

1 Introduction
As the field of glycoscience continues to expand, various new aspects of synthetic carbohydrate chemistry have begun to emerge. In recent years, investigations of the mechanistic aspects of the glycosylation reaction have often been overlooked. Thus, it has not been since the major efforts by Isbell's [1] and Lemieux's groups [2] that these basic mechanistic studies were undertaken. However, with the various opposing and reinforcing electronic effects that simultaneously exist in carbohydrate molecules, it has proven quite difficult to understand the factors behind the stereoselectivity of glycosylation (for selected references, see [3]). Thus, only recently have the mechanistic studies of chemical glycosylation again witnessed a renaissance [4]. To this end, a number of investigative studies have recently been performed using simplified model carbohydrate mimetics, allowing for the study of only particular effects without the interference of others [5]. For instance, Woerpel and coworkers studied a series of sugar mimetics [6], which in turn, provided new insight into the conformational preferences of glycosylation intermediates, and other independent studies have further supplemented these conformational mechanistic studies. From these studies, it is becoming increasingly apparent that the conformational changes undergone by the glycosyl donor during the reaction (i.e., flattening of the anomeric center, conformational reorientation to provide charge stabilization, anomerization or participation) can have dramatic influence over the reaction outcome. Justification for these findings has been supported both experimentally and computationally [7].
Along these lines, we assumed that many of these mechanistic studies could be simplified if a general synthetic route to sugar mimetics possessing a conformationally controlled pyranose ring, was available. While a number of research groups have performed total asymmetric syntheses of natural carbohydrates and derivatives thereof [8], to the best of our knowledge, no synthesis of a conformationally restricted tetrahydropyran designed to mimic the conformational preferences of many glycopyranosides has been achieved through the use of a bulky substituent to limit conformational fluxuation. As the equatorial proclivity of the t-butyl substituent has been well established in prior equilibrium studies of substituted cyclohexanes (Scheme 1), it was subsequently chosen as the ideal substituent to effectively restrict unwanted conformational changes. As such, we set out to obtain the t-butylated pyranose derivative wherein the t-butyl group would reside at the carbon opposite to the anomeric center (position C-4 using carbohydrate nomenclature or C-3 for the heterocycle numbering; as depicted in Scheme 2, A).
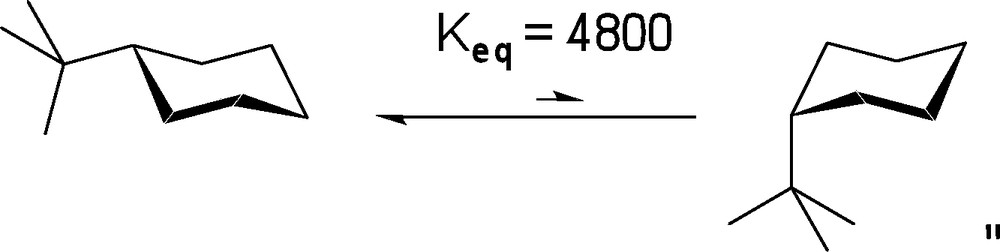
Conformational restriction using t-butyl substituent on cyclohexanes.
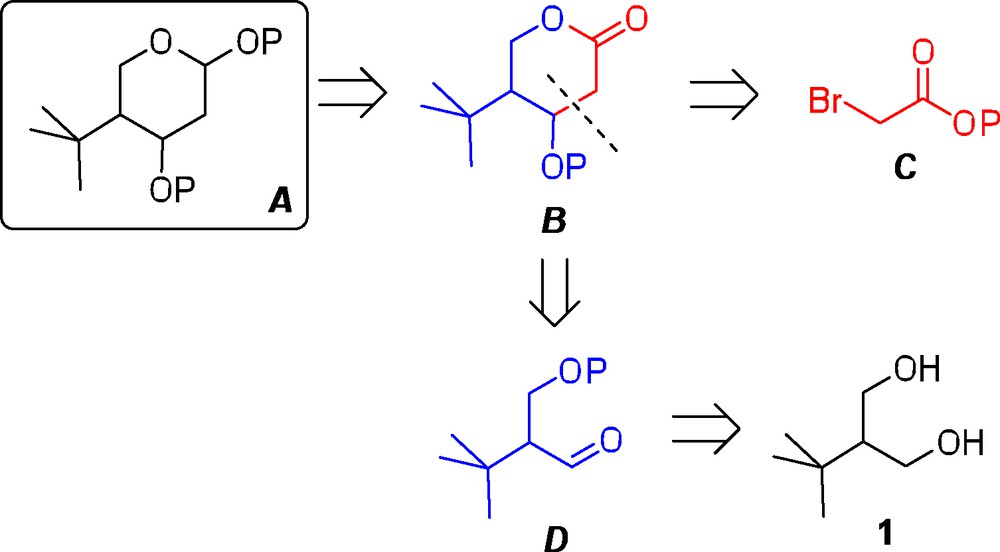
Retrosynthetic analysis for the synthesis of a generic 4-tert-butylated pyranose A.
