1 Introduction
Glucuronan is an anionic polysaccharide excreted by the Sinorhizobium meliloti M5N1CS mutant strain (NCIMB 40472). It is a homopolymer of β-d-glucopyranosyluronic acid with 1 → 4 linkages, variably O-acetylated at C-3 and/or C-2 position depending on the Mg2+ concentration in the culture medium [1]. Oligoglucuronans were easily obtained by using a glucuronan lyase from Trichoderma sp. GL2 [2]. The catalytic mechanism of the enzyme has been identified as a β-elimination, leading to a double bond formation between C-4 and C-5 on the non-reducing unit of generated oligomers. These oligosaccharides have been shown to elicit plant natural defences [3].
The practical use of these compounds as plant defence elicitors requires the addition of adjuvants in the formulation in order to facilitate the penetration of these polar compounds through hydrophobic membranes covering leaves. As natural bacterial glycolipids have been reported to act as activators of defence responses in plants [4], the introduction of alkyl chains on oligoglucuronans is a challenging modification that would decrease their polarity and should change their interaction with plant cells. In this paper, we report the microwave- (MW) assisted alkylation of a model glucuronic acid (GlcA) trisaccharide (dp3), isolated by reversed phase chromatography from the oligosaccharide mixture obtained by enzymatic degradation [5]. Different conditions were assayed, and the structure of the product analyzed by NMR spectroscopy and mass spectrometry.
2 Results and discussion
Recently, we have described the glycosylation/esterification of GlcA with different alcohols under MW irradiation in the presence of different Lewis acids [6]. As was previously reported using classical methods [7], MW activation of GlcA led to d-glucofuranosidurono-6,3-lactone derivatives. GlcA pyranosides could be prepared by developing a two step synthesis: first, GlcA was converted to 2,3,4-tri-O-acetyl-d-glucopyranurono-6,1-lactone, then this lactone was reacted with different alcohols in the presence of a Lewis acid. When SnCl4 was used, disubstitution occurred, giving alkyl (alkyl d-glucopyranosid)uronate derivatives.
In oligoglucuronans, the formation of the furanuronolactone under Lewis acid catalysis would be prevented, because the HO-4 is involved in the interglycosidic bond. Only the GlcA at the reducing end could give a pyranurono-6,1-lactone, assuming that the reaction conditions do not affect glycosidic bonds. Moreover, this could only be possible if the lactonisation was faster than both glycosylation and esterification.
The dp3 was chosen as a model compound to study the substitution. Firstly, the reaction was carried out in methanol using one equivalent of SnCl4 per monosaccharide unit. After 10 min under MW irradiation at 65 °C, no reaction was observed. This is probably due to the low solubility of the oligosaccharide in methanol, and to the fact that the reaction temperature is limited to the boiling point of methanol. Therefore 1-butanol was used instead of MeOH (Scheme 1) and the reaction was performed at 100 °C. After 10 min, the mixture was completely homogeneous and the consumption of the starting dp3 was followed by ESI-MS.
Reverse phase HPLC (MeOH-H2O) of the reaction mixture showed a major product at a retention time of 41.7 min (Fig. 1). After purification by preparative HPLC, ESI-MS (positive mode) was performed (Fig. 2). Surprisingly, the peak observed at m/z 831 corresponds to the molecular ion [MNa]+ of a dp3 bearing five butyl chains.
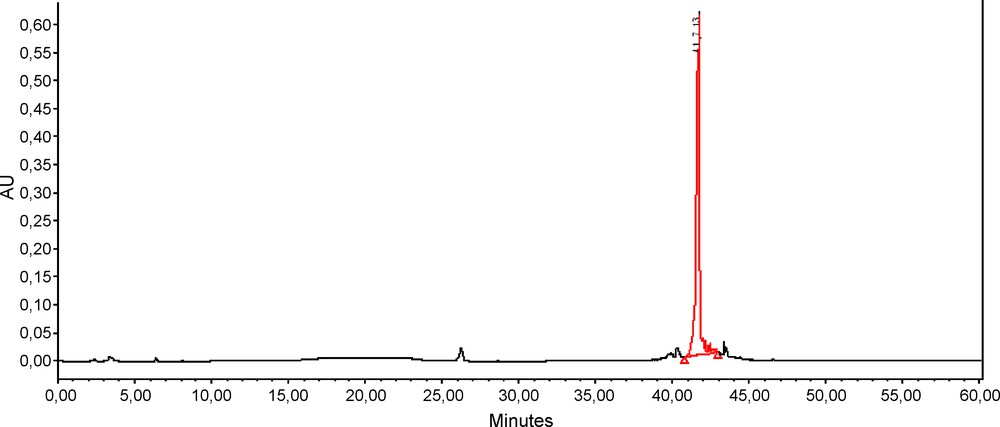
HPLC purification of the reaction trisaccharide/butanol/SnCl4 on a Prevail C-18 column (22 mL/min, 100% H2O for 40 min then 100% MeOH to 60 min, light scatering detector).
