

1 Introduction
Nanoparticles have attracted growing attention over the last two decades due to their various applications in many areas ranging from energy to biology. Among them, luminescent metallic and semiconducting nanoparticles hold a particular status in relation to their unique electronic and optical properties. Metallic nanoparticles have driven the blooming of plasmonics and proven of foremost interest for various applications including catalysis, therapy and diagnosis while semiconductor-based nanoparticles (i.e., quantum dots) show size-dependent luminescent properties and have gained overwhelming popularity in many areas including nonlinear optics, optoelectronics and biological imaging. A number of them however raise toxicity and biodegradability issues. In this context, Fluorescent Organic Nanoparticles (FONs) have emerged as a complementary alternative towards ultra-bright luminescent nanoparticles with readily tunable composition and properties [1–3]. These nanoparticles of fully organic nature result from the self-assembly of organic dyes and offer an attractive route for “bottom-up” engineering of optical properties by rational design of the molecular structure of the chromophoric subunits. In this respect, several structural features (electronic distribution, polarity, geometry, topology, adjunction of bulky pendant groups…) of the molecular subunits play a prominent role as they determine the nature and strength of intermolecular interactions (π-stacking, electrostatic interactions, Van der Waals interactions…) operating between chromophoric subunits within FONs. Such interactions may influence both the conformation (as is the case in planarized systems) [4] as well as the relative orientation and positioning of chromophoric subunits. This in turn modulates the through-space and electronic coupling between chromophores which can significantly affect the luminescence properties. As an example, if linear and compact dipolar chromophores (such as push-pull chromophores of the D-π-A type which show strong intramolecular charge transfer (ICT) transitions) are used, dipole–dipole interactions can be deleterious to luminescence as they favor anti-parallel close packing [5]. Such an arrangement of chromophoric subunits can also strongly influence the two-photon absorption (2 PA) properties leading to a reduction of the 2 PA molecular response [6]. However the use of dipolar chromophoric subunits built from propeller-shaped donating moieties (with triphenylamine as the prototypical D subunit) [7–11], basically prevents the anti-parallel arrangement of the dipolar chromophoric subunits in the condensed phase and promotes shifted arrangement instead [8–10]. This strategy led to FONs exhibiting giant one- and two-photon brightness (up to 108 M−1 cm−1 and 106 GM for green-emitting FONs of about 35 nm in diameter) [8,9]. Furthermore by playing on the nature of the π-conjugated system as well as on the strength of electron-withdrawing end groups, FONs showing luminescence tunable from the visible to the NIR region as well as improved colloidal stability and remarkable chemical and photostability have been achieved [8,9]. This demonstrated that molecular engineering of the chromophoric building blocks also allow to tune the surface properties of resulting FONs, providing evidence for the potential of the bottom-up approach based on the use of polar and polarizable chromophoric subunits. Such FONs have proved to afford biocompatible ultra-bright luminescent contrast agents for in vivo angiography by two-photon imaging [10] as well as photostable NIR emitters for single particle tracking, thus offering attractive substitutes to toxic QDs [11]. These FONs yet typically show lower fluorescence quantum yields as compared to their chromophoric subunits in chloroform solution while the emission wavelength is similar. We posited that this behavior is related to the shifted anti-parallel orientation of dipolar chromophoric subunits within FONs (as is observed in the microcrystalline state) [8,10], resulting in a reduction of the radiative decay rates and in the onset of competing non-radiative deactivation processes. Based on this assumption, we decided to investigate symmetrical chromophoric derivatives where basically two short dipolar chromophoric moieties having a triphenylamine as the electron-donating group (EDG) and either a carbaldehyde or a strong dicyanovinyl as the electron-withdrawing group (EWG) are connected via a fluorene moiety that acts as a connecting rigid axis linking the two dipoles while allowing conformational freedom (Fig. 1). Depending on the conformation favored when such derivatives are confined to close proximity, different electrostatic interactions are expected to come into play: dipole–dipole interactions are substantial only between confined chromophores in the syn conformation while chromophores in the anti conformation show vanishing dipole moment. Molecular confinement of dyes within organic nanoparticles is thus expected to influence the molecular conformation and subsequent molecular packing of the chromophoric subunits. This is of major importance as it was shown – in the case of confined dipolar chromophoric subunits – to have a major influence on the peak 2 PA response (with both negative and positive cooperative effects being reported depending on the relative orientation of the dipolar subunits in multichromophoric molecular architectures) [6,13,14]. In the case of “articulated” bis-dipolar systems as dyes 3–4,a–b, we also expect different contributions to the 2 PA response depending on the molecular conformation (dipolar and quadrupolar contribution for the syn conformation, but only the quadrupolar contribution for the anti conformation). Hence, the different symmetry and flexibility of the chosen dyes as compared to simple “linear” push-pull chromophores open an interesting route for taking advantage of interchromophoric interactions for both enhancing the luminescence (in particular, if confinement also favors planarization of the conjugated system as described for sterically hindered systems showing AIEE, i.e., aggregation-induced emission enhancement [12]) and influencing the 2 PA response of the molecular subunits within FONs to achieve enhanced 2 PA absorption. The present route thus offers an attractive way to investigate the combined influence of symmetry and flexibility of chromophoric subunits built from dipolar modules on the confinement-induced effects on both luminescence and two-photon absorption, aiming at achieving enhanced two-photon brightness for bioimaging applications.