2 Results and discussion
For the retrosynthetic analysis, it was envisaged that the final tetrahydropyran A could be accessed from the corresponding lactone precursor B by sequential reduction and trapping of the cyclic hemiacetal through the introduction of a temporary substituent (Scheme 2). Next, the lactone intermediate B could be formed by a coupling reaction between an α-bromoester C and a suitable aldehyde precursor D. In turn, the necessary aldehyde D should then be readily accessible from the commercially available 1,3-diol 1.
In accordance with the retrosynthetic analysis, we began from the commercially available 2-tert-butylpropane-1,3-diol 1, and subsequently reacted it with p-methyoxybenzaldehyde dimethyl acetal in the presence of CSA to yield the cyclic acetal 2 (Scheme 3). The latter was then subjected to a reductive acetal opening in the presence of DIBAL-H, to yield racemic compound 3 in 95% (over two steps). Next, alcohol 3 was subjected to the Swern oxidation protocol (oxalyl chloride and DMSO) to yield aldehyde 4, which corresponds to the generic aldehyde D of Scheme 2, in 90% yield. At this stage, a CC bond forming coupling reaction was necessary, and as such, the Reformatsky reaction utilizing compound 4 and ethyl bromoacetate was found to be appropriate. This coupling was performed in the presence of Zn (dust) followed by treatment with BF3Et2O to give the resulting β-hydroxy ester 5 in 78% yield, as a mixture of diastereomers (4.3:1) with the antiproduct being the major diastereomer.

Synthesis of the acyclic five-carbon synthon 5.
Subsequently, the p-methoxybenzyl (pMBn) substituent in compound 5 was removed under acidic conditions, TFA in aqueous CH2Cl2, whereupon the resulting free hydroxyl spontaneously cyclized, displacing the ethoxy substituent, to afford β-hydroxylactone 6 (Scheme 4). As an unwanted elimination of the β-hydroxyl group was also anticipated as a competing side reaction, the reaction rate was controlled by a slow addition of TFA in very dilute reaction mixture (see experimental part for details). As a result of this precaution, no elimination byproduct could be detected, and the desired lactone 6 was obtained in 92% yield. At this point, the 3,4-cis and 3,4-trans-diastereomers were separated by column chromatography to afford compounds 6a and 6b in 77% and 15% yields, respectively (noting that upon cyclization, carbohydrate numbering was assumed). Thus, the C-3 hydroxyl group of compounds 6a and 6b were then silylated using TMSCl in pyridine, to obtain the corresponding TMS-ethers 7a and 7b, related to the generic lactone intermediate B (Scheme 2) in 99% and 82% yields, respectively. This intermediate protection of the β-hydroxyl group was found essential to ensure the success of the subsequent lactone reduction, which otherwise led to a complex mixture of unidentifiable products. Thus, with the hydroxyl group protected, lactones 7a and 7b were successfully reduced into their corresponding aldehydes, which were isolated as the acetylated hemiacetals upon successive treatment with acetic anhydride in pyridine. As a result, over two steps, target compound 8a was isolated exclusively as the 1,4-trans derivative in 80% yield, whereas compound 8b was isolated as a 2:1 mixture of 1,4-trans and 1,4-cis isomers in 81% yield. This complete stereoselectivity seen in the case of 8a is presumably due to the presence of 1,3-diaxial interactions, which suppress formation of the 1,4-cis isomer, and are not present in derivative 8b.
