

1 Introduction
Excited-state intramolecular proton-transfer (ESIPT) fluorescent dyes, attributed to their intrinsic peculiar four-level cyclic proton-transfer reactions, are a family of intensively investigated functional materials for their wide applications in chemosensors, electroluminescent materials, laser dyes, biochemistry, UV-photostabilizers as well as the peculiar photophysical process [1–3]. It is well known that ESIPT molecules are normally more stable as enol forms in the ground state and more stable as keto forms in the excited state. An extremely fast four-level photophysical cycle (E-E*-K*-K) occurs immediately after photoexcitation of the intramolecular H-bonded molecules, mediated by the intramolecular H–bonds. Therefore, an abnormally large Stokes shift without self-absorption is detected and no self-absorption is detected [1], even in the solution with relatively higher concentration. For their good photostability and no self-absorption properties, ESIPT compounds are expected to be potential intrinsic luminescent materials.
Recently, it has been investigated that specific structures such as J-aggregation with a head-to-tail arrangement and dendritic architecture with site-isolation or restriction of rotation and torsion of the intermolecular bond, by greatly blocking the nonradiative channel, could effectively suppress the self-quenching and thus will notably enhance the luminescence intensity in their aggregate state or solid state. This kind of peculiar enhanced emission in the solid state was the so-called aggregation-induced emission enhancement (AIEE) [4,5].
In this review, we summarize AIEE phenomena of ESIPT compounds and some extended application of the system.
2 Aggregation-induced emission enhancement (AIEE) of excited-state intramolecular proton-transfer (ESIPT) compounds
The first example of AIEE of ESIPT in our research was the compound N,N’-bis(salicylidene)-p-phenylenediamine (p-BSP). p-BSP is one of salicylideneaniline (SA) type and a symmetric aromatic Schiff base molecule from its structural formula (Fig. 1) [6].
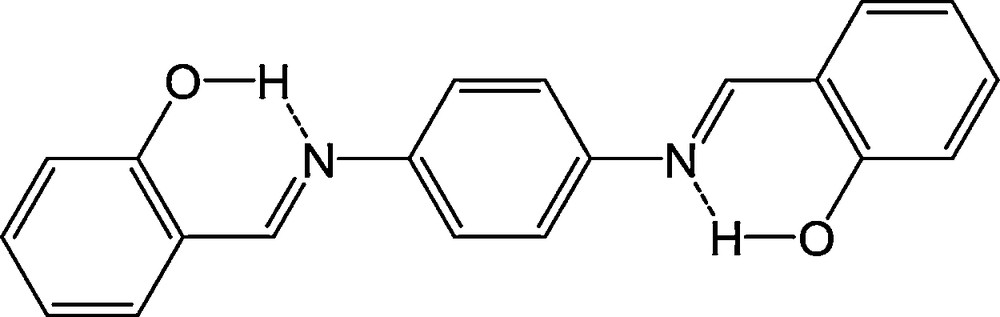
The structure of p-BSP.
The fluorescence spectra of p-BSP nanoparticles (aggregates) in water and methanol dilute solutions are shown in Fig. 2a. p-BSP in methanol shows one peak with the maximum at 535 nm and a flat and very weak roof from 480 to 425 nm. Different from that of the methanol solution, the fluorescence emission of nanoparticles has only one peak with the maximum at 546 nm and is aging-time dependent. p-BSP shows a large change of fluorescence intensity from disperse molecules in methanol to the nanoparticles in water. The relative quantum yield (Φf) increases from 0.0011 in dilute solution to 0.060 in the nanoparticle solution. The maximum of Φf for p-BSP nanoparticles is more than 60 times higher than that of the dilute methanol solution. With increasing aging time, intensities of the emission decrease monotonically but the peak positions are consistent. The Φf decreased in turn from 0.060 to 0.055, 0.051, 0.048, 0.040, and 0.025. Fig. 2b contains the excitation spectra of nanoparticles with various aging times. The shapes of most excitation spectra are very analogous to their absorption spectra.
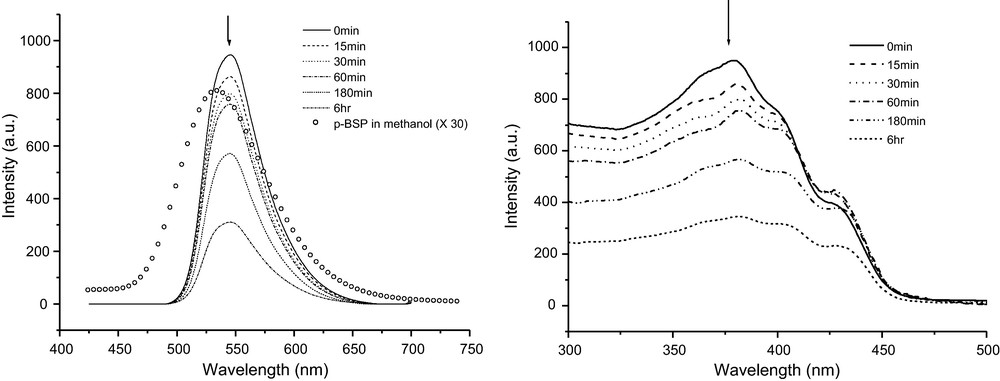
a (left): Fluorescence emission spectra of p-BSP nanoparticles (1.0 × 10−5 mol/L in molecule) in water at different aging time and p-BSP in methanol solution (1.0 × 10−5 mol/L, multiplied by a factor of 30). b (right): Fluorescence excitation spectra of p-BSP nanoparticles (1.0 × 10−5 mol/L in molecule) in water at different aging time.
A novel class of 2-(2’-hydroxyphenyl)benzothiazole-based (HBT-based) excited state intramolecular proton transfer (ESIPT) compounds, N,N’-di[3-Hydroxy-4-(2’-benzothiazole)phenyl]isophthalic amide (DHIA) and N,N’-di[3-Hydroxy-4-(2’-benzothiazole)phenyl]5-tert-butyl-isophthalic amide (DHBIA) has been feasibly synthesized (Fig. 3) and the properties of their nanoparticles in THF/H2O mixed solvent were investigated. Both compounds were found to exhibit AIEE in solid state. On identical experimental conditions, the emission of DHBIA aggregates increased more remarkably than that of DHIA, as shown in Fig. 4 [7].

