

1 Introduction
Dendrimers [1] constitute nowadays a very attractive field of research in nanosciences, with more than a half of all papers about dendrimers published in the last five years (ca. 7000 publications since 2005). Indeed, after being attractive for their aesthetic structure constituted of branching units emanating radially from a central core (Fig. 1), a number of potential applications have now emerged, in particular as catalysts, for the elaboration of nanomaterials, and even in the field of nanomedicine. Most dendrimers are constituted of organic fragments, and have in particular a nitrogen atom at each branching point, as in the case of the very popular PAMAM (Polyamidoamine) dendrimers [2]. However, besides organic dendrimers, heteroatom-containing dendrimers also possess interesting properties, particularly phosphorus-containing dendrimers [3]. The very first example was described by Engel et al. in 1990 (polyphosphonium dendrimers) [4], and in 1994 we have described the first neutral phosphorus dendrimers [5], and published more than 230 papers in this field to date.
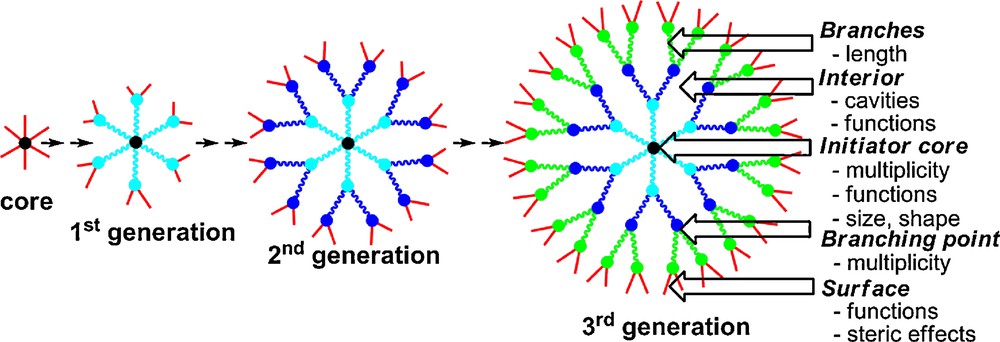
Schematization of the synthesis of dendrimers and description of their main components and characteristics.
This review will focus on the role of phosphorus for the elaboration of dendrimers [6] and of various highly sophisticated dendritic structures [7], and we will emphasize the prominent role played by 31P NMR for their characterization, with numerous examples taken from our own research activities.
2 Phosphorhydrazone dendrimers
2.1 Synthesis and characterization of their internal structure
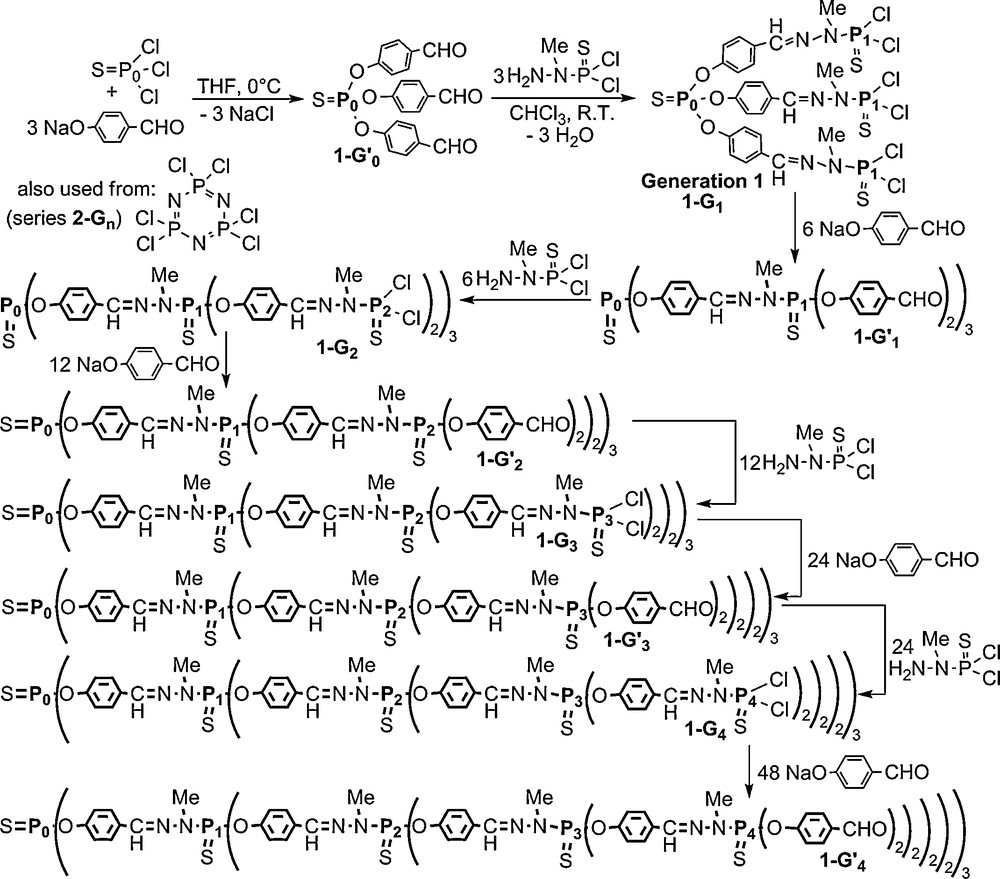
The first type of phosphorus-containing dendrimers that we synthesized has a tetracoordinated phosphorus atom at each branching point. The central core was first P(S)Cl3 (thiophosphoryl trichloride) [5] but it was later replaced by N3P3Cl6 (hexachlorocyclotriphosphazene) [8], which allows increasing more rapidly the number of branches. These dendrimers are built by the repetition of two steps: the nucleophilic substitution of Cl by 4-hydroxybenzaldehyde, and the condensation of aldehydes with the phosphorhydrazide H2NNMeP(S)Cl2 (Scheme 1). Each level of branching points creates a new generation (denoted G for P(S)Cl2 terminal groups, G′ for aldehyde terminal groups); when using P(S)Cl3 as core, this synthesis was carried out up to the 12th generation 1–G12, which is still the highest generation of well defined dendrimers known to date [9], and up to generation 8 when starting from the cyclotriphosphazene core (2–G8) [8]. Both steps are quantitative and necessitate only a very slight excess of reagents (1–3%) which is easily removed by washings; furthermore, the sole by-products are NaCl and H2O. This synthesis is currently carried out in the lab at a 10 g-scale (and in a semi-pilot at a 400 g-scale).
The characterization of dendrimers is never trivial, due to their repetitive structure [10]. In the case of phosphorus-containing dendrimers, the presence of phosphorus atoms at each branching point allows the perfect control of the structure by 31P NMR. Indeed, it is well known that 31P NMR is suitable to characterize even highly sophisticated phosphorus derivatives [11]. In the case of dendrimers, their repetitive structure might hamper any characterization since the chemical environment is strictly identical at all layers. However, due to a slightly different density at each layer, the angles around phosphorus atoms should be slightly modified to optimize the space occupation. This geometric factor, as well as the global environment should induce a slightly different chemical shift for each layer. This is indeed what is observed, as shown in Fig. 2 (in this figure as well as in all the other figures displaying 31P NMR spectra, 1H decoupling was applied). The upper most spectrum is obtained for the fourth generation 1–G’4, and displays five different signals, with the expected intensity for the single phosphorus of the core P0, the three P1 of the first layer, the six P2 of the second layer, the 12 P3 of the third layer and the 24 P4 of the fourth layer [5].
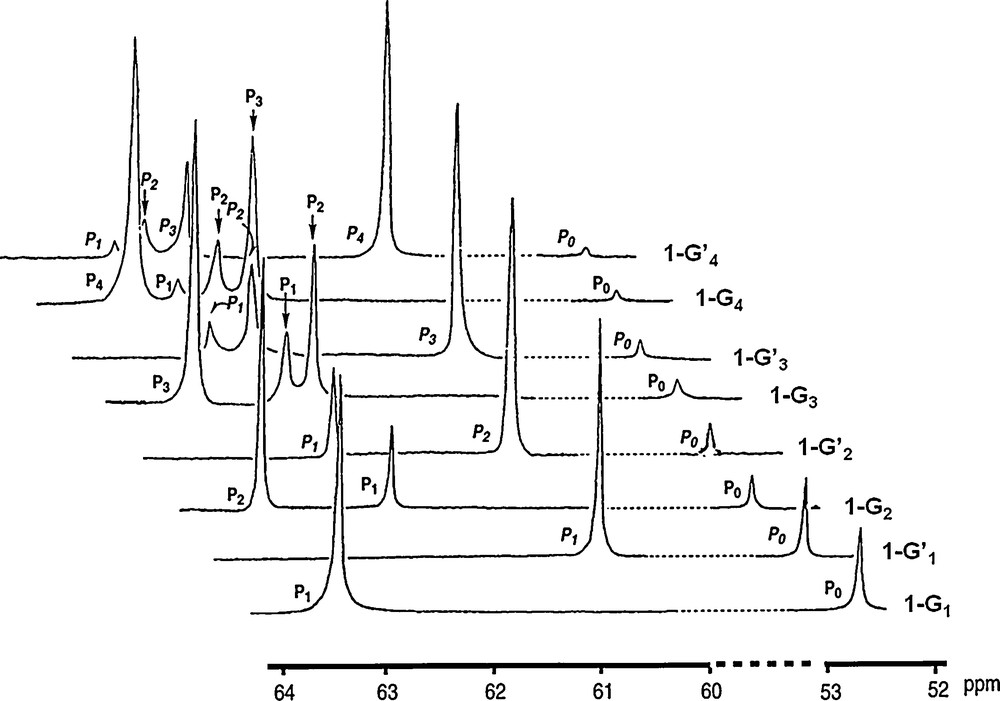
31P NMR spectra of all generations of dendrimers 1-Gn (see Scheme 1 for numbering of P atoms).
Furthermore, 31P NMR is able to monitor the advancement of the reaction during the synthesis. Indeed, the substitution reaction on P(S)Cl2 terminal groups induces first a deshielding of the signal from ca. 63 ppm to ca. 69 ppm when the first substitution occurs, affording P(S)Cl(OC6H4CHO) terminal groups. Then the second substitution induces a shielding when the second Cl reacts to afford P(S)(OC6H4CHO)2 terminal groups at ca. 61 ppm. Thus, any incomplete substitution is easily detectable by such a method. This fact is not really surprising since the substitution reactions occur directly on phosphorus; more surprisingly, the condensation reactions with the phosphorhydrazide that occur at seven bonds from phosphorus are also easily monitored by 31P NMR, and induce a deshielding of the signal from ca. 61 to ca. 62.5 ppm, enabling also to detect indirectly the presence of unreacted aldehydes, which can be also controlled by 1H NMR and IR spectroscopy.
The signal of the core is easily detectable up to generation 6 (1–G6); in this case, there is a single phosphorus atom at the core and 96 phosphorus atoms on the surface [12]; thus, the sensitivity of the 31P NMR technique is better than 1%. All the synthetic process can be monitored in this way by 31P NMR up to the highest generation (generation 12). Of course, in this case not all layers are detectable, but at least the last four layers give reliable signals for which the same shielding and deshielding effects pointed out for small generations are also observed, as shown in Fig. 3 [9]. The fact that the 31P NMR signals of several preceding generations (layers) is observed in all cases allows us to ascertain the purity of these dendrimers, more easily and precisely than for any other type of dendrimers, for which 1H NMR only displays overlap of signals and 13C NMR is not sensitive enough.
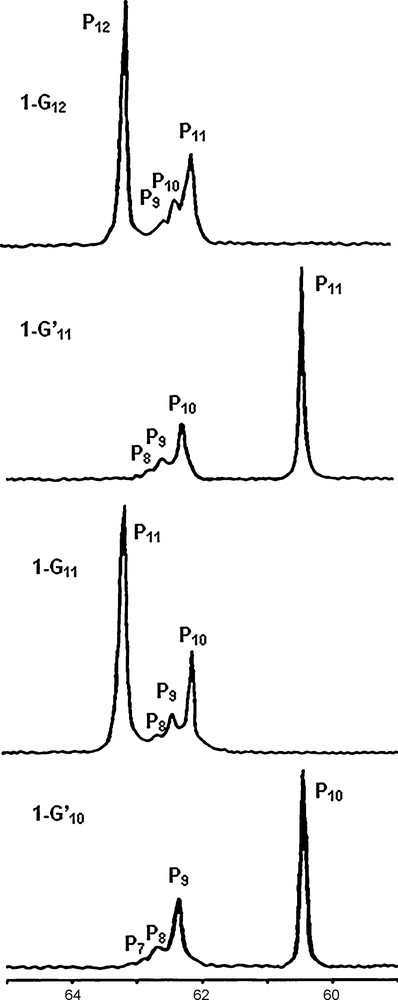
31P NMR spectra of the highest generations of dendrimers, from generation 10 to generation 12 (1-G12). The numbering of the generation and of phosphorus atoms is identical to the one shown in Scheme 1.