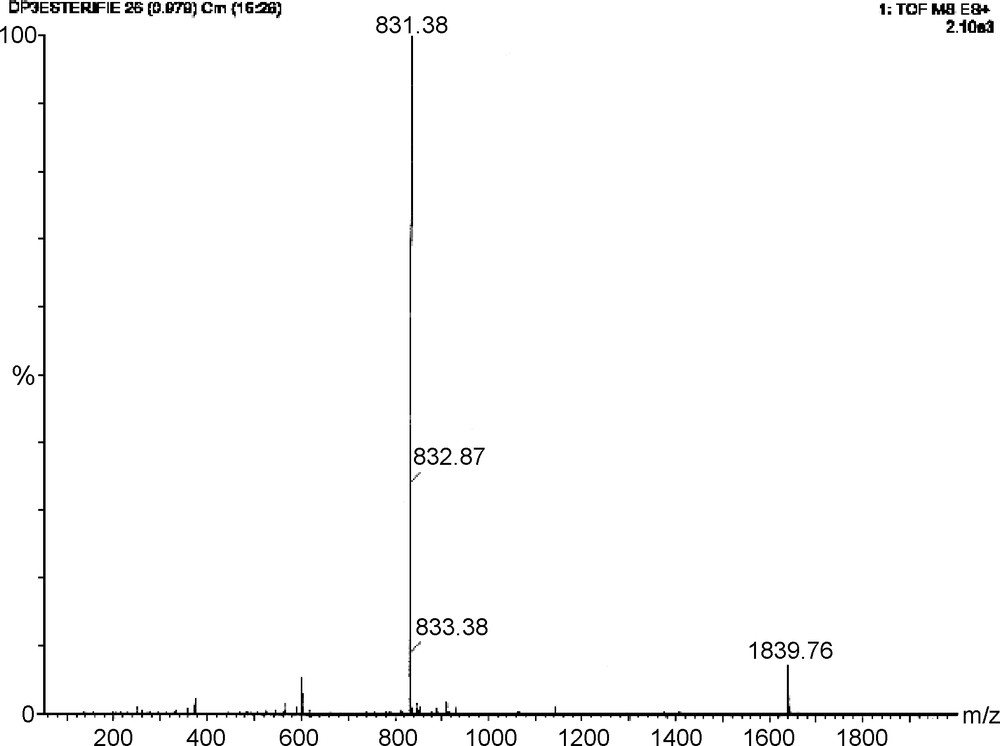
ESI-MS spectrum (positive mode) of the main product (retention time 41.7 min) isolated by HPLC.
We concluded at this point, based on our previous results [6] with the GlcA 6,1-lactone/SnCl4/MW, where simultaneous glycosylation and esterification occurred, that the three carboxyl groups were converted into butyl esters, and, in addition, the butyl glycoside was formed. Further analysis has been necessary to determine the position of the fifth butyl chain.
13C NMR spectra of starting and modified dp3 are shown in Figs. 3 and 4, respectively. The signals corresponding to butyl chains are clearly observed at δ 32.2–30.4 (CH2), 19.3–19.1 (CH2), 14.0–13.8 (CH3) ppm. C-6 signals from saturated GlcA units appeared shielded by 2–3 ppm, overlapping that of unsaturated GlcA. The resonance assigned to C4Δ at δ 111.3 ppm in starting dp3 is clearly seen at δ 111.6 ppm in the product, indicating that a double bond is still present.