The synthesis of DHBA and DHBIA.

Photographs of DHIA (a) and DHBIA (b) with concentrations both of 5 × 10−6 mol/L in THF dilute solution (left) and in THF/H2O mixture (right, THF: H2O = 38:62 (v/v)) under irradiation at 365 nm.
3 Understanding the emission properties of excited-state intramolecular proton-transfer (ESIPT) compounds
Enhanced emission in organic nanoparticles is an interesting phenomenon and may have some potential applications. The reasons for these phenomena were interpreted in terms of the intra- and intermolecular effects exerted by fluorophore aggregation. The increase of rigidity in a molecule can lead to the decrease of vibration in a molecule, in addition to probably decreased internal conversion of excited molecules, and may increase the emission. Intermolecular effects in conjugated chromophores are correlated with aggregation structures such as H- and J-aggregates, etc.
In p-BSP, both intra- and intermolecular effects may affect the enhanced emission of p-BSP nanoparticles. In the good solvents such as THF and methanol, the p-BSP molecule is nonplanar and may undergo some conformational changes. The two halves of the molecule are much less coupled due to the free rotation around the single bond. As mentioned above, p-BSP in solution exhibits photochromism due to ESIPT. When THF and water are mixed, p-BSP molecules aggregate together due to their poor solubility in water. Rotations around a single bond are restricted. It is hard to form the photochromic species. These would indicate that photochromism disappears in nanoparticles. In addition, the p-BSP in crystal is a planar symmetric molecule, so the intermolecular aggregation would induce planarization and rigidity in water. Molecular mechanics force field computations confirm this presumption. The optimized geometry of a single p-BSP molecule shows two phenyl rings of salicylidene are perpendicular to a third phenyl ring in the molecule. When the second p-BSP molecule is added to the system, the two molecules may influence each other and result in a more planar structure than a single molecule in water. We noticed that emission of nanoparticles decreased with increasing aging time. Intermolecular charge transfer induced self-quenching is considered a possible reason for this [6–8].
Therefore, the enhanced emission of p-BSP is attributed to the combined effects of molecular planarization, restricted molecules motions, and J-aggregate formation and the latter two may be the main factors. When the crystal forms at the longer aging time, the intermolecular charge transfer between the molecules is increased. It enhances the nonradiation relaxation of the excited energy. A relatively smaller emission than at the beginning is then detected for the sample with the longer aging time.
For DHIA and DHBIA, in molecular dispersed dilute solutions, two end-substituted HBT units of the molecules could rotate freely around the single bonds and the radiant decay would be effectively quenched by this kind of intramolecular torsion. While in the aggregate state, the intramolecular rotation and torsion were greatly impeded and therefore the nonradiative decay channel was effectively restricted, which in turn populated the irradiative state of the excited molecules and resulted in a great increase of fluorescence. Hence, it was easy to understand why the molecularly dispersed dilute solutions of DHIA and DHBIA were so weakly luminescent while their nanoparticles and aggregates were highly emissive.
The noteworthy discrimination of DHIA and DHBIA in the fluorescence spectra and their aggregate shapes allowed us to hypothesize that DHIA and DHBIA might present different aggregation modes. Also, such differences could possibly facilitate the discriminated degrees in their emission enhancement, about eight times between these two compounds. Thus, it was necessary for us to carry out deeper investigations into the discrepancy in the aggregation modes. Molecular modeling on the basis of molecular mechanic calculation was applied for further analysis. To enlarge conjugation for minimizing thermal energy in the system, two possible arrangements (face-to-face/head-to-tail) were possessed by aggregates of DHIA and DHBIA molecules. In a parallel face-to-face aggregation, full ð-ð stacking was the primary alignment for molecules of DHIA (Fig. 5a). The distances between molecules were 3.5 Å, which was a common interval in planar aromatic system in crystal stacking. Molecules of DHIA had, somewhat, the tendency to aggregate head-to-tail as well, but with enhanced thermal energy. For the coexistence of two ways of aggregation, the face-to-face packing was dominant. However, for DHBIA, due to the bulky t-Butyl groups, the molecules were not able to possess an energy-favored full parallel π–π stacking. Forcing two molecules parallel to each other, the intermolecular distances will arrive at more than 5 Å, which indicated a much weaker intermolecular interaction and the corresponding crystal could not be formed. Thus the face-to-face aggregation, in energy optimization, is not the favored packing fashion for DHBIA. When adopting a head-to-tail aggregation (Fig. 5b), the distance between two HBT subunits of two adjacent molecules would be restrained at 3.4 Å, which indicated an intensified π–π stacking between the tail HBT arm of one molecule and the head HBT arm of another molecule. Meanwhile, the whole molecular structure became more planar in this stacking mode. With such a head-to-tail aggregation, microscopically one dimensional molecular stacking could be observed for DHBIA aggregates. Their simulated aggregation fashions correlated well with distinguish growth tendency of DHIA and DHBIA particles in SEM observations, respectively [7].
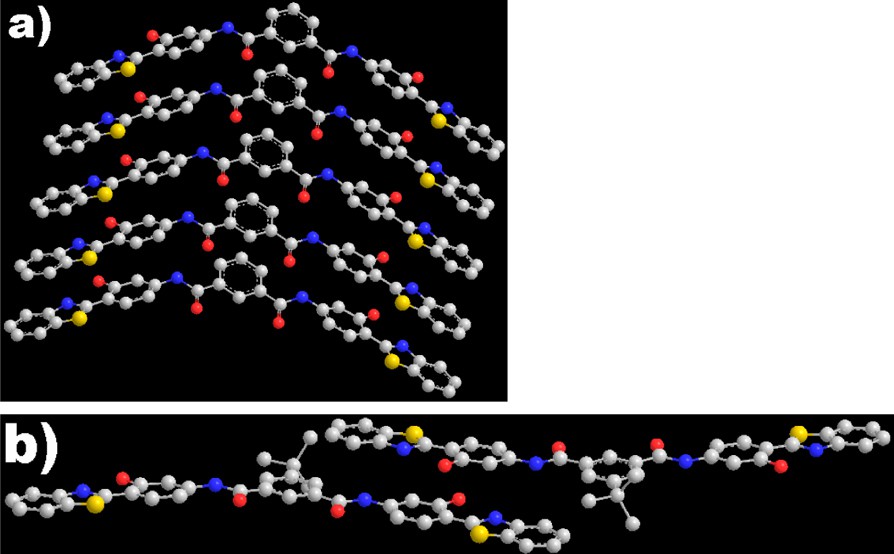
Simulated stacking modes of DHIA (a) and DHBIA (b) molecules utilizing π–π stacking of HBT subunits.