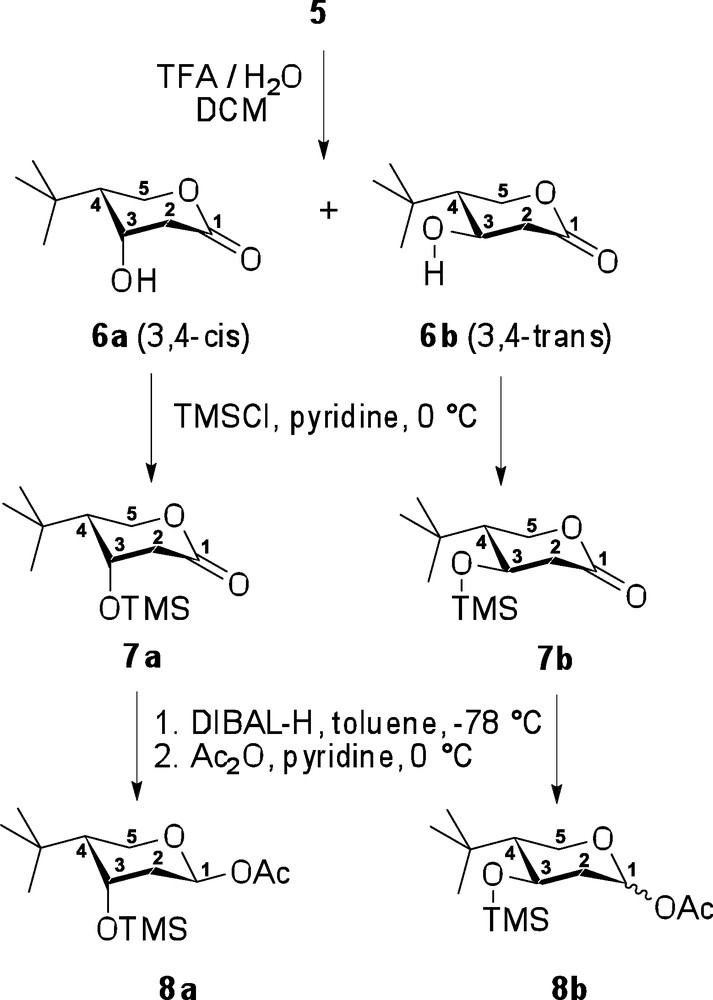
Completion of the synthesis of two diastereomers 8a and 8b.
3 Conclusions
Herein we have reported a concise synthetic route to access conformationally stable sugar mimetics. An important feature of these obtained glycomimetics, is the bulky t-butyl functionality at the C-4 position, which imposes a chair conformation as the lowest energy conformer, making these compounds ideal pyranose mimetics. If required, this route could also deliver non-racemic products by the known enzyme-catalyzed desymmetrization of compound 3 [9].
4 Experimental
General: Column chromatography was performed on silica gel 60 (70–230 mesh), reactions were monitored by TLC on Kieselgel 60 F254. The compounds were detected by examination under UV light and by charring with a visualization solution of 3.4% H2SO4, 1.0% AcOH, 2.5% p-anisaldehyde dimethylacetal in ethanol. Solvents were removed under reduced pressure at < 40 °C. CH2Cl2 was distilled from CaH2 directly prior to application. Methanol was dried by refluxing with magnesium methoxide, distilled and stored under argon. Acetonitrile and toluene were distilled over CaH2 under argon atmosphere and was refluxed for 2 h before application. THF was distilled over sodium using benzophenone as indicator under argon atmosphere and was refluxed for 2 h before use. Anhydrous pyridine was obtained from Sigma-Aldrich and used as is. Optical rotations were measured at ‘Jasco P-1020’ polarimeter. Melting points were measured at Thomas Hoover capillary melting point apparatus. Unless noted otherwise, 1H-n.m.r. spectra were recorded in CDCl3 at 300 MHz (Bruker Avance), 13C-n.m.r. spectra were recorded in CDCl3 at 75 MHz (Bruker Avance) or at 125 MHz (Bruker ARX-500). HRMS determinations were made with the use of JEOL MStation (JMS-700) Mass Spectrometer.