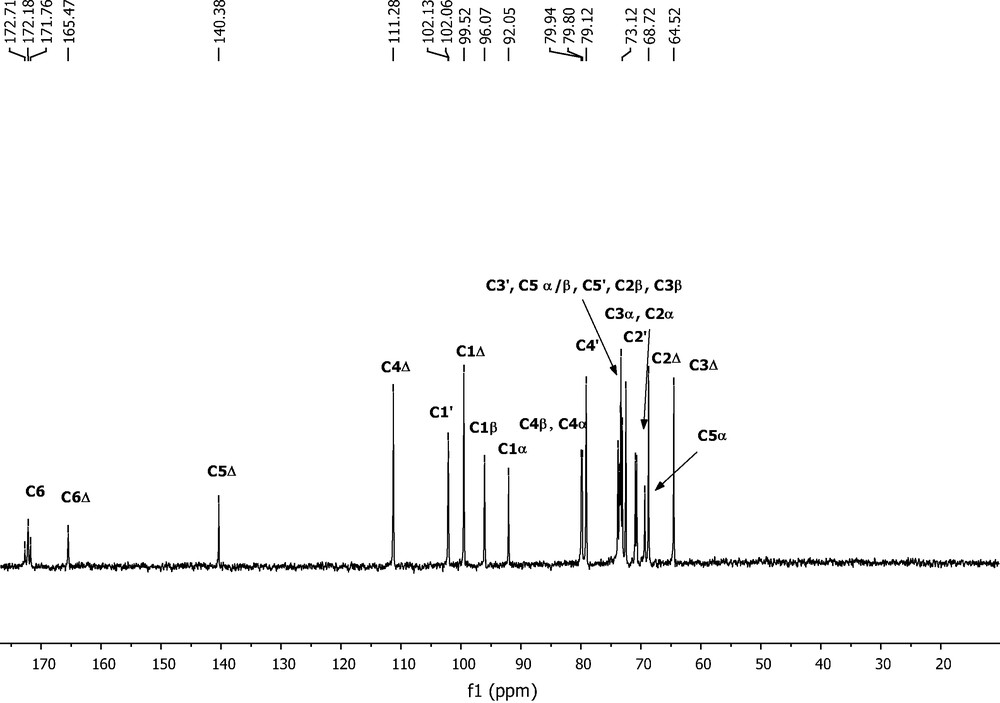
13C NMR spectrum of starting trisaccharide.

13C NMR spectrum of modified trisaccharide.
The deshielding observed for C1β (from 96 to 103 ppm) and C1α (from 92 to 98 ppm) indicates anomeric substitution. Therefore glycosylation and esterification have occurred. As the additional chain did not affect the double bond, it must be a substituent of one hydroxyl as a butyl ether. The position of substitution remained to be determined, and ESI MS-MS spectrometry in both negative and positive modes was performed.
The MS-MS spectrum in negative mode of the molecular ion m/z 807 [M − H]− (Fig. 5), showed the fragmentation at the glycosidic bonds m/z 287 (C1) and m/z 519 (C2), suggesting the presence of an additional butyl chain at the non-reducing end [8]. The ion at m/z 213 indicating a loss of butanol (C4H10O, m/z 74) and the fragment ion at m/z 231 a loss of a butylene (C4H8, m/z 56) from the C1 peak confirmed this assignment. The fifth butyl chain must therefore be either at O-2 or O-3 of the non-reducing end unit.
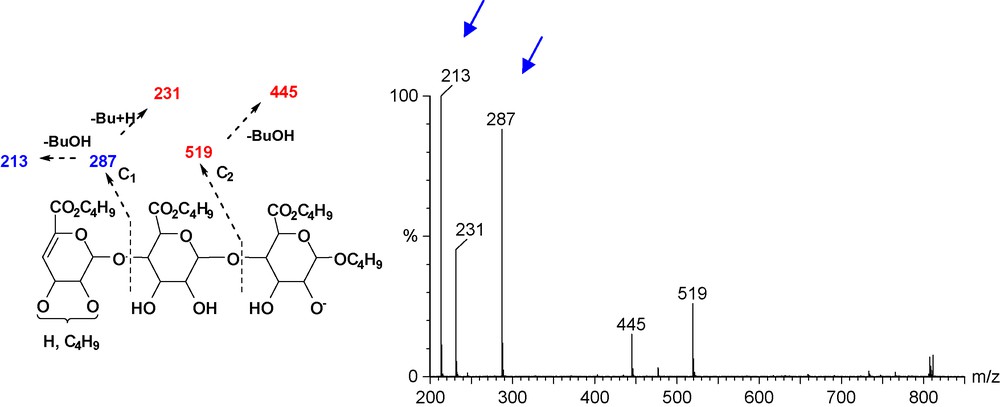
ESI (negative mode) MS-MS of m/z 807 [M − H]−.