At this step, it is important to emphasize that, even if in many cases MALDI-TOF mass spectrometry is believed to be able to ascertain the purity of dendrimers, it does not give reliable results in the case of phosphorhydrazone-containing dendrimers. Fragmentations and rearrangements, in particular at the level of the hydrazone linkages are observed [13]. These fragmentations are due to the laser used to induce desorption of the dendrimers. Fig. 4 displays the result obtained after irradiation at 337 nm (same wavelength than the laser) and analyzed by size exclusion chromatography (SEC). The very important broadening of the SEC signal shows the fragmentations and rearrangements of the structure, and proves that mass spectrometry techniques using an UV laser destroy the structure of these dendrimers.
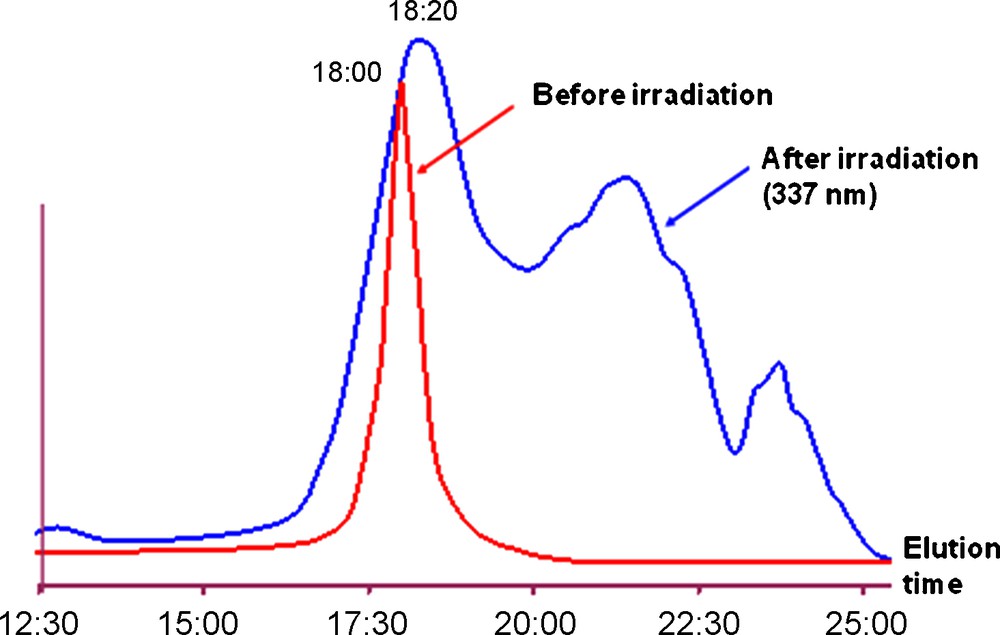
Purity of dendrimer 1-G’4 (see Scheme 1) before and after irradiation at 337 nm and analyzed by SEC.
Besides perfectly regular dendrimers, there exists also some layer-block dendrimers [14], i.e. dendrimers which have at least two types of repeat units within the cascade structure, and organized as layers. We described the first regular layer-block dendrimer 3–G4, which was built with the alternation of PS and PO units [15]. It was obtained by replacing H2NNMeP(S)Cl2 by H2NNMeP(O)Cl2 at some specific layers, using the same method as that shown in Scheme 1. In particular, the P1 and P3 layers possess PO groups, whereas the P0, P2, and P4 layers possess PS groups. The largest compound in this series is the fourth generation 3–G4 shown in Fig. 5. The presence of both phosphoryl and thiophosphoryl groups affords even clearer 31P NMR spectra, compared with the fully thiophosphorylated dendrimers shown in Fig. 2. In particular, the signals of the phosphoryl and thiophosphoryl groups appear in two very different areas. Furthermore, the difference between the chemical shift of P(X)Cl2 and P(X)(OC6H4CHO)2 is much larger for XO (Δδ ∼ 25 ppm; see the difference for P1 between 3–G1 and 3–G’1 or the difference for P3 between 3–G3 and 3–G’3) than for XS (Δδ ∼ 3 ppm). For the generation 4, all signals appear in the expected area for the P0S core (56.2 ppm), the two phosphoryl layers (−5.2 ppm for P1O, −5.9 ppm for P3O), and the two thiophosphoryl layers (62.4 ppm for P2S, 63.1 ppm for P4S) (Fig. 5).
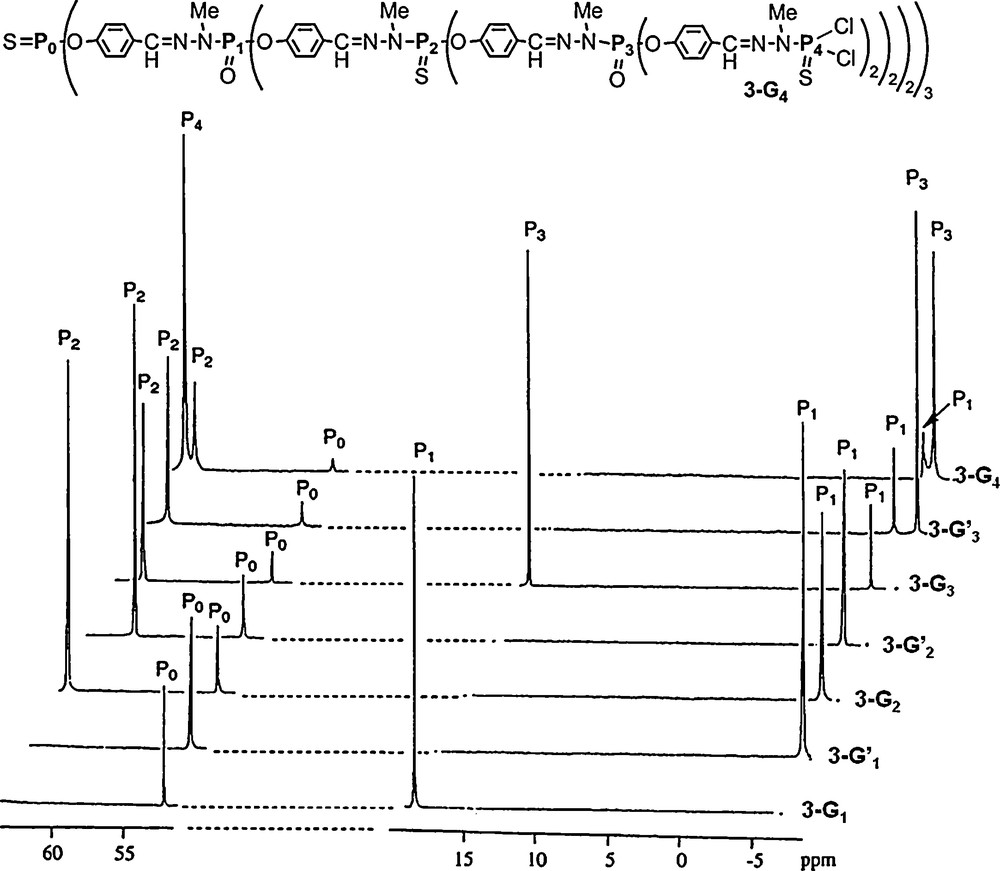
31P NMR spectra of layer-block dendrimers (PS and PO) from generation 1 to generation 4 (3-G4).
The presence of PO groups is interesting for 31P NMR characterization, and also because P(O)Cl2 terminal groups have a higher reactivity than P(S)Cl2 terminal groups. However, this higher reactivity is accompanied by a higher chemical sensitivity, in particular to hydrolysis. Thus, we generally prefer using PS instead of PO derivatives, particularly when searching for potential applications of dendrimers. Other types of modifications of the structure can be introduced, for instance in the branches by replacing hydroxybenzaldehyde by the azobenzene HO–C6H4–NN–C6H4–CHO [16], but also by varying the type of core, using for instance an octafunctional phthalocyanine [17].
2.2 Reactivity of the terminal groups of dendrimers
We have described numerous examples of reactivity on the terminal groups of the dendrimers synthesized as shown in Scheme 1, i.e. dendrimers possessing either P(S)Cl2 or CHO terminal functions. Indeed, most of the properties of dendrimers are related to their types of terminal groups, and the two types of functional groups that we obtain directly during the synthetic process are among the most versatile and reactive functions in phosphorus chemistry and organic chemistry, respectively. We will give here only a few selected examples, which demonstrate also the usefulness of 31P NMR to monitor the reactions and assess their completion.
Starting from the aldehyde terminal groups, we have for instance grafted phosphonate groups on 1–G’n dendrimers, using a mixture Et3N/(EtO)2P(O)H (without solvent). The 31P NMR spectrum of the resulting compound appears surprisingly complex. Indeed, for the first generation 4–G1, beside the singlet corresponding to the phosphorus of the core (51.8 ppm, P0), we observed two signals centered at 21.3 ppm, corresponding to P(O)(OEt)2, and three signals centered at 62.0 ppm, corresponding to the phosphorus of the first generation P1 (Fig. 6A). At first glance, these signals could be due to the formation of three diastereoisomers (one racemic and two meso forms) for each branch of the dendrimer, as the addition of PH groups to aldehydes creates unsymmetrical carbon centers. However, the relative intensity for each set of signals (1:2:1 for δ = 62.0 ppm and 1:1 for δ = 21.3 ppm) and the line separation seems to be in agreement with the presence of a triplet and a doublet, with a coupling constant of 3.9 Hz, which should correspond to the coupling of P1(S) with P(O) through seven bonds! This surprising result prompted us to verify this hypothesis by selective phosphorus-decoupling NMR experiments. The selective irradiation of the signal at 62.0 ppm clearly induces the transformation of the signal at 21.3 ppm from a doublet to a singlet (and vice versa) (Fig. 6B) [18]. These experiments confirm the existence of the 7JPP coupling constant in compound 4–G1. Such a long range coupling constant was very rarely reported in the literature and concerns generally through-space couplings which are unlikely for compound 4–G1 for steric reasons.
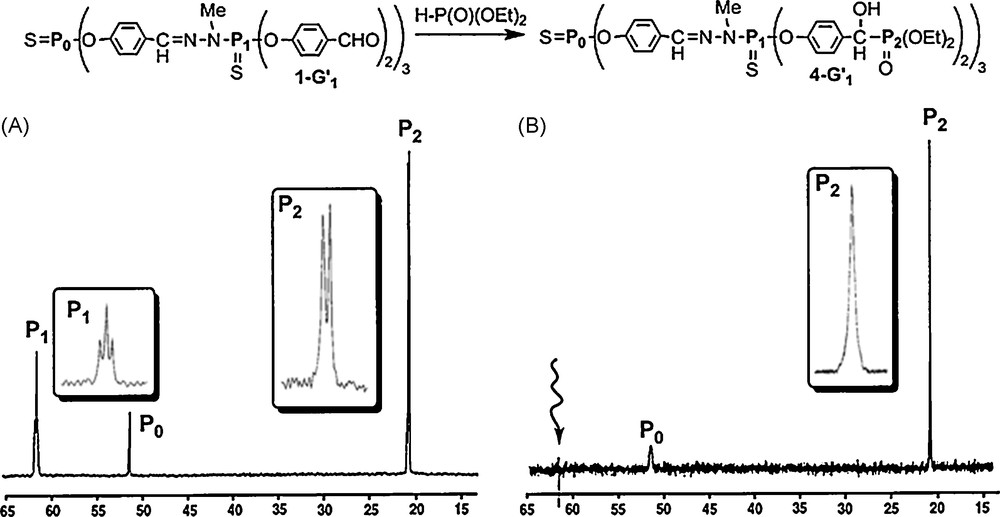
Addition reaction on aldehyde terminal groups. (A) 31P NMR spectrum of the first generation 4-G1. (B) 31P NMR spectrum when irradiating selectively P1.