2-tert-Butyl-1,3-O-p-methoxybenzylidenepropane-1,3-diol (2). Similar to that of a published procedure [10], p-anisaldehyde dimethyl acetal (12.9 mL, 75.8 mmol) was added to a solution of 2-tert-butylpropane-1,3-diol (1, 5.0 g, 37.9 mmol) in anhydrous acetonitrile (20 mL) under argon. Camphorsulfonic acid (0.44 g, 1.9 mmol) was added and the resulting reaction mixture was refluxed for 30 min, whereupon it was cooled to rt and stirred for additional 1.5 h. After that, the reaction mixture was neutralized by addition of triethylamine and concentrated in vacuo. The residue was crystallized by the addition of hexanes. The clear flaky crystals were filtered off, rinsed with hexanes and dried in vacuo to afford the title compound 2 in quantitative yield. Analytical data for 2: Rf = 0.56 (ethyl acetate/hexanes, 1/4, v/v); 1H-n.m.r.: δ, 0.90 (s, 9H, C(CH3)3), 1.84–1.95 (m, 1H, CHC(CH3)3), 3.71–3.78 (m, 5H, OCH2, OCH3), 4.22–4.28 (dd, 2H, J = 4.3 Hz, J = 11.6 Hz, OCH2), 5.30 (s, 1H, CHPh), 6.82–7.39 (m, 4H, aromatic) ppm; 13C-n.m.r.: δ, 27.6 (× 3), 30.6, 43.6, 55.4, 69.4 (× 2), 101.4, 113.8 (× 2), 127.5 (× 2), 131.3, 160.1 ppm; HR-FAB MS [M + H]+ calcd for C15H23O3+ 251.1647, found 251.1653.
2-tert-Butyl-1-p-methoxybenzyloxypropan-3-ol (3). Similar to that of a published procedure [10], a solution of compound 2 (5.39 g, 21.7 mmol) in anhydrous toluene (50 mL) was flushed with argon and cooled to 0 °C. 1 M solution of DIBAL-H in hexane (38.6 mL) was added and the resulting reaction mixture was allowed to warm to rt. After 1 h the reaction mixture was cooled to 0 °C and carefully quenched by addition of MeOH (∼ 2.0 mL) until the gas evolution ceased. The mixture was allowed to warm to rt, whereupon sat. aq. potassium sodium tartrate (50 mL) was added and the resulting mixture was stirred for 1 h. After that, the organic layer was separated and the aqueous layer was extracted with ethyl acetate (4 × 5 mL). The combined organic phase was dried over Na2SO4, filtered and concentrated in vacuo. The crude residue was purified by column chromatography on silica gel (ethyl acetate–hexane gradient elution) to obtain the title compound 3 as an oil in 95% (from compound 1). Analytical data for compound 3: Rf = 0.35 (ethyl acetate/hexanes, 1/3, v/v); 1H-n.m.r.: δ, 0.86 (s, 9H, C(CH3)3), 1.59–1.67 (m, 1H, CH), 3.16 (d, 1H, OH), 3.52 (dd, 1H, CH2aOCH2Ph), 3.63 (dd, 1H, CH2aOH), 3.70–3.77 (m, 5H, CH2bOCH2Ph, CH2bOH, OCH3), 4.34–4.43 (dd, 2H, OCH2Ph), 6.82 (d, 2H, aromatic), 7.22 (d, 2H, aromatic) ppm; 13C-n.m.r.: δ, 28.2 (× 3), 31.5, 49.8, 55.2, 64.0, 72.0, 73.2, 113.8 (× 2), 129.2 (× 2), 130.0, 159.2 ppm; HR-FAB MS [M + Na]+ calcd for C15H24O3Na+ 275.1623, found 275.1616.