On the other hand, the MS-MS in positive mode of molecular ion m/z 831 [MNa]+ (Fig. 6) showed intraglycosidic fragmentation. Both 0.2X2 (m/z 603) and 0.2A1 clearly indicate that the additional butyl chain is on the allylic position.
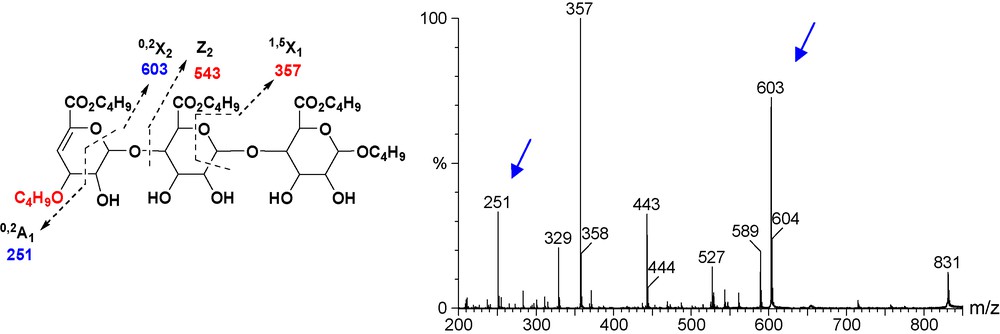
ESI (positive mode) MS-MS of m/z 831 [MNa]+.
Previously, we have observed that for the glucurono-6,1-lactone, different results were obtained by changing the Lewis acid catalyst. In order to investigate this point, the reaction was performed in the presence of FeCl3. The same main product (m/z 831) was obtained.
The 1H NMR spectrum (Fig. 7) of the product is complex, as the presence of different stereoisomers was observed. In addition to anomers α and β, the resonance corresponding to H-4 of the insaturated non-reducing end appeared as two different signals, suggesting that partial epimerization at the allylic position C-3 had occurred.
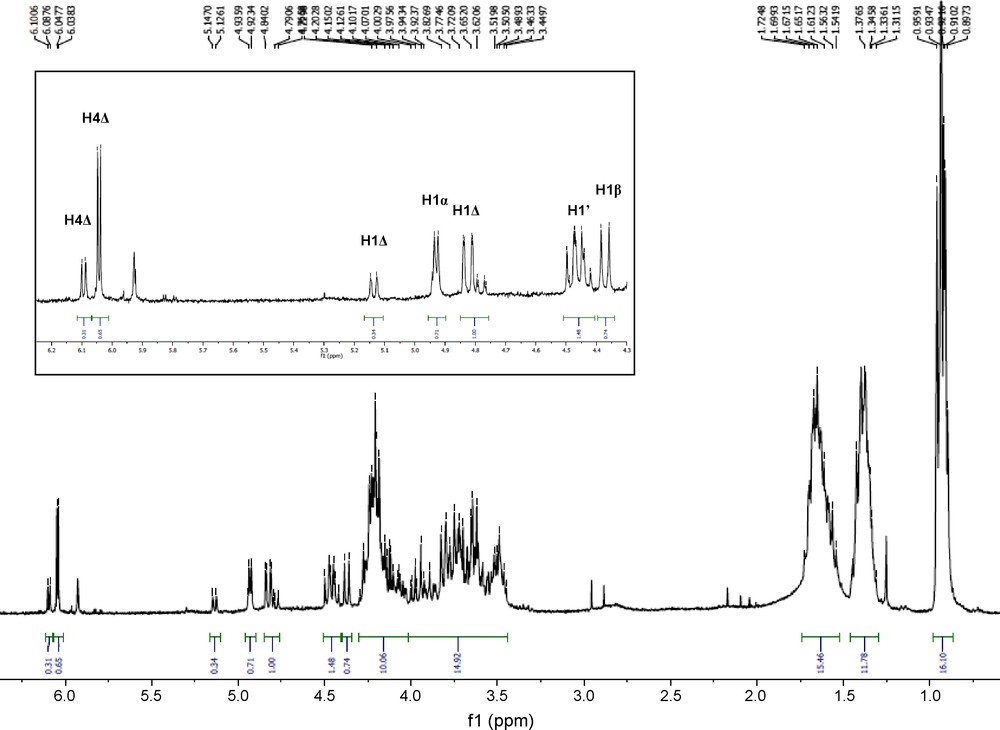
1H NMR spectrum (CDCl3) of modified dp 3.