Moreover, since the proton transfer process could be inhibited by an intermolecular H–bond formed with surrounding solvent molecules, the ESIPT compound is microenvironmentally sensitive. Thus the emission characteristics of ESIPT compounds are expected to be microenvironment dependent. Some ESIPT compounds may exhibit dual luminescence bands that can be adjusted with various solvents or even circumambient pressure, temperature conditions. A recent discovery in our group shows that a triple-luminescent ESIPT compound (SalHBP, shown in Fig. 6) may give rise to a nearly white emission, simultaneously emitting blue, green, and yellow light under high pressure and tunable multiband emission property of luminescent ESIPT compound in different solvents with different polarity provides a possible route for fabricating a white-light-emitting source with a single component luminescent material [9].
Structure of SalHBP (intra-intra EE).
The fluorescence spectra of SalHBP in meTHF at various pressures are shown in Fig. 7. At 1 atm, the emission of SalHBP solutions was very weak and showed two peaks with maxima at 420 and 538 nm. When the pressure was increased to about 5 kbar, the emission at 420 nm increased by almost ∼3 times, which was a rarely observed phenomenon. Until the pressure increased to ∼31 kbar, the fluorescence intensity reached its maximum and afterward began to decrease. At the long wavelength region, a new shoulder emission peak around 500 nm was formed at higher pressure, which could not be detected at ambient pressure. This peak increased immediately after the pressure was enhanced and became obvious at 15.9 kbar and further increased until 31.2 kbar. In the lower energy region, the peak around 538 nm at ambient pressure also enhanced slowly with pressure. With higher pressure up to 38.9 kbar, the peak obviously increased and prevailed the fluorescence of the ∼500 nm peak. Note that the iso-emissive point around 530 nm was detected from 31.2 kbar to 54.2 kbar; this implies a conformational transition between two fluorescent species. As can be seen, the emission at ∼538 nm exhibited a completely different pressure-dependent effect with respect to the ∼500 nm emission.

Fluorescence emission spectra of SalHBP in meTHF under various pressures (excitation wavelength: 355 nm).
On the basis of special structure, SalHBP molecules were expected to exhibit three possible emission bands, separately, coming from EE, KE, and EK tautomers. For the emission at 420 nm, the mirror-image relationship and small Stokes shift between the emission and absorption bands indicated that the emission was originated from the EE excited-state form. As for two emissions at the longer wavelength region, the large Stokes shifts revealed that both of them could be assigned to the proton-transfer excited-state characters. As is already known, ESIPT fluorescence of HBT and SA had been reported to be located at ∼500 and ∼530 nm, respectively. Therefore, emission at ∼500 nm of SalHBP would originate from KE species and another emission at 538 nm would be from EK species. The assignments of these three emissions were confirmed by the analysis of emission and excitation spectra of the SalHBP solution at different temperatures. Thus, changes of SalHBP emission under various pressures could be depicted as follows. With the increase in pressure, the EE emission was enhanced first and then the KE emission increased, and finally the KE emission decreased with increase in the EK emission.
The fluorescence spectra of SalHBP in various solvents are shown in Fig. 8. Unlike the case of absorbance in the solvents (almost no change in different solvents), SalHBP exhibited a significant change in both fluorescence shape and intensity with different polarity of the solvent. In meTHF solution, SalHBP showed two peaks with maxima at ∼416 and ∼538 nm, respectively, but only one emission band at ∼538 nm was observed in MCH solution. Similarly, SalHBP also exhibited two emission bands in protic solvent-methanol, ∼416 and ∼538 nm, respectively. But the emission in the high-energy area increased more than one order of magnitude compared to that of the meTHF solution. With decreasing temperature, the fluorescent intensities of SalHBP in the three solvents presented a monotonous enhancement. The locations of the emission were almost unchanged while there were some differences in spectral shapes from different solvents. In an alcoholic mixture, the main emission band was around 410 nm and it was enhanced only about four-fold from 280 to 140 K. Moreover, a remarkable increase of a weak emission band at ∼500 nm was observed when the temperature was below 180 K. From an observable point of view, the SalHBP alcoholic solution presented apparent blue emission over the whole measured temperature range, which was over and above a few cyan characteristics at lower temperature. In nonpolar MCH solution, SalHBP exhibited only a wide broad emission band with a maximum at 495 nm. With a temperature decrease from 280 to 140 K, the intensity of the emission was increased by almost 28-fold. In the case of meTHF solution, SalHBP exhibited an emission band with a peak at about 538 nm. Also, two weak shoulders were observed at around 420 and 500 nm, respectively. At 140 K, the SalHBP solution emitted a brilliant yellowish-green light with a few white light characteristics for its blue light component. As observed above, SalHBP could emit blue (CIE1931 0.20, 0.16) in methanol/ethanol, green (CIE1931 0.32, 0.46) in MCH, and yellowish green (CIE1931 0.33, 0.42) in meTHF without obvious temperature-dependent color changes.