Structure of “articulated” bis-dipolar dyes for the design of FONs with enhanced two-photon brightness.
2 Results and discussion
2.1 Synthesis of dyes 3,4-a,b
Chromophores 3,4-a,b were synthesized according to the synthetic route depicted in Scheme 1. Diphenylamination of 9H-fluorene,9,9-dibutyl-2,7-diiodo or 9H-fluorene, 9,9-bis(3-methylbutyl)-2,7-diiodo was performed by Ullmann condensation of diphenylamine in the presence of copper powder and a base (K2CO3) as described in the literature [15]. We note that the synthesis of compound 2a was previously reported in higher yields starting from 9H-fluorene,9,9-dibutyl-2,7-dibromo and using a Buchwald Hartwig procedure. This however required the use of air-sensitive, difficult to handle and toxic phosphine P(t-Bu)3 [16]. We thus privileged an alternative protocol which is more sustainable. In the next step, a Vilsmeier-Haack formylation of compounds 2a,b conducted in a POCl3-DMF mixture afforded the di-formyl derivatives 3a and 3b in moderate yields. Finally, subsequent Knoevenagel condensations with malononitrile conducted in refluxing ethanol yielded derivatives 4a and 4b in good yields. All derivatives were isolated as pure compounds after purification by column chromatography and fully characterized by 1H and 13C NMR, IR spectroscopy as well as HRMS and elemental analyses. The alkyl chain grafted on the fluorene moiety which extend over and below the fluorene plane are meant to prevent π–π stacking that would lead to fluorescence quenching. Both linear (nBu) and ramified (isopentyl) chains were used to check the possibility of tuning through-space interactions by playing on the topology of these hindering entities of a similar length but different bulkiness. This offers a likely route for further improving fluorescence properties.
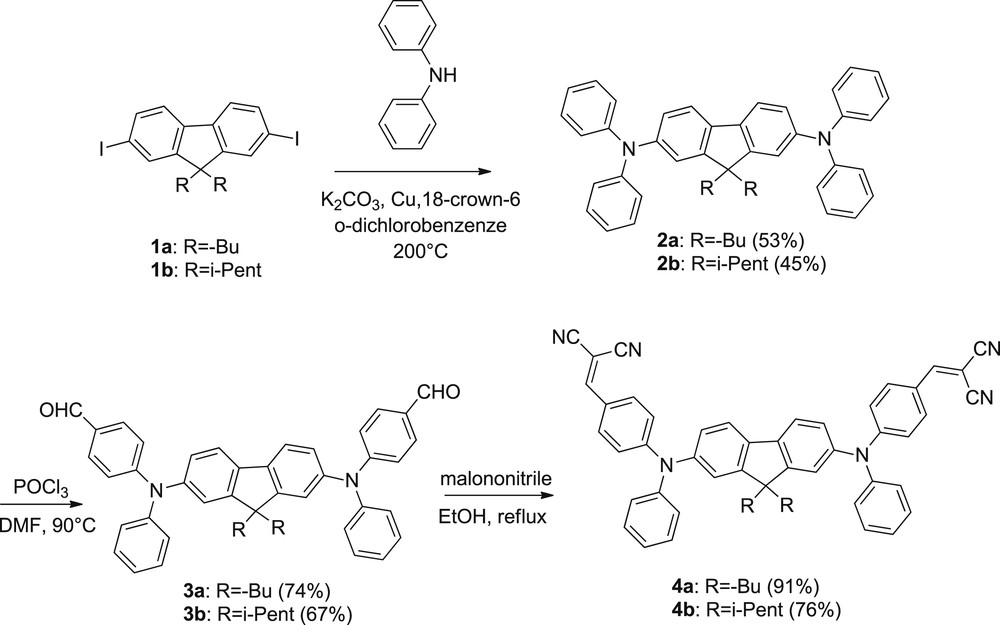
Synthesis of dyes 3–4,a–b.