2-tert-Butyl-3-p-methoxybenzyloxypropanal (4). Following a typical protocol [11] for the Swern oxidation, a stirring solution of oxalyl chloride (4.25 mL, 48.8 mmol) and anhydrous CH2Cl2 (100 mL) was flushed with argon and cooled to −78 °C. A solution of DMSO (6.3 mL, 89.5 mmol) in anhydrous CH2Cl2 (6.3 mL) was added dropwise and the resulting mixture was stirred for 30 min at −78 °C. A solution of compound 3 (8.2 g, 32.5 mmol) in anhydrous CH2Cl2 (15 mL) was added dropwise, and resulting reaction mixture was stirred for 40 min at −78 °C. After that, Et3N (23.3 mL, 166.0 mmol) was added and the resulting mixture was stirred for 1 h at −78 °C, then allowed to warm to 0 °C and stirred for additional 1 h. The mixture was then diluted with H2O (200 mL), and extracted with dichloromethane (4 × 50 mL). The combined organic extract was washed with sat. aq. NaHCO3 (50 mL) and brine (50 mL), then dried over Na2SO4, filtered and concentrated in vacuo. The crude residue was purified by column chromatography on silica gel (ethyl acetate–hexane gradient elution) to allow the title compound 4 in 90% yield as a colorless oil. Analytical data for compound 4: Rf = 0.63 (ethyl acetate/hexanes, 1/3, v/v); 1H-n.m.r.: δ, 0.87 (s, 9H, C(CH3)3), 2.24–2.30 (m, 1H, CH), 3.51 (dd, 1H, 2J = 9.4 Hz, 3J = 4.0 Hz, CH2aOCH2Ph), 3.62 (s, 3H, OCH3), 3.75 (dd, 1H, 2J = 9.5 Hz, 3J = 9.5 Hz, CH2bOCH2Ph), 4.24–4.32 (dd, CH2Ph), 6.72 (d, 2H, aromatic), 7.08 (d, 2H, aromatic), 9.63 (d, 1H, HC = O) ppm; 13C-n.m.r.: δ, 28.3 (× 3), 32.8, 55.2, 61.2, 66.4, 72.9, 113.8 (× 2), 129.2 (× 2), 130.1, 159.2, 204.9 ppm; HR-FAB MS [M + Na]+ calcd for C15H22O3Na+ 273.1467, found 273.1470.
Ethyl 4-tert-butyl-3-hydroxy-5-p-methoxybenzyloxypentanoate (5). Following a reported general procedure [12] for the Reformatsky reaction, BF3-Et2O (4.87 mL, 38.4 mmol) was added dropwise over a period of 1 h to a stirring suspension of aldehyde 4 (4.8 g, 19.2 mmol), zinc dust (6.24 g, 95.4 mmol), and ethyl bromoacetate (6.37 mL, 57.6 mmol) in anhydrous THF (51.2 mL) under argon. During this period, the reaction mixture may warm up rapidly, in which case immediately relieve pressure and cool flask with a cold water bath before continuing the addition of the remaining BF3-Et2O. Upon completion (∼ 1 h), the solid was filtered off, and the filtrate was concentrated in vacuo. The residue was redissolved in dichloromethane (50 mL) and washed with sat. aq. NaHCO3 (10 mL), 1 M aq. HCl (10 mL), and brine (10 mL). The organic layer was then separated, dried, and concentrated in vacuo. The crude residue was purified by column chromatography on silica gel (ethyl acetate–hexane gradient elution) to afford the title compound 5 (cis: trans, 4.3:1) as a colorless oil in 79%. Analytical data for compound 5 [cis (anti)]: Rf = 0.51 (ethyl acetate/hexanes, 1/3, v/v); 1H-n.m.r.: δ, 0.90 (s, 9H, C(CH3)3), 1.14 (t, 3H, OCH2CH3), 1.22 (m, 1H, CH), 2.35 (dd, 1H, 3J = 4.8 Hz, 2J = 15.4 Hz, CH2aC = O), 2.64 (dd, 1H, 3J = 9.0 Hz, CH2bC = O), 3.27 (d, 1H, OH), 3.55 (dd, 1H, J = 3.9 Hz, J = 9.9 Hz, CH2aOCH2Ph), 3.65–3.72 (m, 4H, OCH3, CH2bOCH2Ph), 4.02 (q, 2H, OCH2CH3), 4.39 (m, 3H, CHOH, CH2Ph), 6.77 (d, 2H, aromatic), 7.14 (d, 2H, aromatic) ppm; 13C-n.m.r.: δ, 14.1, 28.7 (× 3), 32.9, 42.7, 51.2, 55.0, 60.2, 67.7, 68.1, 72.8, 113.7 (× 2), 129.1 (× 2), 129.9, 159.1, 172.4 ppm; HR-FAB MS [M + Na]+ calcd for C19H40O5Na+ 361.1991, found 361.1970.