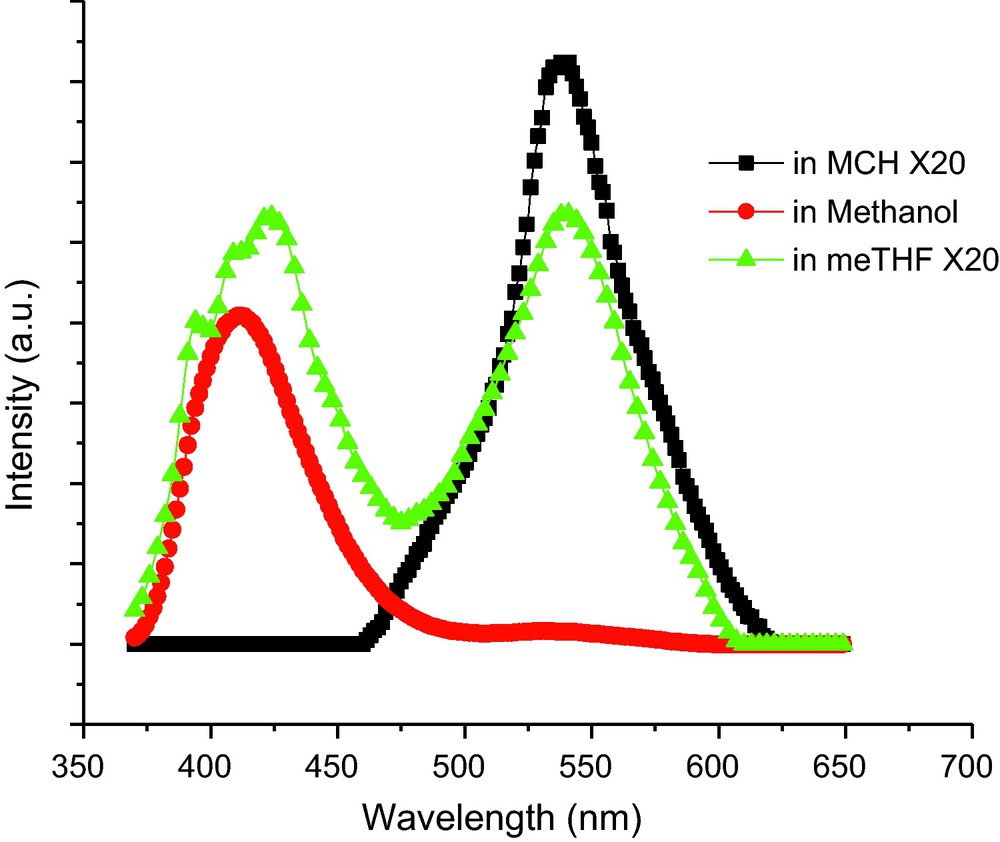
Normalized fluorescence emission spectra of SalHBP (1.0 × 10−5 M) in various solvent at room temperature. Emissions in MCH and meTHF have been multiplied by 20-fold, respectively. Excitation wavelength: 355 nm.
To directly compare the emission of SalHBP in different solvents, the normalized fluorescent spectra are given in Fig. 9. It was obvious that all three spectra were composed of several (at least three) emission bands located at about 420, 490, and 538 nm. The three emission bands were presumed to come separately from excited state EE, KE, and EK tautomers. The locations of the three emission bands showed no apparent shifts in different solvents. It indicated that the solvent did not have an apparent effect on the electronic configuration of the excited state SalHBP molecules, but influenced heavily the population of the three excited state tautomers.
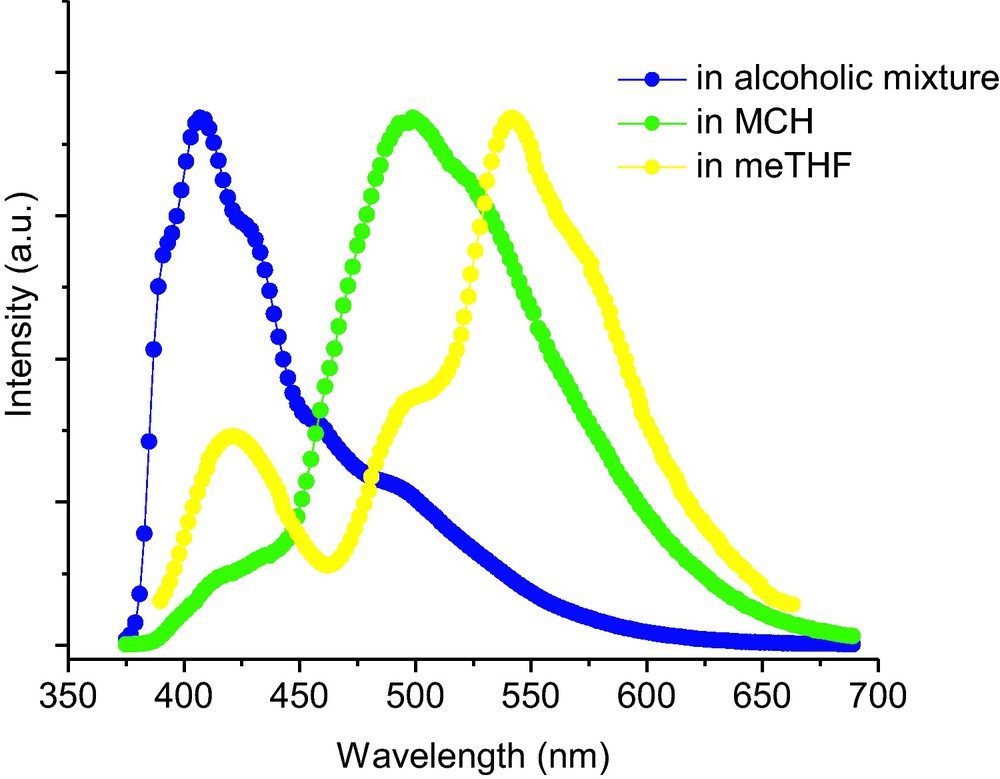
. Normalized fluorescent spectra of SalHBP at 140 K. Excitation wavelength: 355 nm.