Another important reactivity of aldehyde terminal groups is the condensation with various amines and hydrazines. For instance, hydrazine used in large excess affords NH2 terminal groups (5–Gn), which can later on be reacted with Ph2PCH2OH (generated from Ph2PH and (H2CO)n) to afford diphosphino terminal groups (6–Gn) [19]. Such a method can be used also with methylhydrazine. The remaining function can be used to open γ-thiobutyrolactone [20], affording thiol-terminated dendrimers, but also to react with Ph2PCH2OH, affording monophosphine terminal groups [21]. This was the very first example of diphenylalkyl phosphino groups grafted from amino terminated dendrimers, and it has been largely exploited later for the functionalization of PPI [22] and PAMAM dendrimers [23].
These terminal groups have the ability to complex transition metal derivatives such as RuH2(PPh3)4, as illustrated in Fig. 7. Due to the presence of two PPh3 and two PPh2 on each terminal unit, an ABMX-type spectrum is obtained (both for the first and third generation of dendrimers 7–G1 and 7–G3) with numerous P–P coupling constants. The largest coupling constant is between the PPh2 and PPh3 in trans position (2JPBPD = 227 Hz), whereas all the other 2JPP coupling constants are in the range 15–20 Hz (Fig. 7) [19].

Synthesis of dendrimers possessing successively as terminal groups primary hydrazone, diphosphine, and Ru complexes. The lower part displays the 31P NMR spectra of the third and first generations of the Ru complexes (7-G1 and 7-G3).
It must be emphasized that complexes of phosphine-terminated dendrimers [24] and organometallic dendrimers [25] in general are often useful catalysts [26], and can display a very positive dendritic effect (increase of the catalytic efficiency with the generation) [27].
Substitution reactions with phenols or amines are mostly used when starting from the P(X)Cl2 terminal groups (X = mainly S, but also O). The reactivity of each Cl of P(X)Cl2 is different, thus two different substituents can be grafted. If these substituents also possess functional groups, the dendrimers obtained have up to four different terminal functions and were called “multiplurifunctionalized” dendrimers [28]. In some cases, the presence of diastereoisomers can be detected by 31P NMR. For instance, we carried out the reaction of a macrocycle bearing a NHCH2CH2NH2 unit on the second generation dendrimer (8–G2) which possesses 12 P(S)Cl2 end-groups, in the presence of a base (Cs2CO3). Both nucleophilic substitutions took place simultaneously, and we could form the expected five-membered ring adduct. The phosphorus atoms are stereogenic centers and diastereotopic protons could be identified through a 2D experiment. Furthermore, 31P NMR displays a singlet for the P(S) groups of the second generation (δ = 67.99 ppm) and a singlet for the core (δ = 8.37 ppm) as expected, while signal of the first generation appeared as a multiplet between δ = 61.90–63.00 due to the stereogenic P. Such a phenomenon was previously observed at the (n − 1) generation for dendrimers bearing uncontrolled chiral entities [18] as end-groups of the n generation (Fig. 8) [29].
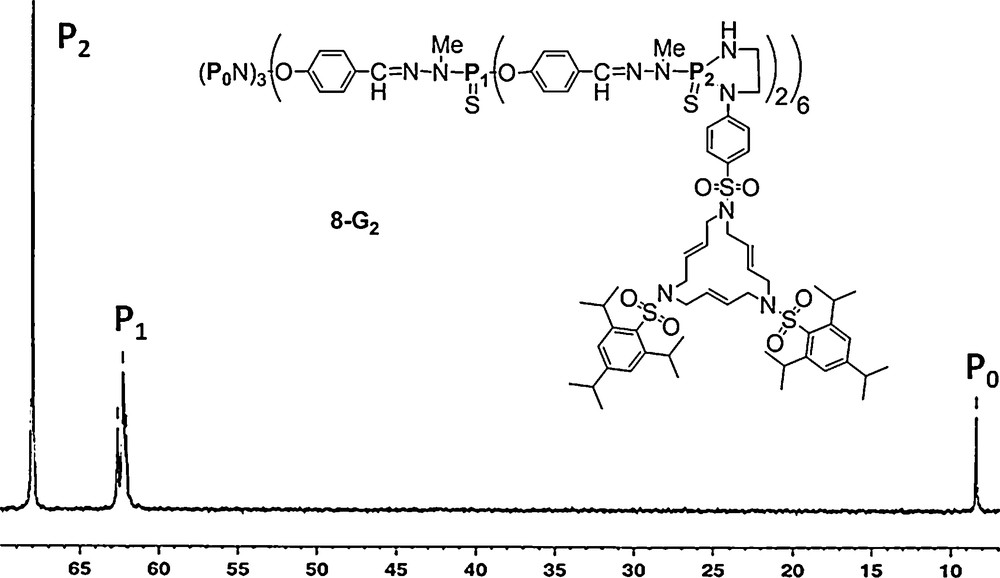
A macrocycle grafted as terminal groups through a diazaphospholane ring on a second generation dendrimer, and its 31P NMR spectrum.
Macrocycles as terminal groups of dendrimers [30] have often interesting complexation properties. The above-mentioned compounds are able to generate platinum nanoparticles and to organize them in organic/inorganic dendritic networks [31]. Related compounds were able to generate palladium nanoparticles, which have catalytic properties and reusability [32]. TTF macrocycles as terminal groups of dendrimers produced sensitive electrochemical sensors able to detect the presence of small quantities of Ba2+ [33]. It must be emphasized that the internal structure of these dendrimers is stable toward electrochemical experiments, and thus enable the grafting of electrochemically sensitive entities, such as ferrocenes [34], as terminal groups [35], but also in the internal structure [36].
3 Synthesis of dendrimers possessing PN–PS linkages inside the structure
3.1 Multistep methods
The method of synthesis of dendrimers shown in Scheme 1 is very powerful, allows an easy modification of the terminal groups, and also the synthesis of layer-block dendrimers as shown in Fig. 5. However, this method does not allow the introduction of real functional groups inside the structure of dendrimers, even if it is desirable to elaborate sophisticated compounds. Here also, phosphorus chemistry offers the very unique opportunity to include chemical functions inside the dendritic structure, able to be activated after the synthesis. For this purpose, we have used the Staudinger reaction between azides and phosphines [37]. Such reactions create PN bonds, which are generally easily cleaved by water (it is a powerful method for obtaining primary amines from azides). However, we have demonstrated that the use of phosphorus azides and particularly thiophosphoryl azides instead of simple organic azides greatly improves the stabilization of the PN bond when included in a PN–PS linkage, due to the delocalization and polarization as P+–NP–S− [38]. The Staudinger reaction is generally quantitative, and generates only N2 as by-product; thus its use for the synthesis of dendrimers should be very powerful.
The first example of the synthesis of dendrimers including such linkage that we have performed necessitates three steps to build one generation. Starting form a hexaaldehyde core 2–G’0, the first reaction is the condensation with methylhydrazine, which affords 9–G’0. The second step is the substitution reaction of the remaining proton of 9–G’0, which allows the grafting of diphenylphosphino groups to yield 9–G”0. The third and last step of the reiterative process is a Staudinger reaction between the diphenylphosphino groups of 9–G”0 and the azide function of a thiophosphoryl azide dialdehyde, which creates the PN–PS linkages and multiply by two the number of terminal (aldehyde) groups, affording the first generation 9–G1 (Scheme 2). The repetition of this sequence of three reactions, using successively methylhydrazine, diphenylchlorophosphine and the phosphoryl azide CD2 provides the second generation 9–G2, then the third generation 9–G3 [39].
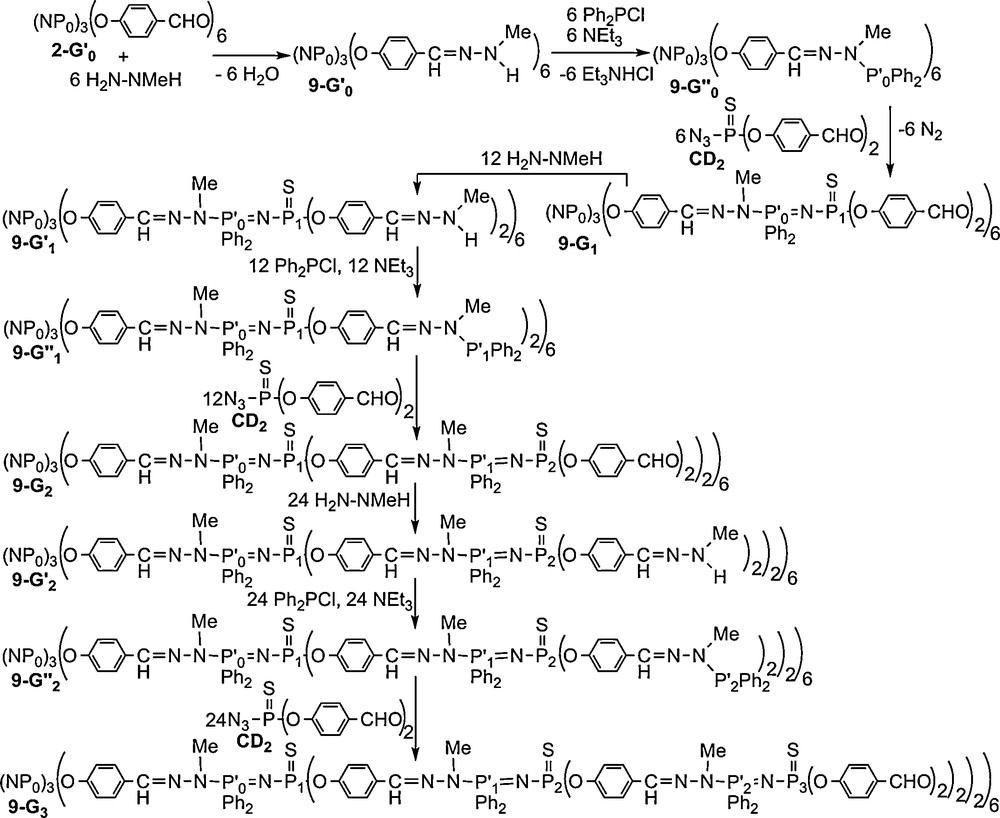
Each step of the synthesis was monitored by 31P NMR, at 81.01 MHz for the lowest generation and 32.43 MHz for the highest generation, this latter field inducing a better separation of the doublets corresponding to the PN–PS linkages (Fig. 9). The spectra of dendrimers 9–G’n (n = 0–2, phosphine terminal groups) display, beside the signals of the inner layers, a singlet at δ = 67.7 ppm corresponding to the aminophosphine end-groups. The Staudinger reaction (9–G”n → 9–Gn+1, n = 0–2) induces in all cases the total disappearance of this signal and the appearance of two doublets at ca. 22 ppm (Ph2P groups) and ca. 48.7 ppm (PS groups) with a coupling constant 2JPP = 34 Hz. Remarkably, and as observed previously for the other series of phosphorus dendrimers, all the layers of phosphorus atoms of the third generation dendrimer 9–G3 are distinguishable. Indeed, the spectrum of 9–G3 consists of one singlet for the three phosphorus of the cyclotriphosphazene core P0 and three sets of two doublets corresponding to the Ph2PN–PS moieties of the first, second and third generations (Fig. 9) [39].
31P NMR spectra of the dendrimers 9-Gn shown in Scheme 2.
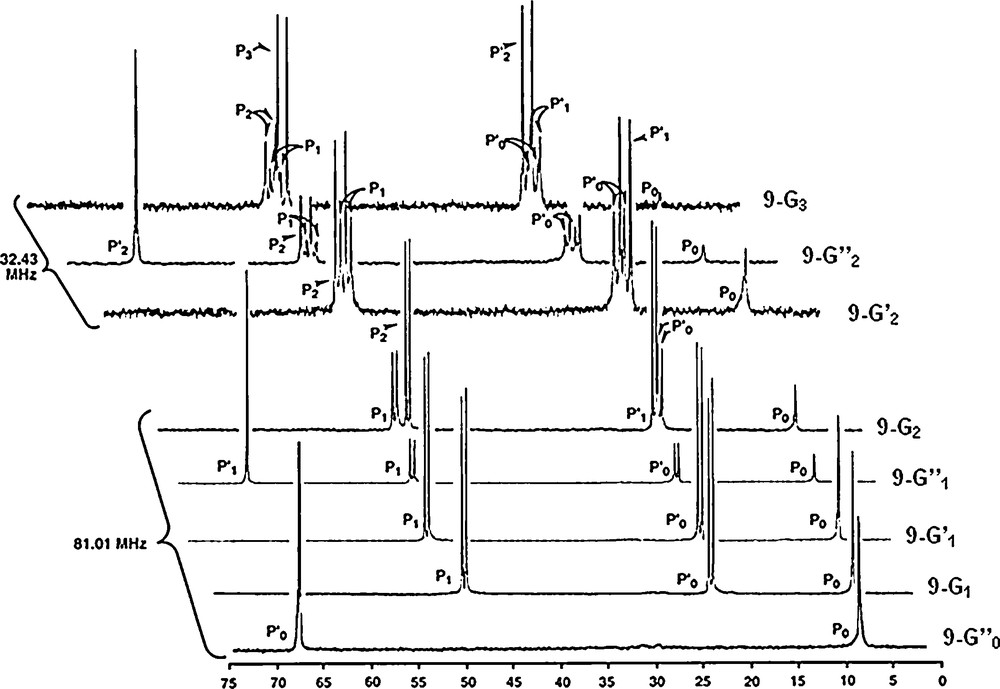
A spectacular use of the Staudinger reaction allowed us to amplify topological differences in a tetraphosphorus macrocycle [40], for which diastereoisomers were expected but never detected. The first step consists in Staudinger reactions between the four azides of the macrocycle 10 and four equivalents of triphenylphosphine monoaldehyde. Such reaction creates four PN–PS linkages (10–G0). A dendron possessing a NH2 group at the core reacts readily with the four aldehyde functions of the macrocycle in a second step, to afford an original example of dendrimer having a macrocycle as the core (10–G2) [41].