Analytical data for compound 5 [trans (syn)]: Rf = 0.43 (ethyl acetate/hexanes, 1/3, v/v); 1H-n.m.r.: δ, 0.89 (s, 9H, C(CH3)3), 1.19 (t, 3H, OCH2CH3), 1.80–1.86 (m, 1H, CH), 2.44–2.47 (dd, 2H, CH2C = O), 3.43–3.50 (m, 2H, OH, CH2aOCH2Ph), 3.62 (dd, 1H, J = 3.9 Hz, J = 11.4 Hz, CH2bOCH2Ph), 3.72 (s, 3H, OCH3), 4.09 (q, 2H, OCH2CH3), 4.23–4.34 (m, 1H, CH2OH), 4.35 (s, 2H, CH2Ph), 6.80 (d, 2H, aromatic), 7.21 (d, 2H, aromatic) ppm; 13C-n.m.r.: δ, 14.1, 29.0 (× 3), 31.7, 39.5, 52.6, 55.0, 60.2, 68.8, 68.9, 72.9, 113.7 (× 2), 129.2 (×2), 129.6, 159.1, 172.7 ppm; HR-FAB MS [M + Na]+ calcd for C19H40O5Na+ 361.1991, found 361.1997.
5-tert-Butyl-4-hydroxy-tetrahydro-2H-pyran-2-one (6a and 6b). Similar to that of a published procedure [13], to a stirring solution of the diastereomeric mixture of compound 5 (5.48 g, 16.2 mmol) in wet CH2Cl2 (400 mL) was added H2O (0.5 mL) followed by the portionwise addition of trifluoroacetic acid (3 × 1.2 mL, 48.6 mmol) over the period of 1 h. The reaction was stirred for 16 h, then neutralized with triethylamine and washed consecutively with sat. aq. NaHCO3 (75 mL) and brine (75 mL). The organic phase was separated, dried over Na2SO4, filtered and concentrated in vacuo. The crude residue was separated by column chromatography on silica gel (ethyl acetate/hexane gradient elution) to afford 6b (15%, white solid) and 6a (77%, white solid). Analytical data for compound 6a [cis]: Rf = 0.42 (methanol/CH2Cl2, 1/19, v/v); 1H-n.m.r.: δ, 0.96 (s, 9H, C(CH3)3), 1.58 (dd, 1H, 3J = 12.0 Hz, 3J = 4.6 Hz, CH), 2.50–2.66 (m, 2H, CH2C = O), 2.95 (bs, 1H, OH), 4.30–4.36 (m, 2H, CHOH, CH2aO), 4.63 (dd, 1H, 2J = 10.8 Hz, 3J = 12.2 Hz, CH2aO) ppm; 13C-n.m.r.: δ, 28.6 (× 3), 31.6, 41.5, 46.8, 63.9, 67.3, 171.3 ppm; HR-FAB MS [M + H]+ calcd for C9H17O3+ 173.1178, found 173.1183.
Analytical data for compound 6b [trans]: Rf = 0.45 (methanol–CH2Cl2, 1/19, v/v); 1H-n.m.r.: δ, 0.92 (s, 9H, C(CH3)3), 1.67–1.73 (m, 1H, CH), 2.51–2.64 (m, 2H, CH2C = O), 3.35 (bs, 1H, OH), 3.95 (dd, 1H, 3J = 10.9 Hz, 2J = 11.3 Hz, CH2aO), 4.20 (m, 1H, CHOH), 4.31 (dd, 1H, 3J = 5.8 Hz, CH2aO) ppm; 13C-n.m.r.: δ, 27.6 (× 3), 31.9, 39.0, 51.8, 65.5, 67.6, 173.2 ppm; HR-FAB MS [M + H]+ calcd for C9H17O3+ 173.1178, found 173.1179.
