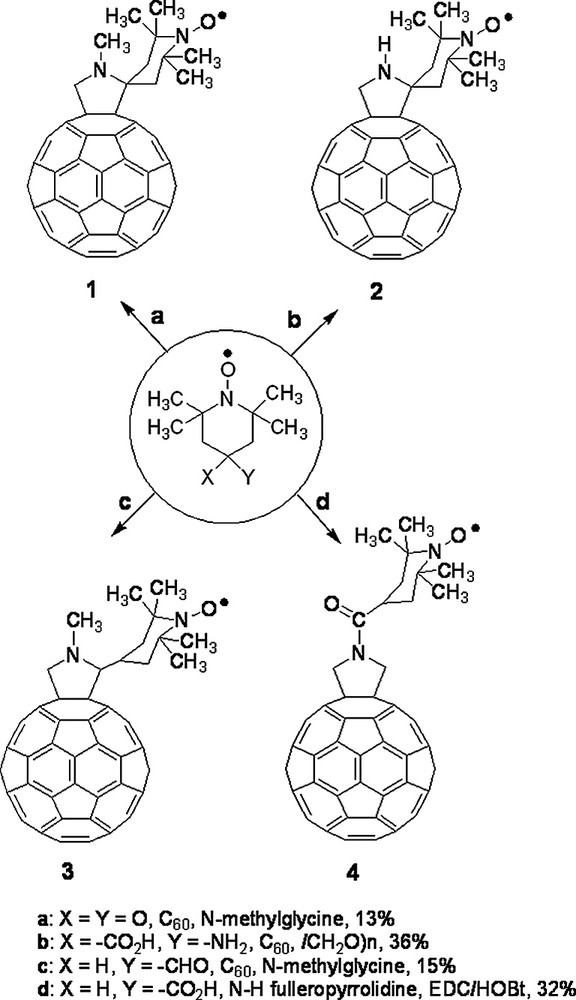
Fullerene-mononitroxides considered in this work.
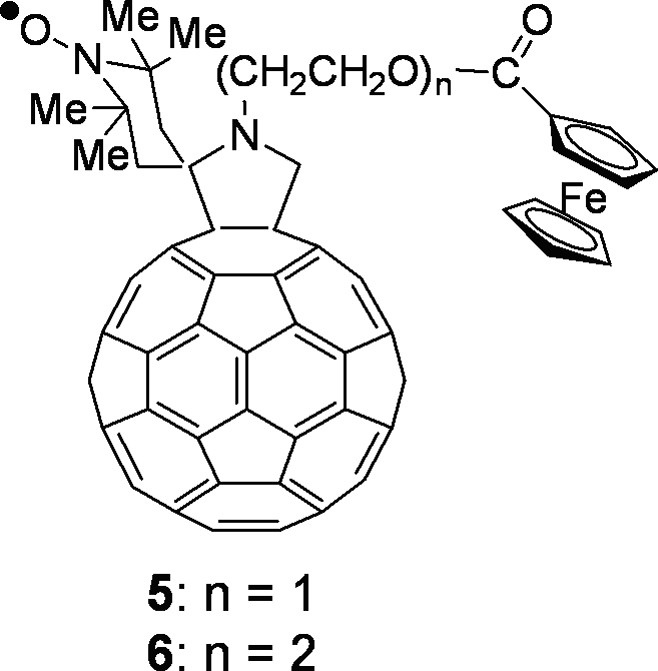
Spin-labeled fullerene–ferrocene dyads.

Fullerene-binitroxides considered in this work.
3 Radical anions
[60]Fullerene has a low reduction potential (–0.94 V vs. Fc+/Fc in 3:1 toluene/CH3CN at –45 °C) [8] and therefore it can be reduced to its radical mono-anion by several reducing agents or by electrolysis [9]. Due to the extensive delocalization of the negative charge over many carbon atoms, the anion is quite stable in the absence of oxygen.
The magnetic nuclei of [60]fullerene that produce hyperfine splittings in the EPR spectra of its anion are the 13C atoms in their isotopic natural abundance. In functionalized fullerenes, the magnetic nuclei could be also those of the addends which, however, present very small hyperfine couplings. Therefore, because of the many non-equivalent 13C atoms that could be potentially present, the 13C hyperfine structure analysis does not give unequivocal results about the electron spin distribution in fullerene anions. We discover that the addition of a nitroxide group to [60]fullerene was a useful way to overcome the above-mentioned problems, thus allowing to map the anion charge distribution through its interactions with the nitroxide unpaired spin density.