31P NMR spectroscopy appears here also as a very useful tool to monitor these reactions. The spectrum of the starting macrocycle displays two sharp singlets (one for the O–P–O linkages, and the other for the N–P–N linkages). The Staudinger reaction induces the appearance of two systems of two sharp doublets, due to two types of PN–PS linkages (PS linked to two O, or to two N). Even at this step (10–G0), it is impossible to detect the presence of any isomer of the macrocycle. In sharp contrast, the grafting of four dendrons has a dramatic influence on the shape of the 31P NMR signals corresponding to the same PN–PS linkages. Indeed, numerous signals are observed, typical of “frozen” structures. Thus, one may attribute the phenomenon observed to the existence of diastereoisomers of the macrocycle, which becomes detectable for the first time thanks to the dendrons (Fig. 10) [41].
31P NMR spectra of a tetraphosphorus macrocycle bearing as R substituents either an azide (10, left), or a bulky triphenylphosphine derivative (10-G0, middle), or a second generation dendron (10-G2, right).
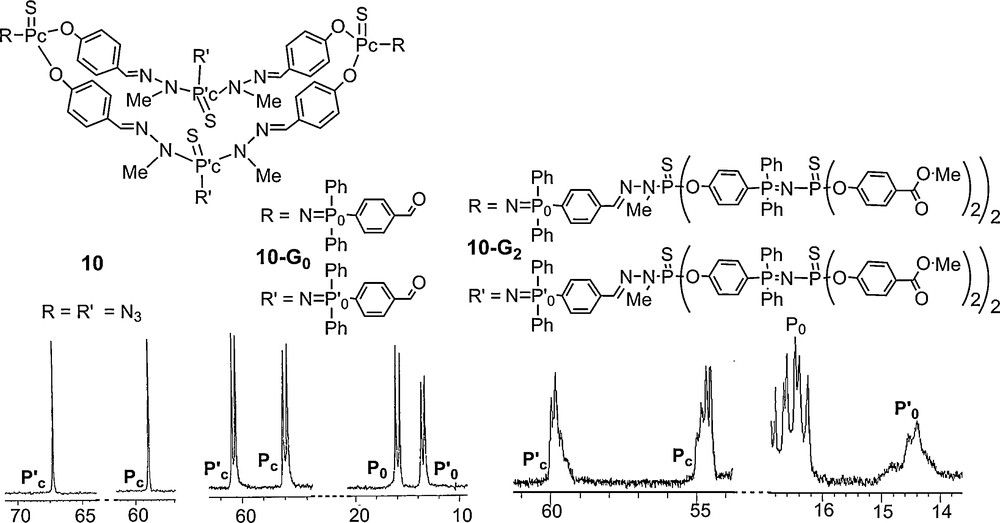
In the previous examples, the PN–PS linkage is used in connection with a phosphorhydrazone linkage, but we have also synthesized phosphorus dendrimers without phosphorhydrazone, using only a protected branched monomer. The CB’2 monomer possesses an azide (C) and two phosphines protected by BH3 (B′). In the first step, this branched monomer is reacted with a phosphine such as PPh3, to create the PN–PS linkages. The second step is the deprotection of the phosphines using DABCO (1,4-diazabicyclo[2.2.2]octane). The free phosphines thus generated are used to react again with the CB’2 monomer, in a repetition of the first step, and so on. In order to increase the number of terminal groups more rapidly, the tetraphosphine CB’4 is used in the last step to afford dendrimer 11–G’3 (Scheme 3) [42]. This method was also applied starting from a triphosphine as core instead of PPh3 [43].

Despite the repetitive structure of this series of compounds and the presence of multiple PN–PS linkages which should generate numerous sets of two doublets, even the 31P NMR spectrum of the third generation 11–G’3 remains interpretable, and all signals could be assigned as shown in Fig. 11. Indeed, besides the large signal corresponding to the free phosphine terminal groups, four sets of two doublets, with the expected intensity are observed, corresponding to the PN–PS linkages of the core, the first, the second and the third generation [42].
31P NMR spectrum of a third generation dendron possessing OC6H4P(Ph)2N–PS linkages as branches.
3.2 Uses of PN–PS linkages for accelerated methods of synthesis of dendrimers
The methods shown in Schemes 2 and 3 are interesting because they provide PN–PS linkages inside the dendritic structure, but they are lengthy processes which necessitates 3 (Scheme 2) or 2 (Scheme 3) steps to multiply by two the number of terminal groups. However, we have discovered a way to use the PN–PS linkages for accelerated methods of synthesis of dendrimers, in which the terminal groups are multiplied at each synthetic step. For this purpose, we have synthesized two types of branched units (AB2 and CD2) which contain two pairs of complementary coupling functionalities. This type of “orthogonal system” implies a set of completely independent class of functional groups, and was never used previously for the divergent synthesis of dendrimers. Furthermore, we choose to use two pairs of complementary functions able to react spontaneously without any activating agent. This last point is highly desirable, since it allows the growing of dendrimers without purification, if each reaction is quantitative. We have shown that the condensation of aldehydes with phosphorhydrazides (Scheme 1) and the Staudinger reaction between phosphine and azides (Scheme 2) are both quantitative, do not necessitate activation or protecting groups, and generate only benign by-products (H2O and N2, respectively). Having in hand these reactions, the goal was to design AB2 and CD2 monomers, one with hydrazine and phosphine functions, the other one with aldehyde and azide functions. The AB2 monomer is H2NNMeP(S)(OC6H4PPh2)2 with A = NH2 and B = PPh2. The CD2 monomer is N3P(S)(OC6H4CHO)2 with C = N3 and D = CHO (already seen in Scheme 2). Starting from a trialdehyde core (type D3), the first step is a condensation reaction with the AB2 monomer, which affords the first generation (12–G1, 6 phosphine terminal groups). The next step is the Staudinger reaction between 12–G1 and 6 equiv of the azide CD2, which affords the second generation 12–G2 (12 aldehyde end-groups). The dendrimer is grown using again the AB2 monomer, then the CD2 monomer (Scheme 4). Thus, the synthesis of the fourth generation 12–G4 having 48 aldehyde end-groups, necessitates only four steps from the D3 core. The compounds obtained are layered dendrimers made of O–C6H4–Z–P(S) linkages, the difference between two layers being the nature of Z, CHN–N(Me) or Ph2PN groups [44].
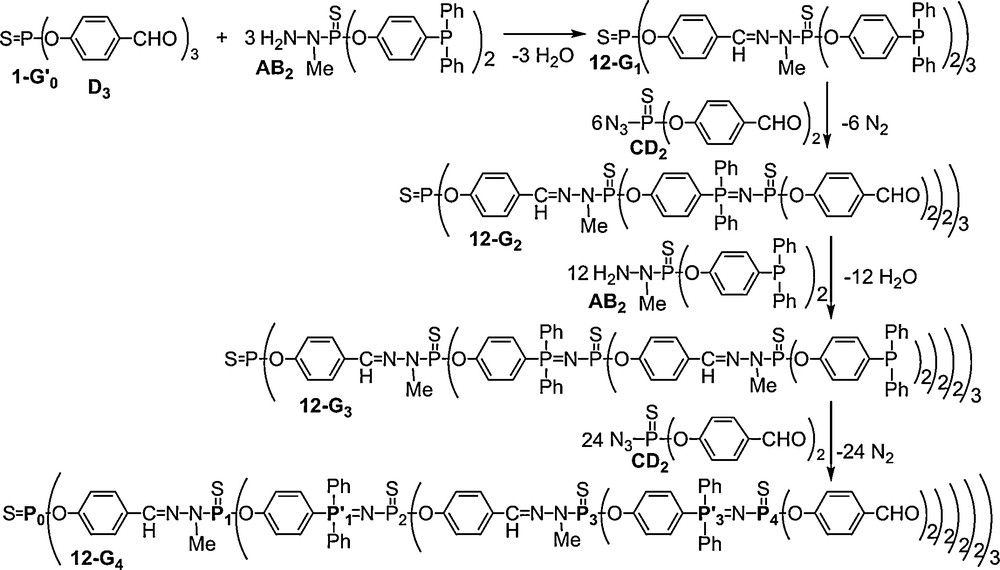
31P NMR is here also the most powerful technique for monitoring these reactions. Indeed, the condensation reaction induces the shielding of the signal of the PS group of the phosphorhydrazide from δ = 67.0 to 61.6 (for 12–G1) or 61.9 (for 12–G3) ppm, whereas the Staudinger reaction induces the appearance of a set of two doublets at δ = 14.3 (PN) and δ = 50.2 ppm (PS) (Fig. 12).
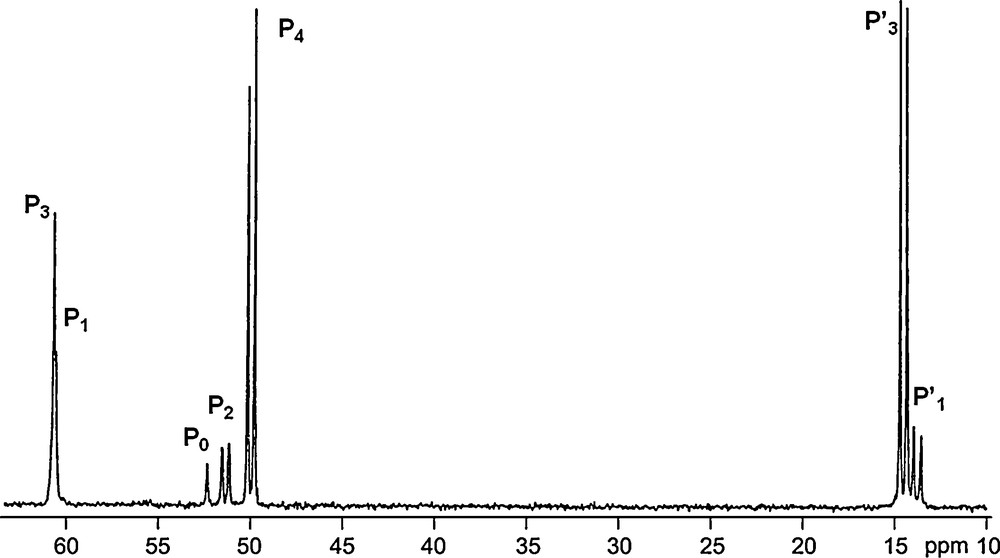
31P NMR spectrum of the dendrimer 12-G4 shown in Scheme 4.
In view of the nice results obtained using this step-by-step process, in which the compounds are isolated at each step, we have carried out a one-pot (but multi-step) experiment to obtain directly the fourth generation starting from the core. Two aspects of the synthesis require particular care: (i) this type of synthesis; (ii) the condensation step is slower during the one-pot process; thus, it is advisable to concentrate the solution before adding the AB2 monomer. The characterization by 31P NMR (but also by size exclusion chromatography) of dendrimer 12–G4 obtained by the one-pot process are very similar to those described for 12–G4 obtained step-by-step, showing that the purity is practically identical in both cases [44].