From the calculated results for various lowest excited singlet state conformations, the emission of SalHBP in different solvents was considered as follows: (1) in methanol, the blue emission was assigned from the excited state tautomer with inter-inter EE2 conformation; (2) in MCH, the green emission with a wide band was from both intra-intra KE and intra-intra EK conformations; and (3) in meTHF, the relatively high polarity of solvent increased the population of conformations with large excited state dipole moment, which induced the red shift of the main emission band compared to the case in MCH. By combining these luminescences in suitable proportions, we can obtain the pseudo white light source with a single dye.
Fig. 10 showed the colorful emission from SalHBP dispersed in copolymer (mole ratio of HEMA:VP is 3:7), PS, and PMMA. A commercial LED (365–375 nm) illuminated weak blue light. A thin transparent polymer layer was coated onto the LED head. Upon illumination, these LEDs generated white light, bluish green light and yellowish green light, respectively. On the chromaticity diagram, these emissions showed CIE coordinates at (0.29, 0.35), (0.26, 0.38), and (0.30, 0.42), in which the white emission was close to that of pure white light (0.33, 0.33).

Photos of: (a) a reference UV LED (365–375 nm) without illumination; (b) the illuminating reference LED; (c) the same LED coated with a thin layer of SalHBP HEMA-VP copolymer solution without illumination; (d) the illuminating LED shown in part (c), (e) the same LED illuminating a coated thin layer of SalHBP PS solution, and (f) the same LED illuminating a coated thin layer of SalHBP PMMA solution.
In PS, the case of SalHBP was similar to that in MCH where two intramoleculer H–bonds of SalHBP exist most of the time, and the emission originated from two ESIPT excited state configurations and presented a green light for low polarity of PS. In PMMA, with higher polarity compared with MCH, PS, and meTHF, PMMA could increase the population of the tautomer with longer wavelength emission. So in PMMA, yellowish green light was observed upon illumination. As SalHBP dissolved in HEMA-VP copolymer, the case was more complicated. Each excited state conformation with different luminescence mentioned above was populated in the copolymer. This phenomenon was also explained by balance between intra- and intermolecular H–bonds. Both HEMA and VP presented a great chance to form an intermolecular H–bond with dissolved SalHBP. But the possibility decreased when monomer was polymerized to polymer for the limitation of free motion of the macromolecules. Suitable limitation resulted in a suitable population of the conformations with different color emission. By tuning the ratio of HEMA to VP in the copolymer, the coated LED generated pseudowhite light that was a combination of all emissions from several conformations of SalHBP, covering the whole visible range from 400 to 700 nm. Apparently, SalHBP can be considered as a potential material for single-component pseudowhite light application.
4 Energy transfer in the aggregation of the excited-state intramolecular proton-transfer (ESIPT) compound
As a general consideration, the doped system with energy transfer is a possible choice to obtain various colorful luminescence materials. Energy transfer is based on the distance dependence between a donor and an acceptor via radiative, nonradiative dipole–dipole, or electron exchange interaction, which results in a decrease of donor emission and an increase of acceptor emission. It is facile to adjust emission color through a variety of donor/acceptor ratios. But most organic light emitting materials exhibit strong nonradiative decay in the solid state, which presents the luminescence quantum efficiency decreasing exponentially with peak broadening and bathochromic shift in comparison with that in dilute solution. This limits the practical application of organic materials in solid-state light-emitting devices. Meanwhile, it is important to develop organic materials with strong luminescence and guest-host systems with highly efficient energy transfer in the solid.
In our work, a series of new ESIPT compounds exhibited obvious enhancement of luminescent intensity in their aggregation solids. We chose an ESIPT compound DHBIA as host material due to its AIEE characteristic and a red light emission compound (2-((1,2-dihydro-1,2,2,4-tetramethylquinolin-6-yl)methylene-amino)-3-((10E)-(1,2-dihydro-1,2,2,4-tetramethylquinolin-6-ylimino)methyl)fumaronitrile))) (MAFN) as the guest molecule due to its solvatochromism. Several kinds of organic nanoparticles were prepared with different doping levels by the reprecipitation method and emissions with different colors were observed [10,11].
Fluorescence spectra of organic MAFN/DHBIA nanoparticles with different doping levels (MAFN:DHBIA: 0–4%) are presented in Fig. 11a. Pure DHBIA nanoparticles showed one peak with a maximum at 510 nm. Different from that of pure DHBIA nanoparticles solution, the fluorescence of doped nanoparticles exhibited two peaks. The peak at 510 nm without location changes in the range of doping was attributed to the emission of the donor-DHBIA, while the other peak that was changed from 620 to 659 nm with increased doping concentration should be ascribed to emission from acceptor-MAFN. The emission of DHBIA was decreasing rapidly with gradual increasing of the doping concentration. As the doping concentration was increased to 4%, the fluorescence of DHBIA was almost quenched, which meant that a complete energy transfer from DHBIA to MAFN occurred. Meanwhile, the doped systems showed a highly efficient antenna effect. When the nanoparticles with 4% doping concentration were excited at 355 nm, the fluorescence intensity of MAFN around 659 nm was 29 times higher than that with excitation at 560 nm, which was irradiated directly to MAFN and shown in the insert of Fig. 11a. Additionally, we could see directly from the photograph that the fluorescence of doped nanoparticles could be tuned from green to red (Fig. 11b) when the doping concentration was increased.