5-tert-Butyl-4-trimethylsilyloxy-tetrahydro-2H-pyran-2-one (7a, cis). A solution of lactone 6a (1.90 g, 11.4 mmol) in pyridine (20 mL) was flushed with argon and cooled to 0 °C. Trimethylsilyl chloride (1.86 g, 17.1 mmol) was added and the resulting mixture was stirred for 30 min. After that, the reaction mixture was concentrated in vacuo, the residue was redissolved in CH2Cl2 (50 mL) and washed with sat. aq. NaHCO3 (10 mL) and brine (2 × 10 mL). Organic phase was separated, dried over Na2SO4, filtered and concentrated in vacuo. The crude residue was purified by column chromatography on silica gel (ethyl acetate–hexane/triethylamine gradient elution) to afford the title compound 7a in 99% yield as white solid. Analytical data for compound 7a [cis]: Rf = 0.42 (ethyl acetate/hexanes, 1/3, v/v); 1H-n.m.r.: δ, 0.11 (s, 9H, OSi(CH3)3), 0.94 (s, 9H, C(CH3)3), 1.56 (dd, 1H, 3J = 12.2 Hz, 3J = 4.7 Hz, CH), 2.47–2.63 (m, 2H, CH2C = O), 4.30–4.34 (m, 1H, CH2aO), 4.38 (bs, 1H, CHOSi), 4.63 (dd, 1H, 3J = 12.0 Hz, 2J = 10.6 Hz, CH2bO) ppm; 13C-n.m.r.: δ, 0.6 (× 3), 28.4 (× 3), 31.4, 41.4, 47.4, 65.2, 67.2, 170.0 ppm; HR-FAB MS [M + Na]+ calcd for C12H24O3SiNa+ 267.1392, found 267.1395.
5-tert-Butyl-4-trimethylsilyloxy-tetrahydro-2H-pyran-2-one (7b, trans). The title compound 7b was obtained as a colorless oil in 82% yield as described for the synthesis of 7a. Analytical data for compound 7b [trans]: Rf = 0.46 (ethyl acetate/hexanes, 1/4, v/v); 1H-n.m.r.: δ, 0.04, (s, 9H, OSi(CH3)3), 0.92 (s, 9H, C(CH3)3), 1.69–1.75 (m, 1H, CH), 2.46–2.50 (m, 2H, CH2CO), 3.89–3.97 (dd, 1H, 2J = 11.8 Hz, 3J = 9.9 Hz, CH2aO), 4.09–4.13 (m, 1H, CHOSi), 4.24–4.31 (dd, 1H, 2J = 11.8 Hz, 3J = 6.0 Hz, CH2bO) ppm; 13C-n.m.r.: δ, 0.7 (× 3), 27.8 (× 3), 32.1, 39.3, 52.3, 66.6, 67.3, 171.6 ppm; HR-FAB MS [M + Na]+ calcd for C12H24O3SiNa+ 267.1392, found 267.1390.