We have reported another straightforward synthesis of dendrimers derived from the same concept, but using CA2 and DB2 monomers, i.e., monomers whose reactive groups are inverted compared to the AB2 + CD2 method (with A, B, C, and D having the same meaning as in Scheme 4). In this case, the core is of type B3 (triphosphine), and the first step is the Staudinger reaction with the CA2 monomer to afford the first generation 13–G1 in one step. Starting from 13–G1, the condensation reaction with DB2 occurs readily overnight at room temperature, to afford the second generation 13–G2. Using again the CA2 monomer to react with 13–G2 affords 13–G3 after heating for 6 days at 35 °C (the same conditions than those used previously for 13–G1). Starting from 13–G3, the fourth generation 13–G4 is obtained by the condensation reaction with the DB2 monomer (Scheme 5).
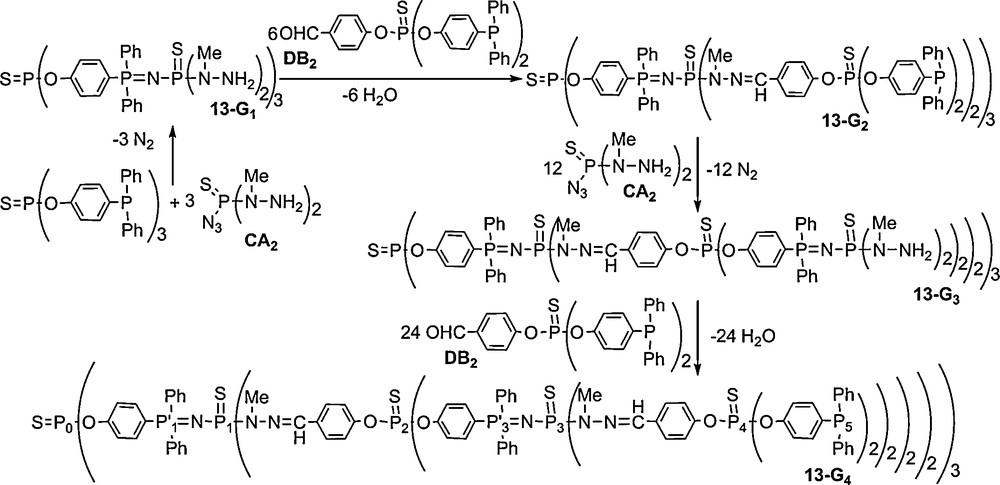
All these compounds (13–Gn) are isolated in nearly quantitative yields, and they are characterized particularly by 31P NMR. Fig. 13 displays the 31P NMR spectrum of the fourth generation (13–G4, 48 phosphine end-groups), in which all the signals corresponding to the eight types of phosphorus atoms are clearly distinguishable, including the signal corresponding to the single phosphorus of the core (P0), and the two sets of two doublets corresponding to the PN–PS linkages of the first and the third generations (ca. 10 ppm for the PN groups (P’1 and P’3) and ca. 57 ppm for the N–PS groups (P1 and P3)). Besides the signal corresponding to the phosphine end-groups (δ = −6.0 ppm), the signals of the thiophosphate groups (P2 and P4) of the internal skeleton appear at ca. 50 ppm [45].
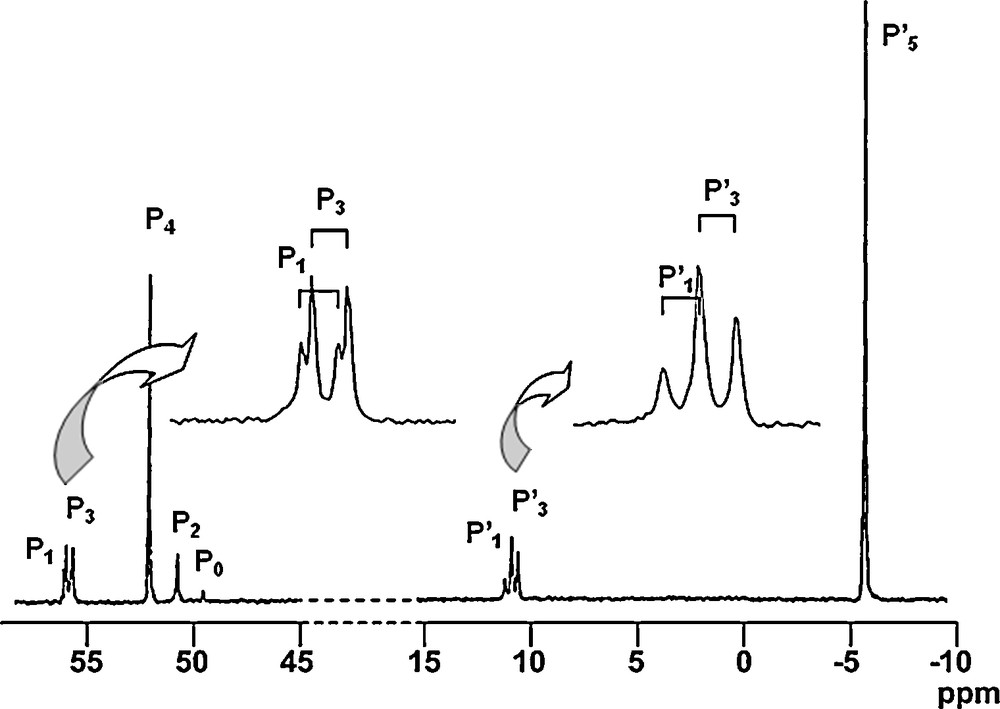
31P NMR spectrum of the fourth generation 13-G4 (see Scheme 5 for the numbering).
The previous approach is very attractive for decreasing the number of steps to attain a given generation, but it does not allow a significant increase in the number of terminal groups. This last point is highly desirable since it is known that properties and applications of dendrimers strongly depend on the nature, but also on the number and density of functional groups on the surface. To address these problems, we have designed highly functionalized new monomers AB5 and CD5, built from hexachlorocyclotriphosphazene (N3P3Cl6). The A, B, C, D functions are the same as previously. The core is the D6 monomer, which is first reacted with the AB5 monomer, to afford the first generation 14–G1 ended by 30 phosphino groups. These functional groups remain accessible to further transformation since the addition of the CD5 monomer allows obtaining the second-generation dendrimer 14–G2, bearing 150 aldehyde groups on the outer shell. Lastly, treatment of this dendrimer with the AB5 monomer leads to the third generation dendrimer 14–G3 bearing 750 phosphino end-groups, and obtained in only three steps (Scheme 6). After the same number of steps, and starting from a hexaaldehyde core, the number of terminal groups is 24 by the method shown in Scheme 1, 12 by the method shown in Scheme 2, and 48 by the method shown in Scheme 3, illustrating the high performance of the AB5/CD5 method to increase rapidly the number of terminal groups [46].
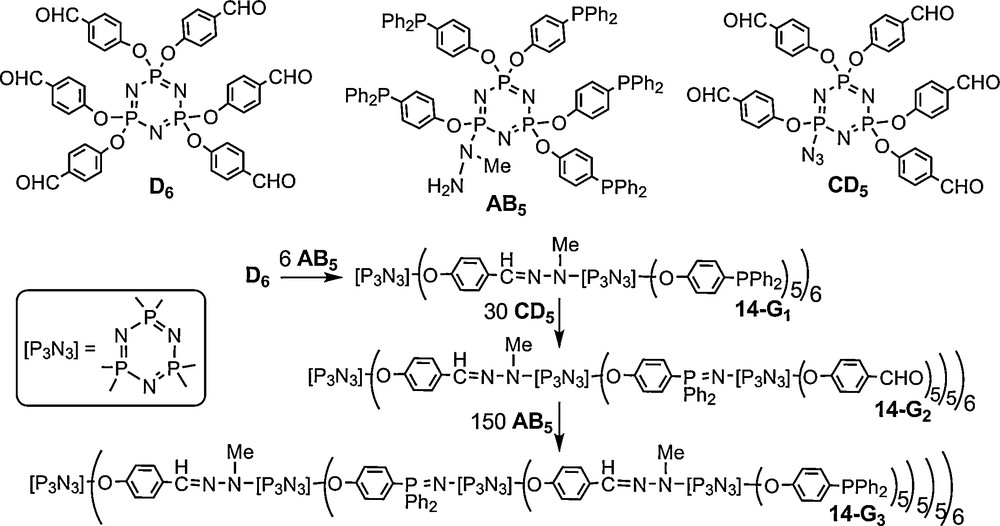
Obviously the AB5 and CD5 monomers can be used in various combinations with the AB2 and CD2 monomers. One example obtained from D6 + 6 AB2 + 12 CD5 is displayed on Fig. 14, together with its 31P NMR spectrum (excepted P1 at δ = 64.4 ppm). Due to non-symmetrical substitutions, the three phosphorus atoms of the cyclotriphosphazenes of the second generation 15–G2 are all different and coupled differently, inducing a very complex 31P NMR spectrum. However, a theoretical 31P NMR spectrum perfectly fits with these complex features, confirming the structure [46].
31P NMR spectrum of the second generation dendrimer 15-G2 possessing 60 terminal groups (the singulet of P1 at 64.4 ppm is not shown).
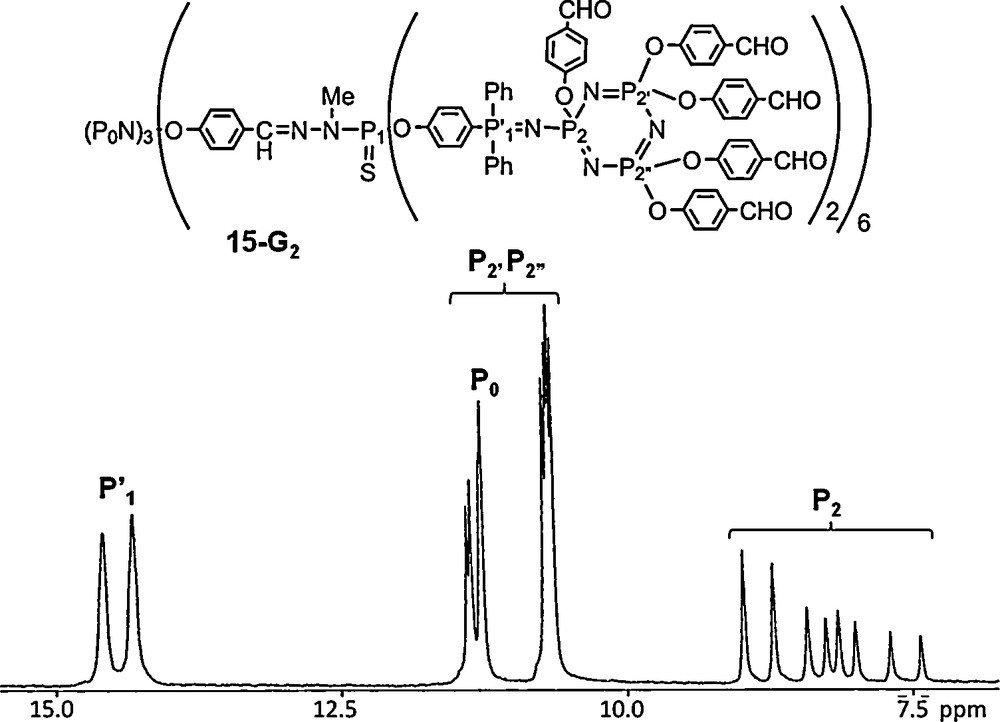
The compounds shown above benefit from the possibility to specifically functionalize hexachlorocyclotriphosphazene. In some recent examples, we have used such a property for the synthesis of “Janus” dendrimers (two types of terminal groups) [47], of dendrimers bearing a fluorophore “off-center” [48], or a fluorophore having two-photon (TP) absorption properties as core [49]. In all cases, the starting reagent is N3P3Cl6. In particular, two units were reacted with a TP fluorophore as shown in Fig. 15 giving rise to compound 16–G0 which displays the expected AB2-type spectrum in 31P NMR. Starting from the remaining P–Cl functions, the synthesis of the bis-dendritic structure was carried out up to the third generation using the method shown in Scheme 1, and in the final step, ammonium groups were grafted as terminal groups, to ensure the solubility in water. The protecting dendrimer shell is indeed successful, leading to nanometric water-soluble fluorescent TP active tracers, having strong TP absorption and strong emission in the blue-visible region in water. The second generation was injected intravenously to a rat and allowed two-photon imaging of the vascular network in the dorsal part of its olfactory bulb [49].
31P NMR spectrum of the precursor of dendrimers having two-photon absorption properties, usable for in vivo bio-imaging.