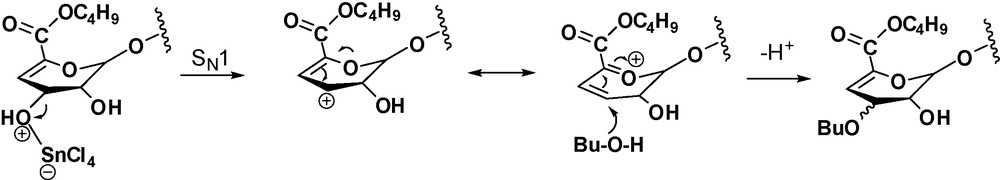
A possible mechanism involves the formation of an allylic cation intermediate that would explain the C-3 epimerisation to give a mixture of α-l-threo/β-d-erythro stereoisomers (Scheme 2). A similar transformation, with epimerization of the allylic hydroxyl of the non-reducing 4,5-insaturated unit has been previously observed on the reaction of oligomannuronates with butanol-methanesulfonic acid [9].
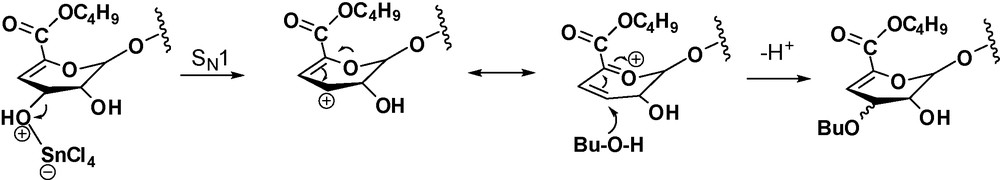
In conclusion, formal alkyl substitution of a glucuronate dp3 was easily performed in one step under MW irradiation, affording a product resulting from simultaneous butanol glycosylation, esterification and butyl ether formation at the allylic position. These results open the possibility of one-pot oligosaccharide modification, readily allowing modulation of the polarity to improve the penetration into the plant.
3 Experimental
3.1 General
All chemicals were purchased from Aldrich or Acros (France). 1H and 13C NMR spectra were recorded on a Bruker at 300 and 75 MHz, respectively. Chemical shifts are given as d values with reference to tetramethylsilane (TMS). Low-resolution electrospray mass spectra (ESIMS) in the positive ion mode were obtained on a Waters-Micromass ZQ quadrupole instrument, equipped with an electrospray (Z-spray) ion source (Waters-Micromass, Manchester, UK). High-resolution electrospray experiments (ESI-HRMS) and MS-MS were performed on a Waters-Micromass Q-TOF Ultima Global hybrid quadrupole time-of-flight instrument, equipped with an electrospray (Z-spray) ion source (Waters-Micromass, Manchester, UK). All solvents were distilled before use. MW irradiation was performed in a CEM Discover® System.
3.2 (4-deoxy-α-l-threo-hex-4-enopyranosyluronic acid)-(1 → 4)-(β-d-glucopyranosyluronic acid)-(1 → 4)-(d-glucopyranuronic acid)
Starting dp3 was obtained as described in [5]. 1H NMR (300 MHz, D2O) δ 6.26 (d, 1H, J = 4.4 Hz, H-4Δ), 5.28 (d, 1H, J = 3.6 Hz, H-1α), 5.22 (d, 1H, J = 3.2 Hz, H-1Δ), 4.73 (d, 1H, J = 8.0 Hz, H-1β), 4.56 (d, 1H, J = 7.8 Hz, H-1′), 4.44 (d, 1H, J = 8.9 Hz, H-5′), 4.15–4.10 (m, 3H, H-3Δ, H-5α, H-5β), 3.96–3.94 (m, 1H, H-2Δ,), 3.90–3.74 (m, 4H, H-3α, H-4α, H-4β, H-4′), 3.70–3.55 (m, 3H, H-2α, H-3β, H-3′), 3.43–3.30 (m, 2H, H-2β, H-2′).