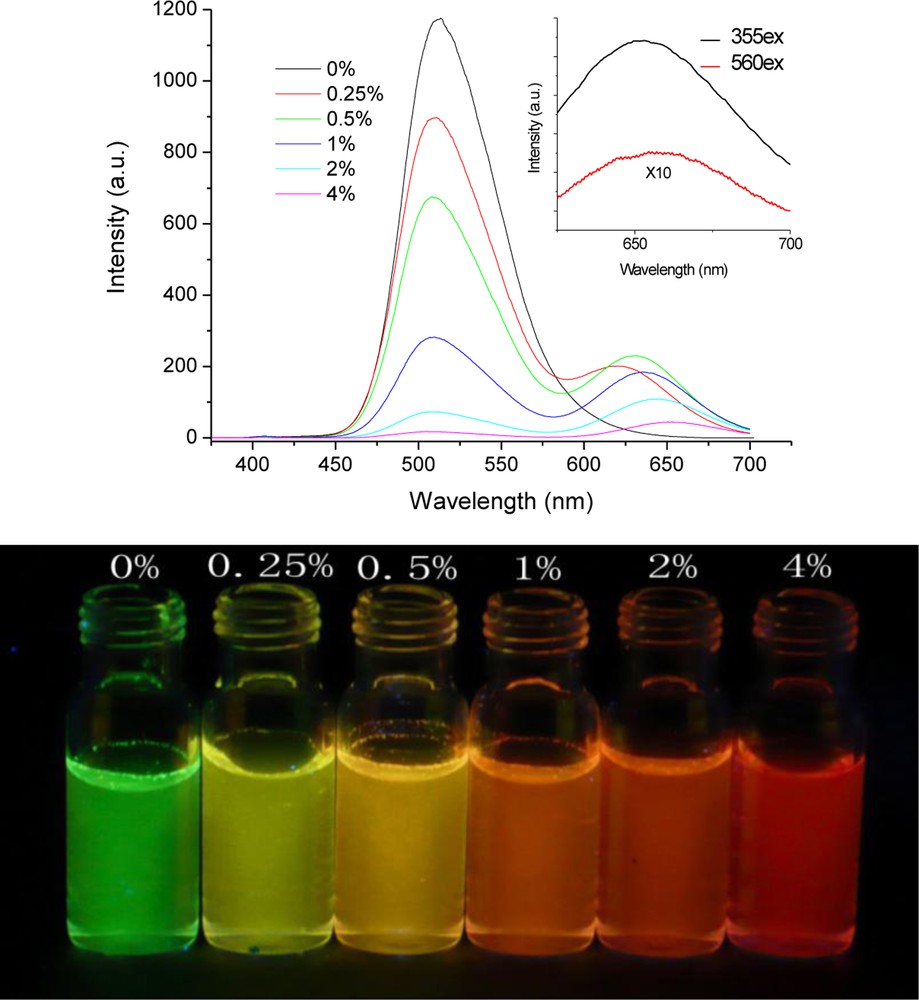
(a) The fluorescence spectra of the nanoparticles with different doping concentrations (excitation with 355 nm). (b) The photograph of nanoparticles under a ultra-violet lamp (365 nm).
The Förster energy transfer mechanism depends on the overlap integral J between the energy donor and acceptor. The two spectra of DHBIA and MAFN had good overlap and that J was calculated as high as 1.05 × 1015 M−1cm−1nm4, which implied that a highly efficient intermolecular energy transfer from DHBIA to MAFN could be expected.
The dependence of the DHBIA fluorescence intensity on the concentration of MAFN showed a positive deviation from the Stern-Volmer linear relation, especially in the high doping concentration range. These results indicated that as the concentration of MAFN was increased, there was a gradual increase in the quenching of DHBIA in doped nanoparticles. These phenomena were so-called “amplified quenching” or “superquenching”. The nanoparticles were considered as systems of densely packed chromophores that showed efficient energy transfer from DHBIA to MAFN. The simple energy transfer model is not suitable to characterize a multi-object system, such as doped nanoparticles and other solid samples. An energy transfer model in nanoparticles was used to analyze these systems. The model involved the effect of multiple quencher species and the statistical distribution of quenchers, and assumed that the fraction of nanoparticles with n quenchers per nanoparticle could be described by the Poisson probability distribution function. The fluorescence quenching results was approximated as:
Fluorescence quenching data could be fitted directly by using the energy transfer model and the experiment data and fit curve were shown in Fig. 12. Fit parameters N and q were 138 and 0.955, respectively, which indicated that 95.5% of the fluorescence of 138 DHBIA molecules would be quenched by a single MAFN molecule. The volume of the DHBIA molecule was calculated as 0.82 nm3. So the fit result meant that one MAFN molecule could quench with high-efficient its surrounding DHBIA located in about 3 nm range. Thus, the Förster energy transfer radius in the doped nanoparticles was estimated to be near 6 nm.

Φ0/Φ of DHBIA versus molar fraction of MAFN in doped nanoparticles. The dotted curve was the fit result. The solid line represented the Stern-Volmer relationship in initial stage.
5 Fluorescent sensor with the aggregation of the excited-state intramolecular proton-transfer (ESIPT) compound
Fluorescent chemosensors with high specificity and sensitivity, ease, and safety of handling have received considerable attention, and a number of fluorescence sensors have been reported that are capable of detecting fluoride ions. The recognition proceeded mostly through hydrogen bonding or Lewis acid coordination, and the sensors could only be operated in organic solvents to detect tetrabutylammonium (TBA+) fluoride rather than inorganic fluoride salts. This incompatibility with aqueous environments is one of the main drawbacks that restrict the application of these sensors. Unavoidable interference from H2PO4−, AcO−, or CN− ions is the other disadvantage. To improve the performance of fluoride sensors, another strategy based on the chemical affinity between fluoride and silicon was developed. However, several tens of minutes or even hours are needed to complete the detection process because low concentrations of these chemodosimeters severely reduce the reaction rate between the fluoride ions and the silyl moieties. However, the low concentration is necessary for general organic dyes to avoid fluorescence quenching induced by concentration effects of self-absorption and self-quenching. More importantly, fluorescence intensity changes are not suitable for direct observation with the naked eye.
We developed a rapid and portable sensor for fluoride ions in aqueous solution [12]. The sensor, which has a high sensitivity, operates through the special affinity between fluoride ions and silicon, and provides two independent modes of signal transduction based on fluoride-dependent changes of fluorescence color (color metric mode) or intensity (power metric mode), respectively. We chose N-(3-(benzo[d]thiazol-2-yl)-4-(hydroxyphenyl) benzamide (3-BTHPB)) as an ESIPT compound. Just like other ESIPT compounds, 3-BTHPB shows two emission bands, which originate from the enol and keto forms at 418 and 560 nm, respectively. The ratio of the two bands is determined by the number of molecules that could undergo ESIPT reactions. We coupled the tert-butyldiphenylchlorosilane with the sodium salt of 3-BTHPB to afford a derivative N-(3-(benzo[d]thiazol-2-yl)-4-(tert-butyldiphenyl silyloxy)phenyl)-benzamide (BTTPB). As expected, BTTPB shows only blue-violet fluorescence, which was almost identical to the emission of the enol form of 3-BTHPB. Upon addition of fluoride ions, the Si–O bond of BTTPB is immediately cleaved, and 3-BTHPB, which exhibits a bright yellow emission in water, is simultaneously released. Therefore, it is logical to expect that the fluorescence color change from blue-violet to yellow upon addition of fluoride ions reflects the extent of the fluoride-induced Si–O bond cleavage. A schematic of the feasible fluorescence sensing mechanism in the fluoride ion detection is shown in Fig. 13.