In the search for dendrimers able to interfere with the human immune system, we have developed a structure/activity relationship and synthesized a series of dendritic structures having phosphonic acid terminal groups. In a particular case, we wanted to study the influence of the density of terminal groups, and thus we have protected successively one to five functions emanating from the cyclotriphosphazene. Fig. 16 displays the compound 17–G0 in which only three Cl among six of the cyclotriphosphazene core are usable to grow the dendritic branches. This compound possesses three differently functionalized phosphorus atoms, and gives rise to an ABM-type spectrum, in perfect correlation with its simulated spectrum [50].

Selective functionalization of the cyclotriphosphazene derivative 17-G0, giving rise to an ABM-type 31P NMR spectrum.
3.3 Reactivity of PN–PS linkages for the elaboration of highly complex structures
As indicated earlier, the PN–PS linkages are polarized with a negative charge on sulfur, and thus they are able to react with strong electrophiles such as alkyl triflates [51]. The alkylation occurs selectively on sulfur atoms included in PN–PS linkages and not on the other PS groups of the molecule [52]. This alkylation induces a weakening of the P–S bond, which can be desulfurized by the nucleophilic phosphine P(NMe2)3, to generate a tricoordinated phosphorus atom [53] able to react again in a Staudinger reaction. By varying the type of functional groups linked to the phosphorus azide, and by repeating the three-step reaction (alkylation, desulfurization, Staudinger reaction), we have obtained a multifunctional oligophosphazene (Scheme 7) and characterized all products, including the largest one by 31P NMR (Fig. 17). Obviously, the spectra are complicated by the presence of numerous P–P coupling constants, which may occur through several bonds as already illustrated in Fig. 6. Interestingly, a possibility of delocalization through several phosphazene linkages may occur in the case of the charged species 19–(PN)x (after alkylation). Indeed, the chemical shift of the phosphorus atom of the Ph3P group of the beginning of the chain (noted P0) is sensitive to the alkylation that occurs on sulphur. Fig. 17 displays the variation of the chemical shift value of all phosphorus atoms for each compound from 18–(PN)1 to 18–(PN)4 and from 19–(PN)1 to 19–(PN)3. The behavior of the P0 atom is totally different from that of P1, P2, and P3 atoms, where chemical shifts vary from ∼50 ppm in the PS form (18–(PN)x) to ∼24 ppm in the P–S–Me form (19–(PN)x) and ∼−15 ppm in the PN form (18–(PN)x+1), and remain almost constant for all the subsequent reactions. In marked contrast, the chemical shift of P0 varies at each step of the building of the oligophosphazene. It oscillates around 17 ppm, with smaller amplitude when the alkylation is remote. The phenomenon remains sensible even for the 18–(PN)3 → 19–(PN)3 reaction, which occurs at seven bonds from P0. Thus, the charge introduced during the alkylation step is delocalized throughout the inorganic phosphazene chain, but with a decreasing effectiveness as the site of alkylation moves away from the beginning of the chain [54].

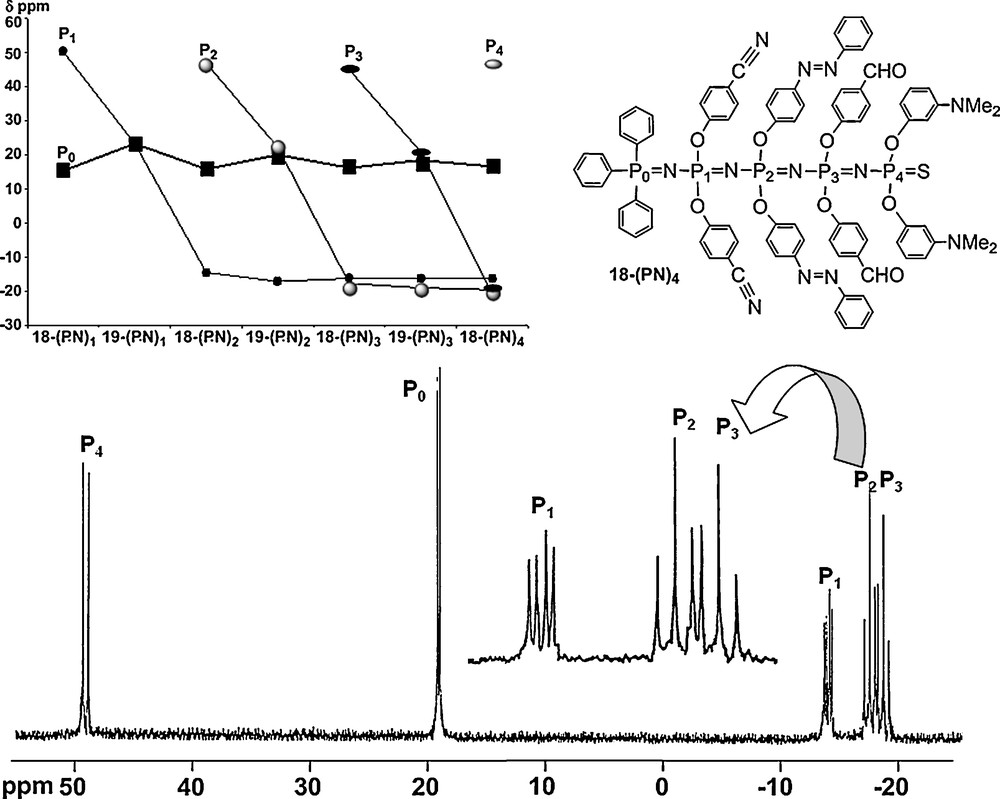
31P NMR spectrum of the linear multifunctionalized polyphosphazene 18-(PN)4, and variation of the 31P NMR chemical shift for all compounds shown in Scheme 7.
The same three-step method was also used for the elaboration of one of the most sophisticated type of dendritic structures known to-date (Scheme 8). It is based on the use of PN–PS internal functional groups of a dendrimer. The synthesis of the central dendrimer is made using the combination of a three-step reaction for the synthesis of the first generation, and the two-steps method shown in Scheme 1 for the growing of the structure. The synthesis of the first generation is carried out using first a condensation reaction between the hexaaldehyde core, then a Mannich-type reaction between the resulting product and six equivalents of the phosphane Ph2PCH2OH, giving an hexaaminophosphane, and finally a Staudinger reaction between this latter compound and the CD2 azide N3P(S)(OC6H4CHO)2 to form the first generation dendrimer, possessing six PN–PS groups. From the first generation, the construction of dendrimers of generation 2 and 3 involves the reiteration of the sequence of two reactions shown in Scheme 1, a condensation reaction between the polyaldehyde with Cl2P(S)N(Me)NH2, followed by addition of the sodium salt of hydroxybenzaldehyde. Starting from this compound 20–G3, the three-step sequence of reactions shown in Scheme 7 was applied, i.e. the alkylation of sulfur by methyl triflate, which occurs only on the PS groups included in PN–PS linkages, the desulfurization with P(NMe2)3, and the Staudinger reaction with the CD2 azide. This sequence of reactions introduces new functional groups (12 aldehydes) inside the structure of the dendrimer 20–[G1@G3]. Starting from the aldehyde internal groups, we have grown new branches inside the dendritic structures, using either the two-step method shown in Scheme 1, or the three-step method outlined in Scheme 8. The synthesis was carried out up to 20–[G4@G3], in which the newly synthesized branches become larger than the initial dendrimer [55].
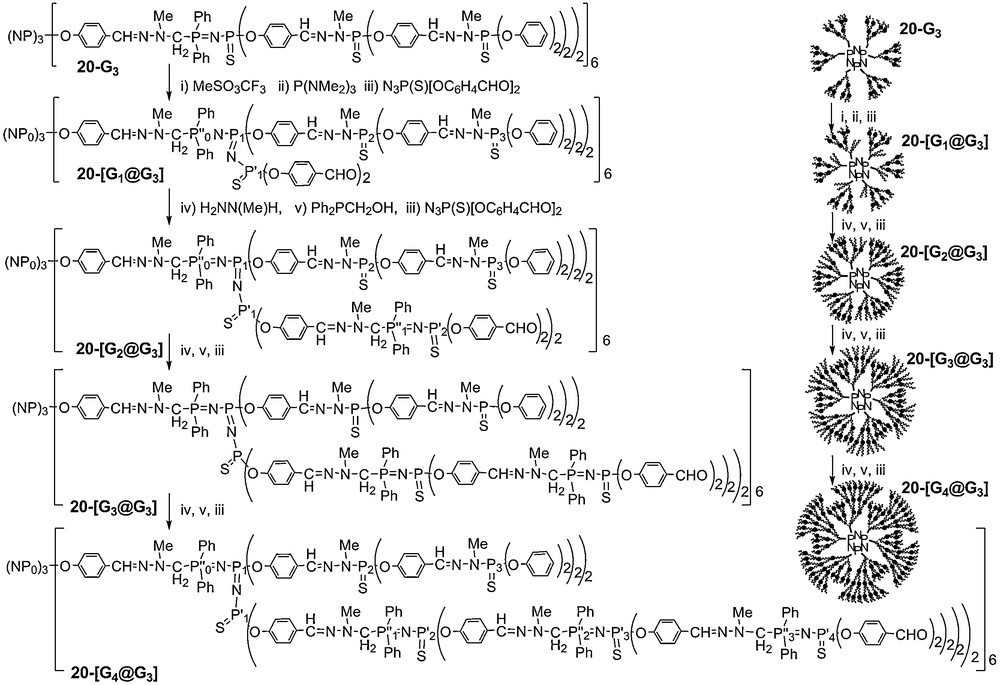
Here also, 31P NMR was used to monitor the construction of these controlled polydendritic structures. N–N–P–O fragments give a singlet, PN–PS linkages give a doublet of doublet (2JPP = 31 Hz); and the PN–PN–PS units give a doublet of doublet for the central phosphorus atom (2JPP = 22 to 23 Hz and 58 to 59 Hz) and a doublet for each of the two other phosphorus atoms (Fig. 18). It must be emphasized that despite the complexity of the structure, all the expected signals can be detected, and with the expected intensity and multiplicity, even for 20–[G4@G3] [55].
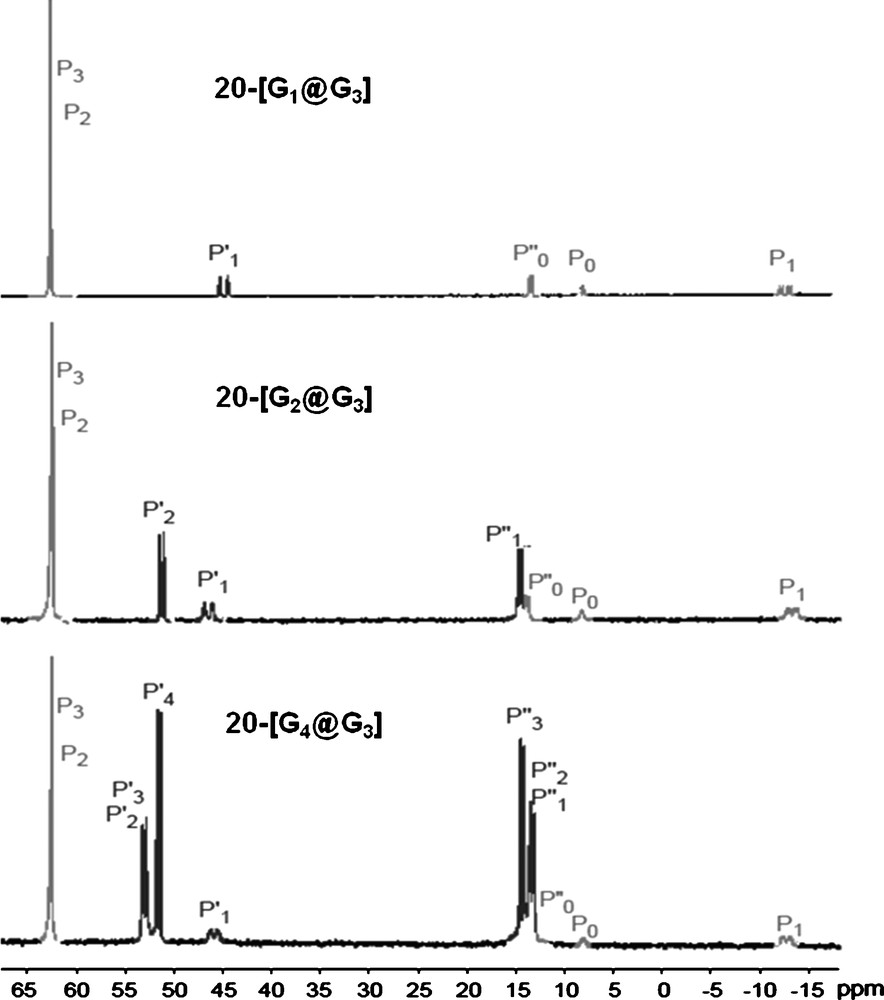
31P NMR spectra of dendrimers 20-[Gn@G3] having two types of internal branches (see numbering in Scheme 8). The light grey signals concern the initial structure.