Fullerene-nitroxide anions of pyrrolidines 1–4 were prepared by reduction with Na or K in 2-methyltetrahydrofuran [3]. Before reduction, all nitroxides gave EPR spectra made of a triplet of lines of equal intensity due to 14N hyperfine interaction. The isotropic hyperfine constant (aN = 15.35 Gauss) and the g factor (g = 2.0061) were typical of nitroxide radicals in apolar solvent (Fig. 2). Further splitting, by the methyl and methylene protons of the piperidine ring, were resolved only for some of the mentioned derivatives.
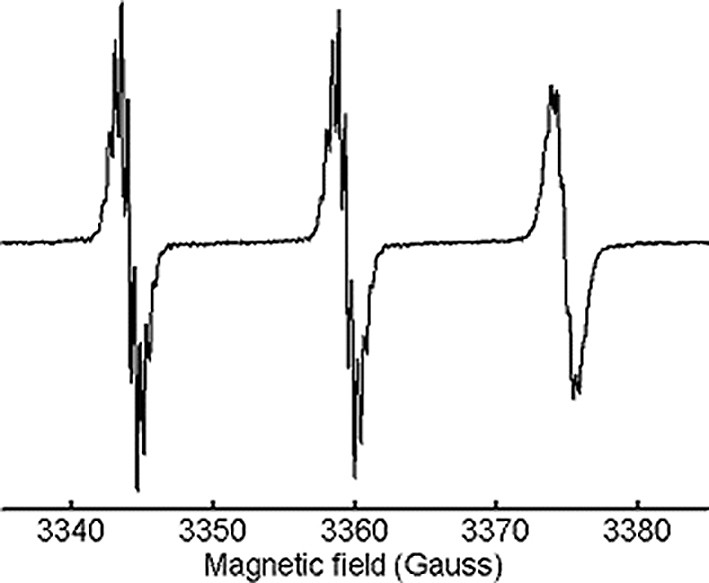
Room-temperature EPR spectrum of 1 in toluene solution.
After metal reduction, an additional 1:1:1 triplet was recorded, with g = 2.0030 and aN = 7.65 Gauss (Fig. 3) that was attributed to a biradical anion species where the unpaired electrons are located on the fullerene and nitroxide moieties, respectively. An aN/2 nitrogen hyperfine splitting has been observed (within experimental errors). This points to an electron exchange interaction J, between the unpaired electrons, much larger than aN [10]. As the reduction proceeds, a single narrow line at g = 1.9999 appeared and eventually was the only one remaining after prolonged contact with the alkali metal. The single line was attributed to the fullerene radical anion after the irreversible reduction of the nitroxide group.

Room-temperature EPR spectra of 1 during the reduction process. Arrows: hyperfine components of neutral 1. Crosses: the signals due to the biradical anion. Points: further reduction product (see text).
The most intriguing conclusion from this study was that the spin distribution in the fullerene-nitroxide radical anions is mostly confined on the equatorial belt of [60]fullerene, as evidenced by the electron–electron and the electron–nucleus dipolar interactions, whose values, obtained from the EPR spectra and theoretical calculations, are in good agreement.
4 Photoexcited states
Upon absorption of UV or visible light, [60]fullerene is excited to a short-lived singlet state, which converts into a triplet excited state by a fast and very efficient intersystem crossing (ISC) process (kISC = 8 × 1010 s–1, quantum yield > 0.9) [11]. This is true also for functionalized fullerenes, but with a triplet quantum yield slightly lower than pristine fullerene [12,13]. The high efficiency of the ISC process, long lifetime and low reactivity of the triplet state make fullerene-nitroxide derivatives particularly suitable for studying a variety of interesting photophysical phenomena.
Several investigations have shown that triplet excited species are quenched in the presence of free radicals, with a mechanism involving the formation of radical–triplet pairs (RTP) [14]. In the pairs, the excited triplet electron spin (S = 1) couples with the radical spin (S = 1/2) generating two states with total spin S = 1/2 (doublet, D*) and S = 3/2 (quartet, Q*), whose energies are separated by the electron exchange interaction J (Fig. 4).