2-Acetoxy-5-tert-butyl-4-trimethylsilyloxy-tetrahydro-2H-pyran (8a, β-cis). Similar to that of the reported procedure [14], lactone 7a (2.65 g, 10.9 mmol) was dissolved in anhydrous toluene (200 mL), cooled to −78 °C, and flushed with argon. DIBAL-H (27.15 mL, 1 M in hexane) was added dropwise and the resulting mixture was stirred for 5 min. After that, the reaction was quenched by addition of ethyl acetate (4 mL) and warmed to rt. Sat. aq. potassium sodium tartrate (100 mL) was added and the resulting mixture was stirred for 1 h. The layers were then separated and the aqueous layer was extracted with ethyl acetate (4 × 50 mL). The combined organic phase was dried over Na2SO4, filtered and concentrated in vacuo. The crude residue was dissolved in pyridine (40 mL), cooled to 0 °C, and flushed with argon. Acetic anhydride (4.3 mL) was added, the resulting mixture was warmed to rt and stirred for additional 6 h. After that, the reaction was quenched by addition of MeOH (∼ 2.0 mL) and concentrated in vacuo. The residue was then diluted with dichloromethane (75 mL), and washed with sat. aq. NaHCO3 (2 × 20 mL) and brine (20 mL). The organic phase was separated, dried over Na2SO4, filtered and concentrated in vacuo. The crude residue was purified by column chromatography on silica gel (ethyl acetate–hexane gradient elution) to afford the title compound 8a in 80% yield. Analytical data for compound 8a [β-cis]: Rf = 0.45 (ethyl acetate/hexanes, 3/17, v/v); 1H-n.m.r.: δ, 0.13 (s, 9H, OSi(CH3)3), 0.89 (s, 9H, C(CH3)3), 1.31–1.37 (m, 1H, CH), 1.56–1.64 (m, 1H, CH2aCOAc), 1.84–1.90 (m, 1H, CH2bCOAc), 2.05 (s, 3H, CH3), 3.82–3.88 (m, 1H, CH2aO), 4.02 (dd, 1H, 2J = 11.4 Hz, 3J = 11.4 Hz, CH2bO), 4.38 (d, 1H, CHOSi), 5.92 (dd, 1H, 3J = 2.4 Hz, 3J = 10.1 Hz, CHOAc) ppm; 13C-n.m.r.: δ, 0.8 (× 3), 21.3, 28.6 (× 3), 31.4, 39.5, 49.2, 63.8, 67.8, 91.9, 169.6 ppm; HR-FAB MS [M + Na]+ calcd for C14H28O4SiNa+ 311.1655, found 311.1656.
2-Acetoxy-5-tert-butyl-4-trimethylsilyloxy-tetrahydro-2H-pyran (8b, α/β-trans). The title compound 8b was obtained as a white solid in 81% yield (α:β = 2:1) as described for the synthesis of compound 8a. Analytical data for compound 8b (β-trans): Rf = 0.50 (ethyl acetate/hexanes, 3/17, v/v); 1H-n.m.r.: δ, 0.10 (s, 9H, OSi(CH3)3), 0.92 (s, 9H, C(CH3)3), 1.38–1.46 (m, 1H, CH), 1.61–1.68 (m, 1H, CH2aCOAc), 2.01–2.04 (m, 4H, CH2bCOAc, CH3), 3.34 (dd, 1H, 3J = 9.0 Hz, 2J = 12.5 Hz, CH2aO), 3.84–3.89 (m, 1H, CHOSi), 3.99 (dd, 1H, CH2bO), 5.62 (dd, 1H, 3J = 9.1 Hz, 3J = 3.2 Hz, CHOAc); 13C-n.m.r.: δ, 0.9 (× 3), 21.3, 29.0 (× 3), 32.0, 39.3, 51.2, 64.1, 68.6, 92.5, 169.9 ppm; HR-FAB MS [M + Na]+ calcd for C14H28O4SiNa+ 311.1655, found 311.1656.
Analytical data for compound 8b (α-trans): Rf = 0.50 (ethyl acetate/hexanes, 3/17, v/v); 1H-n.m.r.: δ, 0.10 (s, 9H, OSi(CH3)3), 0.93 (s, 9H, C(CH3)3), 1.48–1.56 (m, 1H, CH), 1.70–1.79 (m, 1H, CH2aCOAc), 1.87–1.79 (m, 1H, CH2bCOAc), 2.04 (s, 3H, CH3), 3.54 (dd, 1H, 3J = 11.4 Hz, 2J = 11.4 Hz, CH2aO), 3.80 (dd, 1H, 3J = 4.7 Hz, CH2bO), 4.00–4.11 (m, 1H, CHOSi), 6.07 (dd, 1H, 3J = 2.9 Hz, 3J = 3.3 Hz, CHOAc) ppm; 13C-n.m.r.: δ, 0.9 (× 3), 21.3, 29.0 (× 3), 32.1, 38.9, 51.4, 62.7, 67.1, 92.9, 170.1 ppm; HR-FAB MS [M + Na]+ calcd for C14H28O4SiNa+ 311.1655, found 311.1656.
Acknowledgements
This work was supported by awards from the National Science Foundation (CHE-0547566 to AVD) and National Institute of General Medical Sciences (R01-GM076192 to CDS).