This methodology allowed the development of an original and versatile chemistry in the interior of the dendrimers [56]. In particular, we have functionalized the interior with pendant arms bearing zwitter-ions [57], fluorescent groups [58], and various metallic derivatives [59]. For instance, the two different types of branches constituting these compounds have a different reactivity, due to the presence or not of PN–PS linkages. Such behavior is particularly illustrated by the reaction with Au–Cl(tht) (tht = tetrahydrothiophene). Indeed, even with an excess of reagents, gold is complexed only by sulfur included in the PN–PS linkages, and not by the other PS groups of the dendrimer. This fact is clearly illustrated by the 31P NMR spectrum of 21–[AuG4@G3], in which all the PS signals of the PN–PS groups (P’1,P’2,P’3,P’4) are shielded from 53–45 ppm in the free dendrimer to 37–30 ppm in the Au-complex. None of the other signals undergo noticeable changes, including the other PS groups (P2 and P3) (Fig. 19) [60].
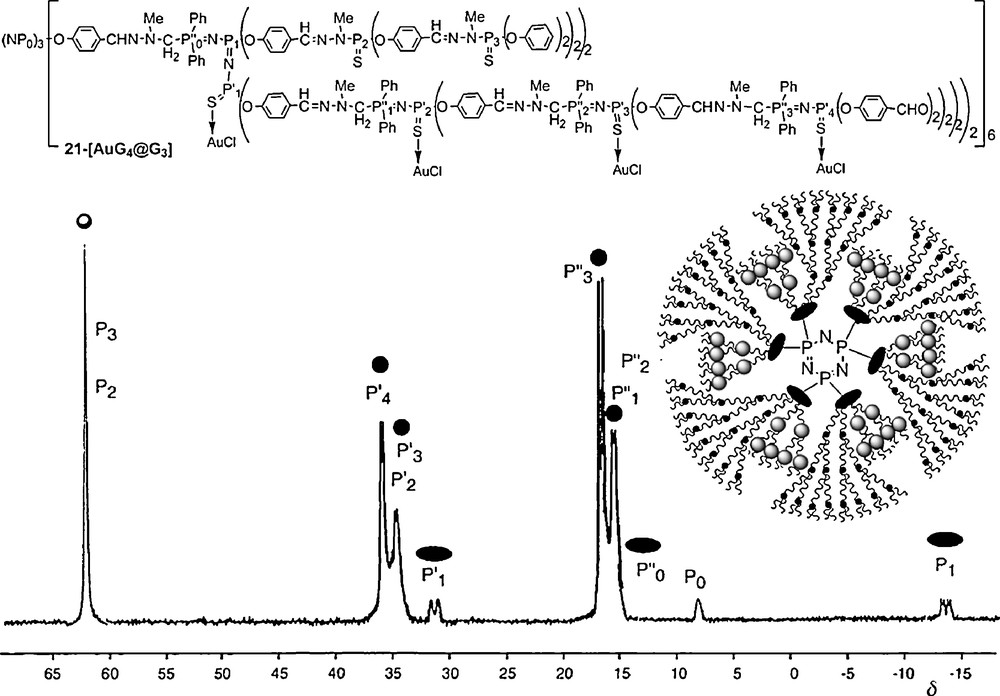
31P NMR spectrum obtained after complexation of the PN–PS groups by Au–Cl (21-[AuG4@G3]).
4 The particular case of dendrons
Dendrons are also called dendritic wedges. They have a fan-shape, generally with one function at the core which can be used for further reactions, for instance to graft them on a multifunctional core to obtain a true dendrimer. Most dendrons are built in a convergent way [61], that is to say that two small dendrons are associated through their core to a branched monomer, to create a larger dendron, and so on. One inconvenience of this method is the poor reactivity of the terminal groups. On the contrary, we have synthesized our phosphorus-containing dendrons by divergent processes, as we do with dendrimers. The advantage is that the terminal groups are always easily functionalized. Of course, the trick is to find a functional group at the core which will not interfere during the synthetic process, but which can be activated later; the PN–PS groups may offer this opportunity. A first example of dendron is shown in Fig. 11, but due to the presence of PN–PS linkages at all levels, we have not tried to test the reactivity of dendron 11–G’3.
We have also synthesized several series of dendrons having only one PN–PS group at the core, as shown for example in Fig. 20. This compound possesses five different types of phosphorus atoms (PO, PN–PS, PS, P–Pd), which should appear in five different areas in the 31P NMR spectrum. This is indeed what is clearly observed for 22–G2 [62].
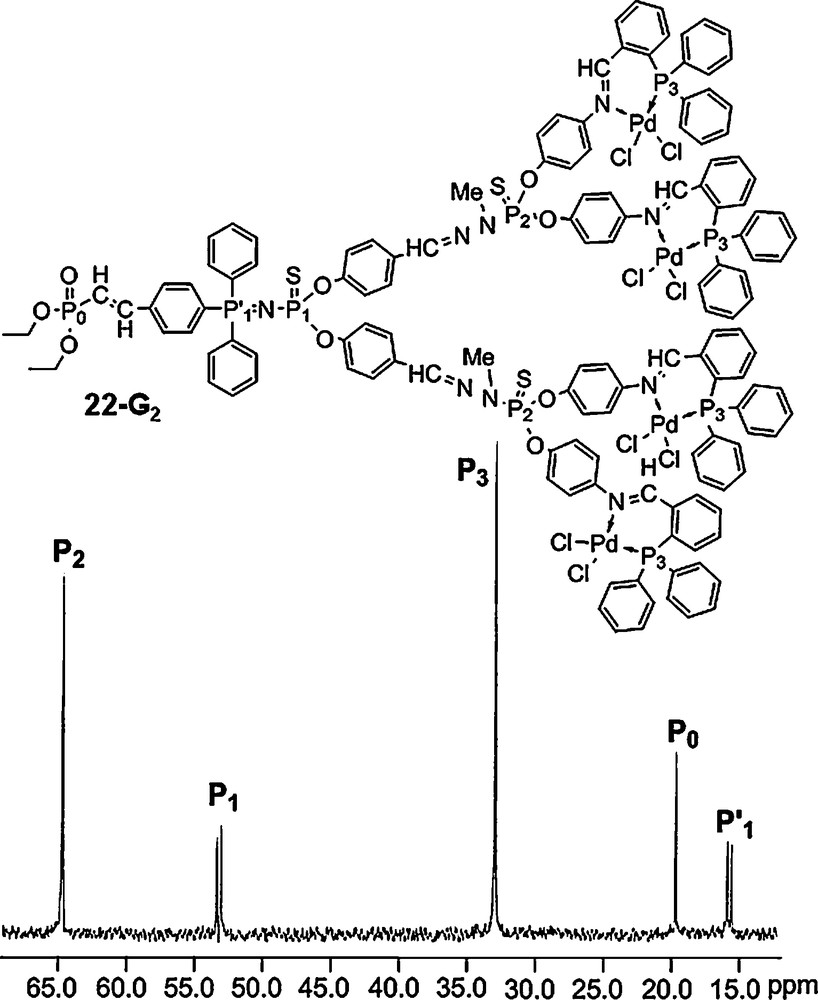
31P NMR spectrum of the dendron 22-G2 possessing five different types of phosphorus atoms.
We have also synthesized a series of dendrons (23–Gn) possessing a vinyl group at the core, directly linked to the PN–PS group. Surprisingly, the replacement of an aryl group by a vinyl group has little influence on the chemical shift of the phosphorus that bears this group (PN; compare P’0 in Fig. 21 to P’1 in Fig. 20; Δδ < 2 ppm), and a larger influence on the remote PS group (compare P0 in Fig. 21 to P1 in Fig. 20; Δδ > 3 ppm) [63].
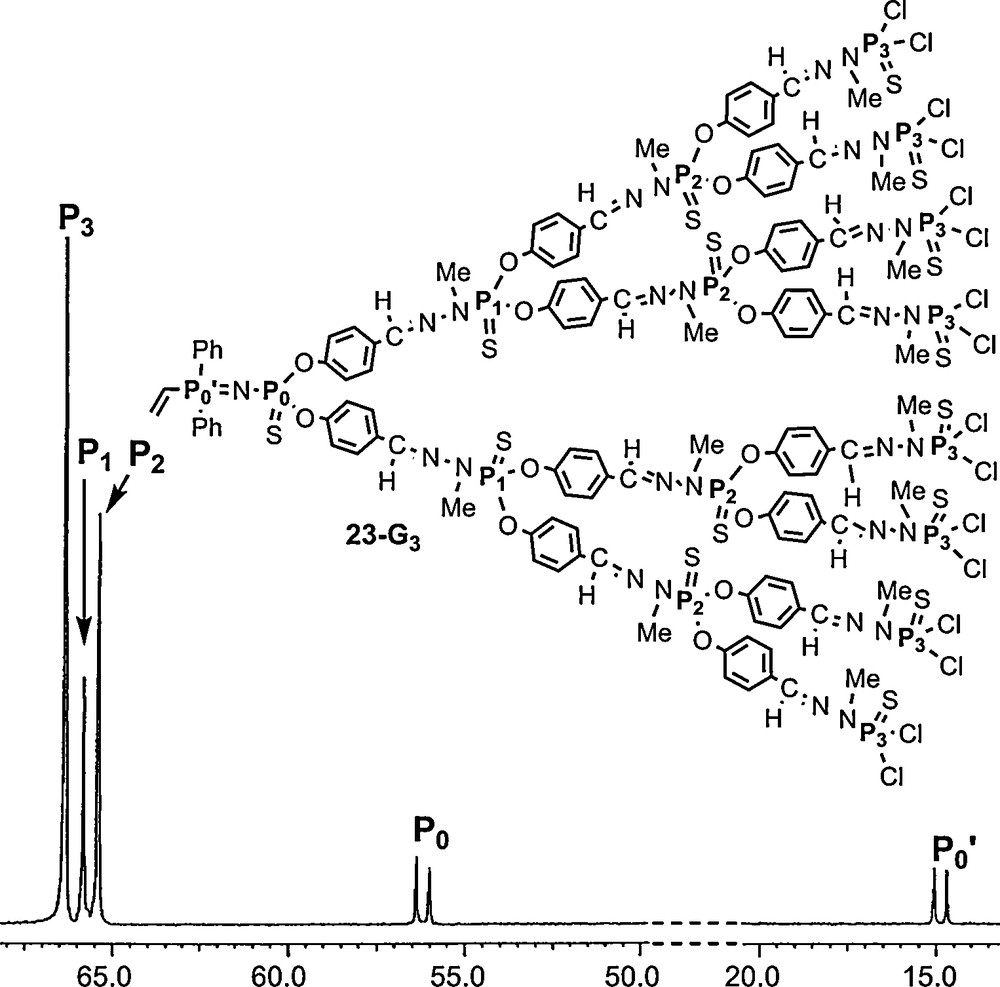
31P NMR spectrum of the third generation dendron 23-G3 possessing a vinyl group at the core.
This fact might be related to an electron withdrawing effect that might enhance the reactivity of the vinyl group. Starting from the second-generation dendron of the series shown in Fig. 21 (23–G2), we have functionalized the surface by phosphine groups. Thus, this dendron (24–G2) possesses two types of functional groups, the activated vinyl group at the core and eight diphenylphosphine as terminal groups. Then we have studied sequentially the reactivity at both levels of the structure. An amino alcohol reacted cleanly via a Michael-type addition to the core, exclusively on the NH2 side to afford 25–G2. This addition induces a deshielding of the signal of the PN group (Fig. 22). In a second step, [Rh(cod)Cl]2 (cod = cyclooctadiene) was reacted with the terminal phosphino groups (26–G2). This reaction is also clearly detected by 31P NMR, by the deshielding of the signal corresponding to the phosphines, and its splitting in two signals due to the coupling with rhodium [64].