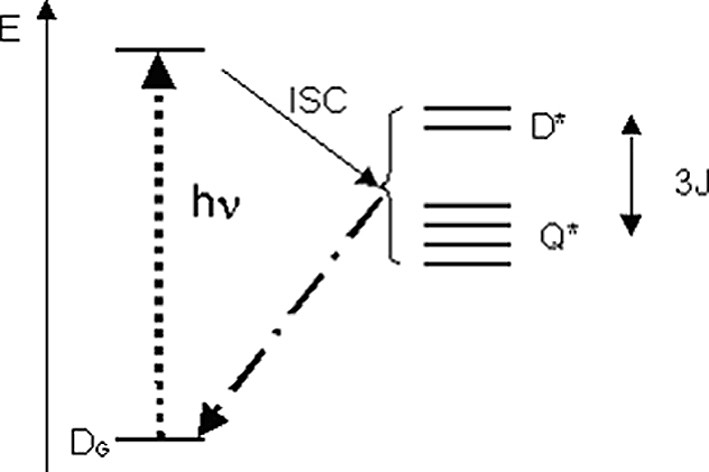
Energy levels of a triplet–radical pair. In the case shown here, the quartet state lies lower than the Doublet i.e. J > 0. The energies are not to scale.
In principle, both D* and Q* can be observed by TR-EPR (TR = time-resolved) in solution, for their characteristic g factors and hyperfine splitting constants. However, for freely diffusing triplet–doublet pairs, the contact time of the partners is usually too short, thus precluding the detection of the coupled states. Their existence was postulated observing the spin polarization effects (see later) on the radical EPR spectrum. With our fullerene-nitroxide derivatives, where the fullerene triplet and the nitroxide radical doublet are in the same molecular structure, the excited quartet state, originated by triplet–doublet coupling, was observed for the first time [3a].
The TR-EPR spectrum of photoexcited nitroxides 1–3 (Fig. 1) in toluene liquid solution consists of six lines (two overlap) belonging to two 1:1:1 triplets with hyperfine separation aN(DG) = 15.2 Gauss, aN(Q*) = 5.5 G and with different isotropic g factors, g(DG) = 2.0061 and g(Q*) = 2.0027 (Fig. 5).
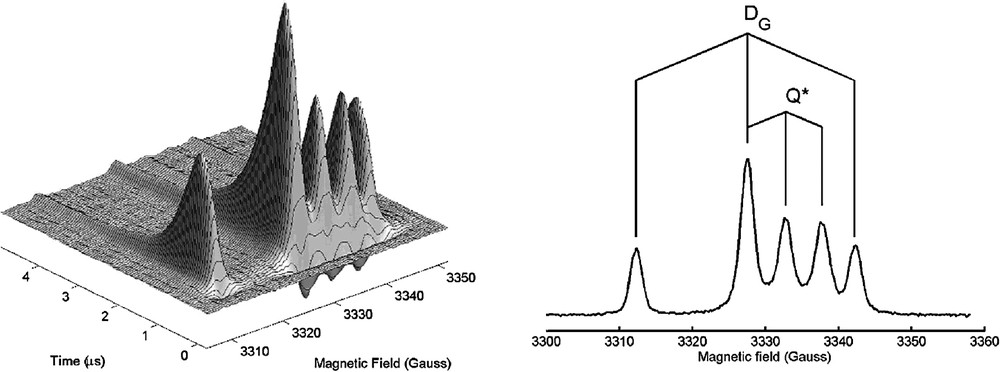
Left: TR-EPR of photoexcited C60-nitroxide derivative 1 in toluene liquid solution at room temperature. The EPR signal is negative (emission) early after the laser pulse, and positive (enhanced absorption) at later times. Right: the spectrum extracted 0.5 μs after the visible laser pulse. DG indicates the doublet ground state, Q* indicates the quartet excited state.
The lines in the TREPR spectra have been assigned to the spin polarized transitions of the ground doublet state DG and of the excited quartet state Q* [3,15]. The hyperfine splitting of the quartet state species is reduced by 1/3 with respect to that of the ground state, as expected if the exchange interaction J is much larger than the isotropic hyperfine coupling of the nitroxide 14N nucleus [16]. In fact, the hyperfine term represents the interaction of the nucleus with a single unpaired electron, which in the quartet state is localized for one-third on the nitroxide and two-thirds on the fullerene sphere. In X-band EPR (microwave frequency about 10 GHz) the central line of the ground state signal and the low-field line of the excited quartet state overlap.