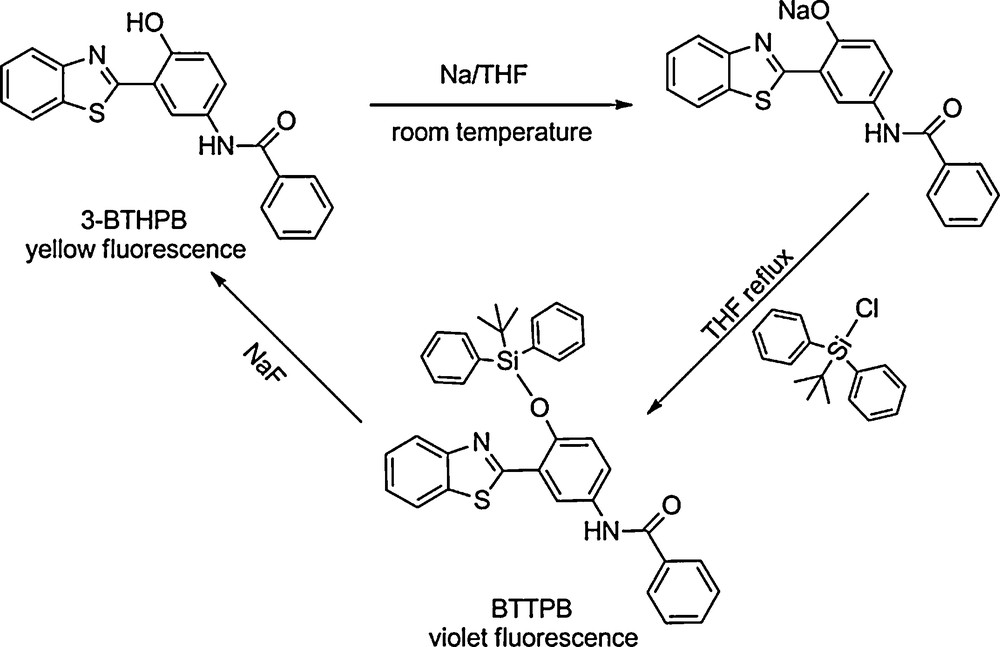
The synthesis of N-(3-(benzo[d]thiazol-2-yl)-4-(tert-butyldiphenyl silyloxy)phenyl)-benzamide (BTTPB), and the sensing mechanism of chemosensor BTTPB for detecting NaF.
Both BTTPB and 3-BTHPB are not soluble in water; to apply them efficiently in an aqueous environment, cetyltrimethylammonium bromide (CTAB) was introduced into the aqueous phase. A stable sensor system was prepared by rapidly injecting a solution of BTTPB in THF (100 μL, 2.0 mM) into a micellar solution of CTAB in water (10 mL, 2.0 mM) under vigorous stirring for 30 s at 20 °C.
UV/Vis absorption and fluorescence spectra were recorded with different amounts of NaF to test the sensing ability of the BTTPB dispersions (Fig. 14a, b). As expected, the addition of fluoride ions to the dispersion resulted in gradual changes in the absorption and emission spectra. Fig. 14b shows the fluorescence spectra of the BTTPB dispersion 200 seconds after addition of NaF with the excitation wavelength at the isosbestic point. The dispersion in the absence of fluoride ions exhibits only one emission band with a maximum at 418 nm, which is assigned to the BTTPB emission. Upon the addition of fluoride ions, the blue-violet emission band decreases and a new emission band located at 560 nm simultaneously appears and increases gradually. The fluorescence color change of BTTPB upon addition of one equivalent of fluoride ions (i.e., 0.38 ppm) is too small to be directly observed with the naked eye, but can be detected by fluorescence spectroscopy. When the fluoride ion concentration was further increased, the change can be seen with the naked eye. As shown in Fig. 14c, 0.95, 1.9, and 3.8 ppm of fluoride ions cause the dispersion fluorescence color to change from blue-violet to violet, pink-purple, and to pink-white, respectively. Therefore we conclude that the BTTPB/CTAB/water colorimetric sensing system is sufficiently sensitive for practical fluoride ion detection.
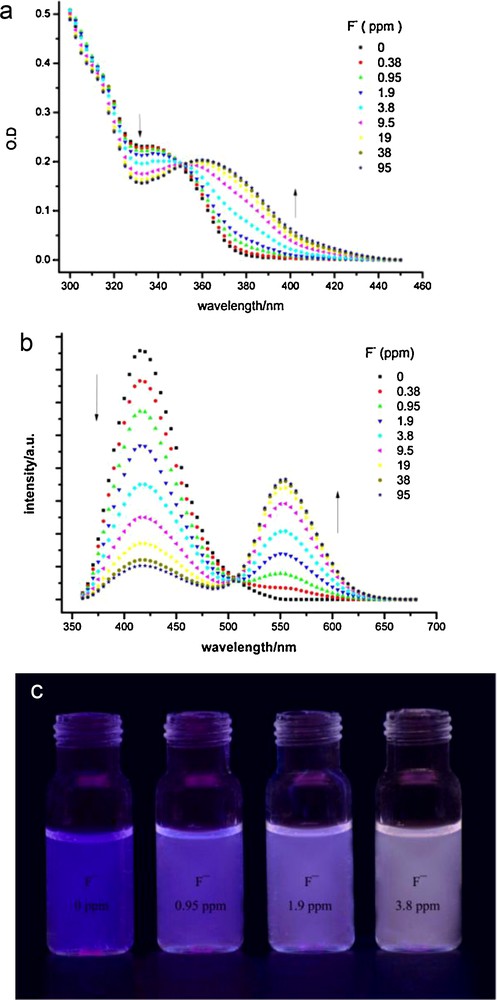
(a) Absorption and (b) fluorescence spectra of the N-(3-(benzo[d]thiazol-2-yl)-4-(tert-butyldiphenyl silyloxy)phenyl)-benzamide (BTTPB) dispersions with aging 200 s after addition of NaF; (c) photos of BTTPB dispersions upon addition NaF (0, 0.95, 1.9, 3.8 ppm, from left to right).