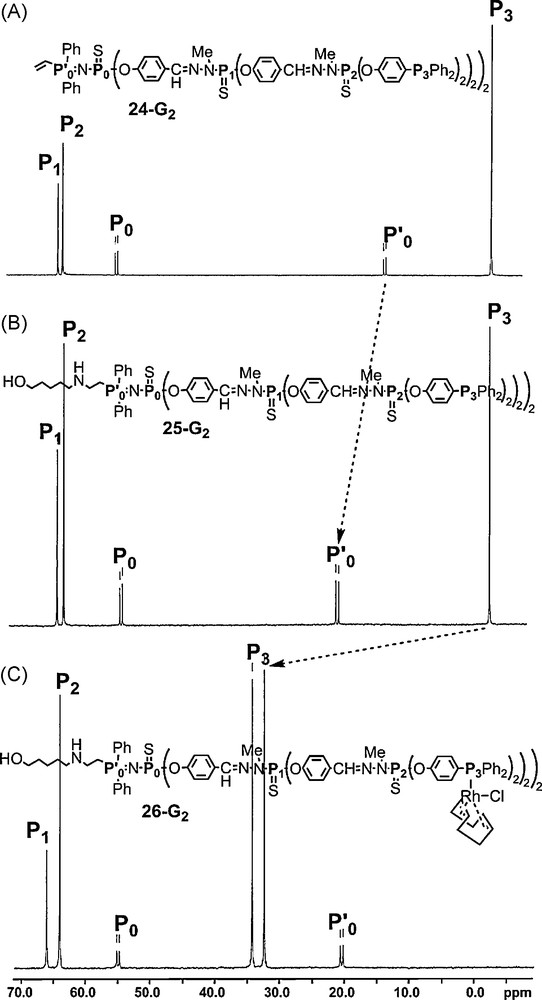
Evolution of the 31P NMR spectrum of a dendron showing first the reaction at the core then on the terminal groups.
These types of dendrons (23–Gn) were also used for the synthesis of sophisticated dendritic structures of type surface-block, layer-block, and segment-block dendrimers. The schematized structures of these compounds are shown in Fig. 23 [65]. Dendrons can also be used for the functionalization of materials as will be seen in the next paragraph.
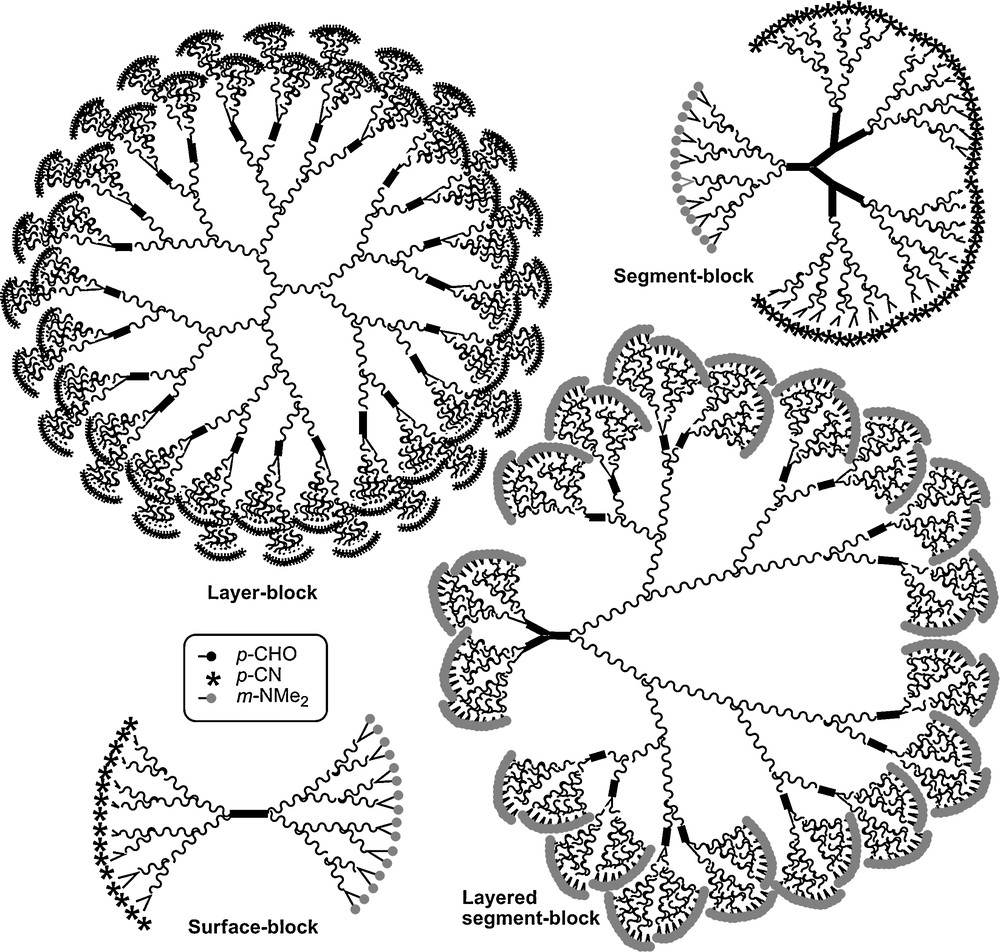
Various types of dendritic structures, all obtained from the type of dendron shown in Fig. 21 (23-Gn).
5 31P NMR spectra of dendrimers in the solid state
Dendrons and dendrimers can be embedded in various types of materials [66] such as titanium oxo clusters [67], but also in silica to create new organic/inorganic materials. 31P NMR can be also performed in the solid state, using the magic angle spinning technique. Obviously, the signals are not expected to be as thin as in solution. We have synthesized a series of dendrons up to the third generation possessing a hydrolysable triethoxysilane as core and used them to obtain a silica xerogel including these dendrons in the material. For this purpose, these dendrons were used in cohydrolysis and polycondensation in THF with 1 mol % of TBAF (tetrabutylammonium fluoride) as catalyst, and various amounts of TEOS (tetraethoxysilane) and water. It appears that the higher the dendron generation, the greater the demanded quantity of TEOS for the gelation. Thus, the gelation of the generation 0 required at least 10 equiv of TEOS, while 140 equiv of TEOS was necessary for the gelation of the third generation. The solid-state 31P NMR spectroscopy provides information concerning the dendron moieties incorporated into silica. The solids prepared from generation 0 (27–G0) display two resonances as expected in the same range as those observed in solution (Fig. 24). For the others, the number of resonances observed from solid state 31P NMR spectroscopy is always inferior to that observed in solution 31P NMR, as shown in particular for the third generation 27–G3 (Fig. 24) [68].
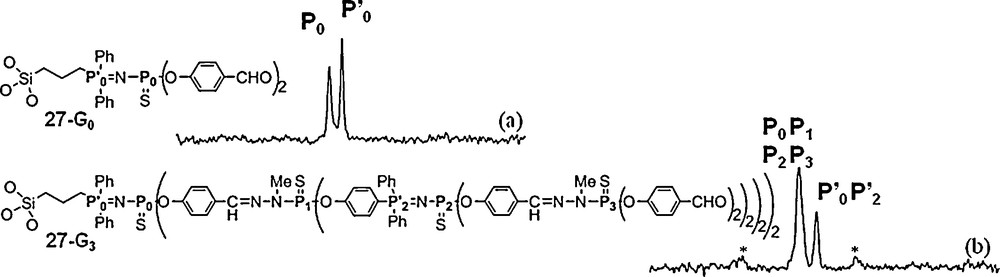
Solid state 31P NMR spectra of a generation 1 (27-G0, (a)) and a generation 3 (27-G3, (b)) dendron embedded in silica. Signals arising from sidebands are marked with asterisks.
We have not only embedded dendrons but also dendrimers inside silica. In this case, a different technique was used, which allowed the elaboration of periodic mesoporous silica of type MCM-41, obtained via the concomitant use of polycationic dendrimers and cationic surfactants, SiO2, NaOH and water. The hexagonal structure of MCM-41 is preserved up to about 26% in weight of dendrimer included in the silica. The cationic surfactant can be selectively removed to liberate the pores, while preserving the non-covalently incorporated dendrimers, which are too large to be removed. However, these dendrimers included in the mesoporous silica are fully accessible through the mesoporous volume to small molecules such as tetrahydrofuran. Fig. 25 displays the structure of the cationic dendrimer 28–G8 which was included in silica. Analysis of suspensions in water of this functionalized silica displays no signal in solution 31P NMR (Fig. 25 A). In contrast, if THF is added to this suspension, a broad signal at 70 ppm corresponding to the dendrimer clearly appears (Fig. 25 B). To be sure that this signal is exclusively due to dendrimers included in the silica, and not to dendrimers in solution, the solution was separated from the solid. No signal was detected for this solution; thus, the dendrimers were not extracted by THF, and the signal really came from the dendrimers included in the silica, showing that they are accessible to THF [69].
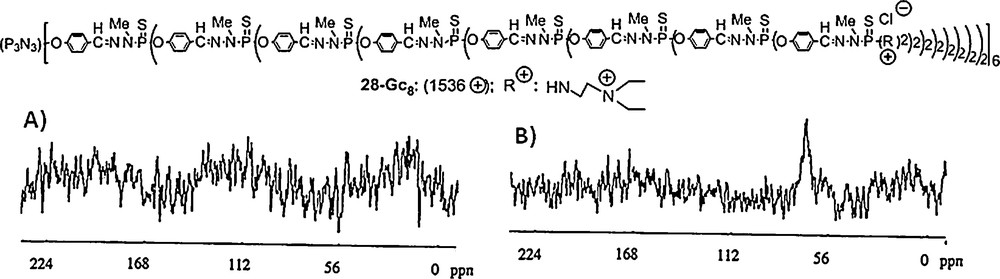
Solution 31P NMR spectra of a suspension of the generation 8 dendrimer 28-G8 embedded in silica. (A) Suspension in water. (B) Suspension in water/THF.
Other types of positively charged dendrimers, particularly those bearing Girard terminal groups (N–NHC(O)CH2NR3,Cl) were shown able to produce an original type of material, “solid water” by gelation at room temperature, even when using small quantities of dendrimers (< 0.25% in weight) [70].
6 Conclusion
We have demonstrated in this review that 31P NMR is an invaluable tool for the characterization of highly sophisticated superstructures such as dendrimers, and also to ascertain their purity. The focus was on the synthesis of dendritic structures, but 31P NMR continues to be useful every day, even now that we are mainly concerned by applications of our dendrimers. Some leads were evoked along this review and can be briefly developed here, along three main axes that are catalysis, materials, and biology.
Most catalytic properties are studied using organometallic derivatives of dendrimers [71]. The main advantage of dendritic catalysts is that they can be easily recovered and reused, thanks to their large size. The phosphorus dendrimers were used in particular for Knoevenagel condensations and Michael additions [72], for Stille couplings [73], for O– and N-arylation and vinylation of phenol and pyrazole [27], for asymmetric allylic alkylation [74], for asymmetric benzoylations [75], for C–C cross coupling reactions [76], for Mizoroki-Heck reactions [32], and in aqueous media [77].
In the field of materials [78], we have seen some examples concerning the incorporation of dendritic structures inside materials [67–69], which were used also for the elaboration of Organic Light Emitting Diodes (OLEDs) from fluorescent dendrimers [79]. However, the simple functionalization of the surface of materials at a nanometric scale by a single layer or by multiple layers of dendrimers can greatly modify the properties of the materials [80]. For instance, nanotubes made of dendrimers were obtained in this way [81], a glass slide functionalized by a single layer of dendrimers bearing oligonucleotides afford sensitive DNA chips [82], and a titanium film functionalized by fluorescent dendrons is a chemical sensor for phenols [83]. In addition, soft matter dendrimers bearing two-photon absorption fluorophores [84] are able to replace hard matter (and generally toxic) quantum dots, in particular for biomedical imaging [49].
Biomedical applications necessitate water-soluble dendrimers [85]. In most cases, this goal is achieved by grafting charged substituents as terminal groups [86], either ammonium or carboxylic or phosphonic acid salts. Ammonium ended dendrimers (in particular those of type 28–Gn) are efficient transfection agents (the ability to induce the insertion of genetic materials inside cells) [87], and have a high anti-prion activity (such as mad-cow disease), including in vivo [88]. Carboxylic acid terminated dendrimers when reacted with amino functionalized biologically active compounds generate “catanionic” species, which display anti-HIV activity [89], or are usable for ocular drug delivery [90], depending on the amino derivative used. However, the most important properties that we have obtained up to now concerns dendrimers ended by phosphonate salts. Indeed, several of them display surprising properties towards the immune blood system: they are able to induce monocytes activation [91], to multiply Natural Killer cells (that play a key role against infections and many cancers) [92], and possess anti-inflammatory properties [93]. It must be emphasized that several of these properties, in particular those related to human immunity, are specific of phosphorus dendrimers, highlighting the importance of this element.
Acknowledgement
We thank the COST Phosphorus Science Network (PhoSciNet, project CM0802).