The excited doublet state was not detected by EPR, because it decays very fast to the doublet ground state by a spin allowed process. The quartet decay to the ground doublet state, which is spin prohibited, occurs because of the partial quartet–doublet mixing induced by the dipolar interaction between the triplet unpaired electrons. Since the four quartet sublevels possess different doublet character, they decay with different rates to the ground state, causing a transient deviation of spin sublevels populations from the thermal equilibrium values, both in the quartet state and in the ground state. This effect is called electron spin polarization and is revealed as an anomalous EPR line intensity: spectra in enhanced absorption (A) or even in emission (E) are observed. In some cases, the pattern is more complex and gives multiplet effects, characterized by the low field hyperfine lines in enhanced absorption and the high field ones in emission (A/E) or vice versa [14]. The spin polarization enhances the EPR sensitivity, and therefore excited species can been detected by EPR even if present in solution at low concentration. The spin polarization eventually decays by electron spin lattice relaxation to the Boltzmann equilibrium value, in times of the order of hundred of nanoseconds to microseconds (Fig. 6).

Energy levels diagram and processes responsible for the electron spin polarization in triplet–radical pairs. On the left side, the population processes after the laser pulse (hν). On the right side, the depopulating processes. The thickness of the lines are drawn approximately proportional to the populations of the levels. Abs. = absorption, Em. = emission.
5 Electron transfer
In the field of photoinduced electron transfer (ET), functionalized fullerenes have been widely exploited as electron acceptor units in combination with a wide variety of electron rich chromophores [17]. [60]Fullerene is often selected for its strong electron acceptor characteristics in both the ground and excited states. Upon photoexcitation, the reduction potential of [60]fullerene and derivatives shifts towards less negative values, both for singlet and triplet excited states (1.44 V vs. SCE for 1C60 and 1.01 V vs. SCE for 3C60 if compared with 0.44 V vs. SCE for ground state C60) [18]. In addition, [60]fullerene spherical structure, and its larger size than that of conventional planar acceptors, accelerates the forward ET while retarding the back ET.
Nitroxide spin-labeling proved to be a useful tool for the EPR study of photoinduced intramolecular ET in donor-acceptor fulleropyrrolidine dyads, such as 5 and 6 (Fig. 7).
The EPR signal of the quartet excited state of 5 and 6, which corresponds to triplet fullerene/doublet nitroxide as reported in the previous section, was substantially quenched relative to a suitably prepared model compound 1 (Fig. 1) without ferrocene. Kinetic parameters of the electron transfer process have been obtained by simulating the time evolution of the EPR signal of both the ground electronic state and the excited quartet state that become spin polarized during the excitation and decay paths [4,19,20]. The distance between donor and acceptor, when the photoinduced electron transfer occurs, has been estimated for both dyads on the basis of the Marcus electron transfer theory [21]. In particular, it has been found that the photoinduced ET process occurs faster for the dyad having the longer bridge in the temperature range studied, through the possible formation of an intramolecular exciplex.
6 Exchange interaction
The electron exchange interaction J is an important parameter that characterizes the properties of paramagnetic systems bearing two or more unpaired electrons. It depends on the relative distance and orientation of the electron spins. The value and sign of J, which is related to ferromagnetic or anti-ferromagnetic coupling of the spins, influences molecular properties such as the electron spin polarization pattern, the excited states decay kinetic, and possible magnetic field effects on the reactivity of transient radical species.
For separated radical species, the radical–radical exchange interaction JR–R takes place exclusively through space, possibly mediated by solvent molecules. Although the exact dependence of JR–R from the distance between the radicals is not well established, usually an exponential decay with distance is assumed. On the other hand, JR–R in covalently linked systems, occurs also through the polarization of the bonds between the spins. This through-bond interaction, which depends on the number and nature of the bonds separating the spins, decreases quite rapidly in the case of single bonds.