13C NMR (75.5 MHz, D2O) δ 172.7, 172.2, 171.8 (C6α/β, C6’), 165.5 (C6Δ), 140.4 (C5Δ), 111.3 (C4Δ), 102.1 (C1′), 99.5 (C1Δ), 96.1 (C1β), 92.1 (C1α), 79.9, 79.8 (C4α, C4β), 79.1 (C4′), 73.8 (C3β), 73.6, 73.4, 73.1, 72.8 (C3′, C5′, C2β, C5β), 72.5 (C2′), 70.9 (C3α), 70.7 (C2α), 69.4 (C5α), 68.7 (C2Δ), 64.5 (C3Δ).
ESI-HRMS [M − H]− calcd. for C18H23O18: 527.0884. Found: 527.0881.
3.3 Butyl (butyl 3-O-butyl-4-deoxy-α-l-threo-hex-4-enopyranosyluronate)-(1 → 4)-(butyl β-d-glucopyranosyluronate)-(1 → 4)-(butyl d-glucopyranosid)uronate and butyl (butyl 3-O-butyl-β-d-erythro-hex-4-enopyranosyluronate)-(1 → 4)-(butyl β-d-glucopyranosyluronate)-(1 → 4)-(butyl d-glucopyranosid)uronate
A mixture of dp3 oligosaccharide (30 mg), 1-butanol (1 mL) and SnCl4 (6 μL) was stirred under argon and irradiated (300 W) at 100 °C for 10 min, and then concentrated to a syrup. RP HPLC purification (C18 Prevail column, 22 mL/min, 100% H2O for 40 min then 100% MeOH from 40.01 to 60 min) afforded the product (18 mg, 41% yield) as a mixture of four stereoisomers α-l-threo/β-d-erythro, α/β. The reaction was also performed under the same conditions but in the presence of 9 mg of FeCl3 and purified as before (16 mg, 36% yield).
1H NMR (300 MHz, D2O) δ 6.10 (d, 0.3H, J = 4.0 Hz, H-4Δ), 6.05 (d, 0.7H, J = 2.8 Hz, H-4Δ), 5.13 (d, 0.3H, J = 6.3 Hz, H-1Δ), 4.92 (d, 1H, J = 3.8 Hz, H-1α), 4.82 (dd, 0.7H, J = 0.9 Hz, J = 8.4 Hz, H-1 Δ), 4.77 (dd, 0.3H, J = 1.3 Hz, J = 7.3 Hz, H-1Δ), 4.50–4.41 (m, 1.5H, H-1′, OH), 4.36 (d, 0.7H, J = 7.8 Hz, H-1β), 1.75–1.5 (m, 10H, CH2), 1.27–1.39 (m, 10H, CH2), 0.97–0.87 (m, 15H, CH3). Only the assigned resonances are indicated.
13C NMR (75.5 MHz, CDCl3) δ 168.6–167.0 (C6, C6’), 161.7, 161.6 (C6Δ), 140.9 (C5Δ), 111.6, 108.7 (C4Δ), 103.8, 103.5, 103.0, 100.9 (C1β, C1′, C1Δ), 98.5 (C1α,), 83.0–82.1 (C4, C4′), 72.4–66.1 (C2, C2′, C2Δ, C3, C3′, C3Δ, C5, C5′, CH2O), 32.2–30.4 (CH2), 19.3–19.1 (CH2), 14.0–13.8 (CH3).
ESI-HRMS [M + Na]+ calcd. for C38H64O18Na: m/z 831.3990. Found: m/z 831.3979.
Acknowledgements
The authors acknowledge the Conseil régional de Picardie (Programme alternatives végétales) for financial support. The authors thank Serge Pilard (plate-forme analytique, institut de chimie de Picardie, université de Picardie Jules Verne) for MS-MS spectrometry and helpful discussion, and David Lesur (laboratoire des glucides) for LC-MS and HPLC analysis.
Appendix A Supplementary data
HSQC spectrum of starting dp3. HSQC spectrum of modified dp3.