2.2 Synthesis and characterization of fluorescent organic nanoparticles (FONs)
Organic nanoparticles built from chromophores 3a, 3b, 4a or 4b were easily obtained using an adaptation of the reprecipitation procedure initially proposed by Nakanishi and coll [2]. The controlled addition of a minute volume of a mM stock solutions of chromophores in THF into a large excess of deionized water readily leads to colored and limpid solutions. This simple protocol conducted at room temperature does not require inert atmosphere and costly experimental conditions and was found to be robust and reproducible. Subsequently to absorption characterization that attested the formation of chromophore-based nanoparticles, the presence of spherically shaped nanoparticles was confirmed by transmission electronic microscopy (TEM). As shown in Fig. 2, dynamic light scattering (DLS) experiments evidenced narrow size distribution with a typical hydrated diameter in the 100 nm range and “dry” radius of about 30 nm (see Table 1). Interestingly, organic nanoparticles made from chromophores 3–4,a–b show very negative ζ potential (around −70 mV) of major promise in terms of colloidal stability.

Top: DLS data for FONs 3a,b (left) and 4a,b (right). Bottom: TEM images of freshly prepared FONs 3a (left) and 4a (right).
Characteristics of FONs made from articulated bis-dipolar dyes 3–4,a–b.
| Dha [nm] | DTEMb [nm] | Zeta potential [mV] | Nc | εmax Φf/NPsd [×108 M−1 cm−1] | σ2max Φf/NPse [×106 GM] | λemmax [nm] | |
| FONs 3a | 87 | 29 | −66 | 11500 | 0.83 | 0.27 | 518 |
| FONs 3b | 99 | – | −76 | (16500) | 1.6 | 0.43 | 514 |
| FONs 4a | 95 | 36 | −69 | 19000 | 0.3 | 0.25 | 661 |
| FONs 4b | 99 | – | −73 | (22000) | 0.3 | 0.32 | 660 |
a Hydrodynamic diameter derived from DLS experiments.
b Diameter of the FONs measured by TEM.
c Number of dye subunits per FONs.
d One-photon brightness.
e Two-photon brightness.
2.3 Photophysical properties of dyes and related FONS
The absorption and fluorescence characteristics of chromophores 3–4,a–b in organic solution and corresponding organic nanoparticles in water suspension are collected in Table 2. As illustrated in Fig. 3, chromophores 3–4,a–b dissolved in an organic solvent show an intense absorption band in the near UV (3a–b) or blue visible region (4a–b). A bathochromic and hyperchromic shift is observed when increasing the electron-withdrawing end group strength (3→4). Chromophores 3a–b show sizeable fluorescence emission in the visible region while chromophores 4a–b show only very weak fluorescence even in low polarity and viscous solvents (and vanishing fluorescence in polar solvents). Chromophores 3a–b show a marked red shift of their emission band upon increasing the solvent polarity and only a very slight shift of their absorption band (Fig. 4). This reveals an emissive excited state having a much larger dipole moment than the ground-state. Chromophores 4a–b show a similar behavior although the absorption band is more red-shifted while the emission band seems less sensitive to solvent polarity (though investigation of fluorescence variation in a large range of solvent polarity is precluded due to fluorescence quenching in polar solvents). Hence chromophores 3–4,a–b, though symmetrical in structure, show a solvatochromic behavior similar to that of a standard push-pull chromophore undergoing an increase of dipole moment upon excitation (such as DANS as a prototypical push-pull chromophore). Such behavior is similar to that reported for specific quadrupolar chromophores showing symmetry breaking in the excited state [17–19], although in the present case the two dipolar subunits are not sharing a common donor core but are instead articulated dipolar subunits connected by a rigid fluorene module.