With the excitation wavelength at 385 nm, no emission was found in the absence of fluoride ions, and the intensity of the yellow emission increased drastically with fluoride ion concentration; this result is indicative of an “off–on” switch triggered by fluoride ions. The detection limit of the BTTPB dispersion system for fluoride ions is measured to be about 100 ppb. This result shows that in power metric mode the dispersion system has a very good sensitivity for the detection of aqueous fluoride ions. The selectivity of the BTTPB dispersion was tested by addition of 500 equivalents of common anions. Only NaF induced an immediate red shift in the fluorescence maximum from 418 to 560 nm. All other anions did not cause any emission intensity changes at 560 nm until 30 min after the addition of each anion. This result indicates that the system is a very good sensor for recognizing fluoride ions over other anions.
Fluorescence sensors for fluoride ion detection are usually solution-based. This approach is inconvenient since the sensors cannot be used as efficient tools under special circumstances, such as for in situ on-site detection. To facilitate the use of our system, we prepared test papers of BTTPB by immersing a filter paper in the solution of BTTPB in THF (2.0 × 10−3 M) and then drying it by exposure to air. For the detection of fluoride ions in water, the test paper was immersed in a fluoride-containing aqueous solution (containing 2 mM CTAB) and then exposed to air to remove water and to avoid further fluorescence color changes. The Commission internationale de l’éclairage (CIE) 1931 (x,y) chromaticity diagram of the test papers after immersion in solutions of NaF with different concentrations for three minutes are shown in Fig. 15.
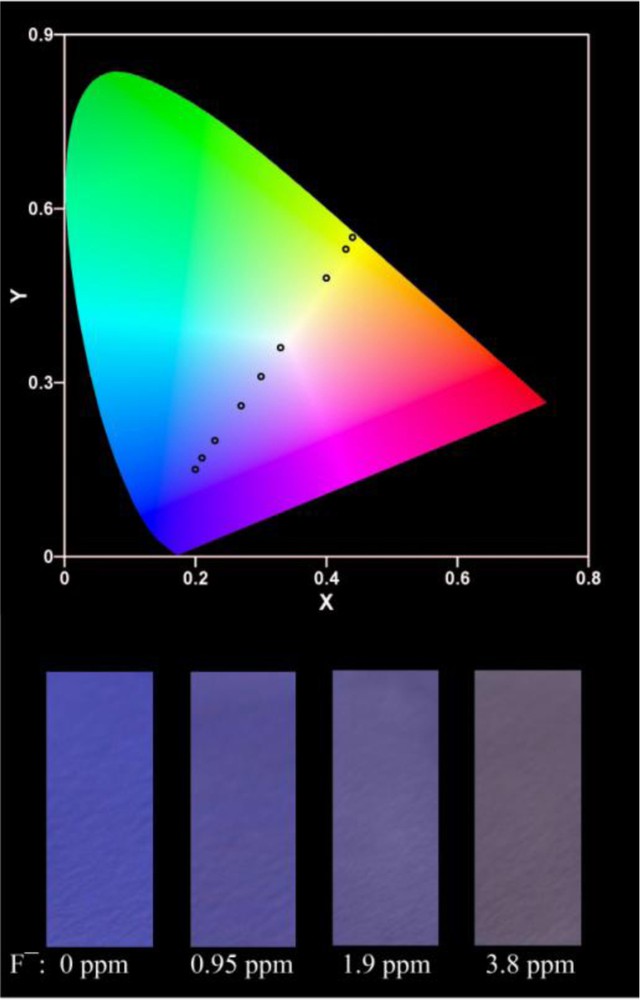
CIE 1931 (x,y) chromaticity diagram of the test paper for detecting NaF with different concentrations derived from fluorescence spectra. From left to right, NaF concentration: 0, 0.38, 0.95, 1.9, 3.8, 9.5, 19, 38, 95 ppm; and photos of the test papers after immersion into different concentrations of NaF.
When the fluoride ion concentration was increased, the color of the test paper changed from blue-violet to bright yellow. The immersion time was optimal for the detection of fluoride ions in the range of the enforced drinking water standard. According to the diagram, it can be easily seen if the fluoride ion concentration in drinking water exceeds the standard. Consequently, the easy-to-prepare test paper can be utilized to roughly and quantitatively detect and estimate the concentration of fluoride ions.
6 Summary
A series of novel ESIPT compounds were designed and synthesized. AIEE phenomena were observed in these compounds when their aggregates were formed in aqueous solution. The AIEE of the molecules were mainly caused by the restricted intramolecular motion as well as larger population of intramolecular H–bonds. Further investigation revealed that the different stacking modes were possibly responsible for different extents in the emission enhancement. Because of the high emissive efficiency of the aggregate, the aggregate is an ideal sample to study the energy transfer in solid state. Quantitatively, analysis of the energy transfer from donor to acceptor in the solid indicates that one energy acceptor could quench the emission of the energy donor in several nanometers. The strong emission in aqueous solution of the aggregates provides a selective and sensitive method to detect aqueous fluoride anion based on the specific affinity between fluoride and silicon. All the results demonstrate that the ESIPT compounds would be novel optical functional materials in the solid state.
Acknowledgements
We are grateful for the financial supports of National Natural Science Foundation of China (Grant Nos. 20703049, 20733007, 20873165, 50973118), the National Basic Research Program (2007CB808004, 2009CB930802) and Chinese Academy of Sciences.