The exchange interaction JR–R was studied for two types of fullerene-nitroxide derivatives, namely, the bisadducts group 7–11 (Fig. 8) where two 2,2,5,5-tetramethylpyrrolidine-1-oxyl moieties are placed at precise distance on the fullerene spheroid [6], and the fulleropyrrolidine monoadduct 12, in which two TEMPO radicals are covalently linked at positions 2 and 5 of the pyrrolidine ring [7]. The magnitude of JR–R was measured by computer simulation of the EPR spectra and the sign was obtained by recording 14N ENDOR spectra.
Five isomeric bisadducts (out of the eight that in principle can be produced for symmetry reasons) have been isolated and characterized.
The EPR spectra characteristics of the investigated binitroxides (7–12) in their ground state are strongly affected by JR–R. With the exception of the equatorial bisadduct 11, JR–R is comparable to the nitrogen hyperfine coupling aN and could be evaluated precisely. Furthermore, it was found that the absolute value of JR–R for binitroxide 12 decreases as the temperature increases. This was accounted for by the temperature dependent average of contributions from conformations in which JR–R has different signs. Interestingly, for bisadducts 7–11, which have a rigid structure, JR–R was found to be independent from the temperature.
It was found that JR–R is negative (antiferromagnetic coupling) for bisadducts trans-1, trans-2 and trans-3, while it becomes positive in the trans-4 and equatorial isomers. The JR–R sign was correlated with the overlap between the unpaired electron wave function of the two radicals, which depends on the relative orientation of the two nitroxides [7,22]. The knowledge of the factors influencing JR–R is of particular interest because it is related to other electronic couplings involved in important processes such as ET [23].
7 Higher spin states
The successful detection and investigation of the excited quartet states prompted us to study higher spin species obtained upon photoexcitation of fullerene-nitroxides.
Upon photoexcitation of binitroxide 12 in frozen toluene solutions, a strong polarized TR-EPR spectrum was recorded and attributed to an excited quintet state (S = 2) originating from the coupling of the fullerene triplet excited state (S = 1) with the two nitroxide spins S = 1/2. The quintet nature of the excited state was inferred through spectral simulations and by measuring the transient nutation frequency [20]. The spin polarization has been accounted for by the annihilation of the fullerene excited triplet caused by the interaction with the nitroxide spins.
For biradicals 7–11, groups of spin-polarized EPR lines, alternate in absorption and in emission (A/E) or E/A, were recorded upon photoexcitation in liquid solutions. This behavior was accounted for by assuming that the nitroxide spins quenches the excited triplet localized on [60]fullerene through a triplet–triplet annihilation process. Biradicals 7–11 behave as stable spin-correlated radical pairs whose singlet spin levels become selectively overpopulated by the triplet annihilation process. At low temperature in frozen glassy matrix, biradicals 7–11 show TR-EPR spectra characteristics of the fullerene triplet with no coupling to the nitroxide groups [24].
On the basis of these results, we investigated a binitroxide-methanofullerene 13 (Fig. 9).

Methanofullerene-binitroxide.
In this molecule, the strong exchange coupling between the photoexcited fullerene triplet and the biradical moiety allowed us to detect for the first time a fully resolved excited quintet state in liquid toluene at room temperature [25].
Beside the possibility to study photophysical processes involving the excited states, we proved that nitroxide labeled fullerene derivatives provide interesting systems, where spin multiplicities and hence magnetic properties could be controlled by light.
8 Concluding remarks
Labeling functionalized fullerenes with stable nitroxide radicals is a convenient and practical route that allowed us to gain a deeper insight into the fullerene anions and triplet excited state spin distributions by EPR. The nitroxides have been shown to actively participate in the photophysical processes yielding, after laser excitation, a variety of high spin molecular states, such as quartet and quintet states, that were detected for the first time. Our results show that nitroxide labeling paved the way to the investigation of structural and photophysical properties of complex fullerene molecular structures and their photophysics. In this connection, we expect that in a near future newer and more elaborated fullerene–polynitroxide systems will play a significant role in the field of photoswitching and photocontrol of molecular spin and molecular magnetism.
Acknowledgements
We thank all friends and collaborators whose names appear in the references. We express our gratitude to G. Scorrano and M. Prato for their fundamental contribution during the development of our common research on the chemistry of the fullerenes. We gratefully acknowledge financial support from MIUR (PRIN 2004035502, FIRB RBNE01P4JF, FIRB RBAU017S8R) and CNR (ITM-CNR, Padova).