Photophysical data of dyes 3–4,a–b dissolved in THF and as self-aggregated molecules within FONs in water.
| λabsmax [nm] | εmax [104 M−1 cm−1] | λemmax [nm] | Stokes shift [103 cm−1] | Φfa | FHWM [cm−1]b | τ [ns]c | 2λabsmax [nm] | λ2PAmax1 [nm]d | σ2max1 [GM]e | λ2PAmax2 [nm]d | σ2max2 [GM]e | |
| 3a/THF | 382 | 5.6 | 530 | 7.3 | 0.11 | 4400 | 2.2 | 764 | 800 | 62 | 740 | 144 |
| FONs/H2O | 381 | 4.8 | 518 | 6.9 | 0.15 | 3900 | 1.6 (35%) 5.0 (65%) | 762 | 820 | 107 | 740 | 157 |
| 3b/THF | 381 | 5.3 | 530 | 7.4 | 0.10 | 4400 | 1.8 | 762 | 800 | 74 | 730 | 145 |
| FONs/H2O | 385 | 4.3 | 514 | 6.5 | 0.23 | 4100 | 1.7 (30%) 5.3 (70%) | 770 | 820 | 80 | 740 | 113 |
| 4a/THF | 453 | 7.8 | 670 | 7.1 | <0.01 | 4400 | 0.23 | 906 | – | – | – | – |
| FONs/H2O | 458 | 7.4 | 661 | 6.7 | 0.02 | 3300 | 0.6 | 916 | 940 | 727 | 900 | 656 |
| 4b/THF | 452 | 7.9 | 667 | 7.1 | <0.01 | 4400 | 0.22 | 904 | – | – | – | – |
| FONs/H2O | 459 | 7.3 | 660 | 6.6 | 0.02 | 3400 | 0.8 | 917 | 970 | 526 | 890 | 719 |
a Fluorescence quantum yield.
b Full width at half maximum of the emission band.
c Fluorescence lifetime.
d Two-photon absorption (2 PA) band maximum.
e Two-photon absorption cross-section at λ2PAmax (1 GM = 10−50 cm4 s photon−1).

Normalized absorption and emission spectra of chromophores 3–4,a–b in solution in CHCl3 (left) and of corresponding FONs in H2O (right).
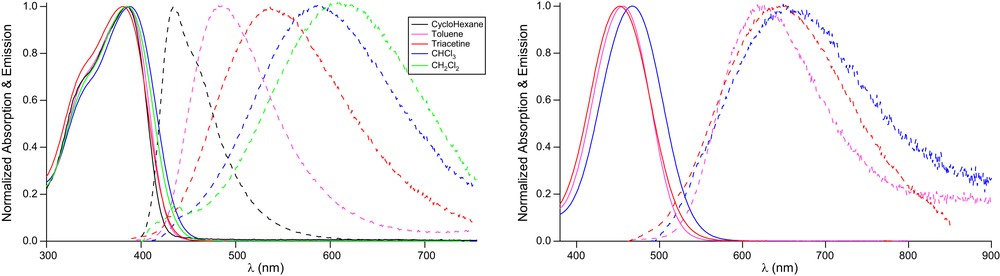
Solvatochromism of dyes 3a (left) and 4a (right).
All organic nanoparticle suspensions made from dyes 3–4,a–b were found to be fluorescent, confirming the formation of FONs. Strikingly, FONs made from chromophores 3a–b show enhanced fluorescent properties as compared to molecules dissolved in THF (Table 2), evidencing a typical AIEE emission behavior whereas FONs made from chromophores 4a–b are slightly fluorescent, contrary to their molecular counterpart in organic solvents, indicating a typical AIE behavior. Hence “articulated dipolar” dyes, although quite different in design from prototypical hindered chromophores [20–22] showing the AIE phenomenon, also lead to aggregation-promoted fluorescence enhancement. This demonstrates that the implanted engineering strategy-based on the control of dipole–dipole interactions by assembling two push-pull chromophoric modules within an articulated symmetrical structure (where the fluorene module plays the role of a rigid spacer also acting as a through-bond coupler) and on the presence of the alkyl chain acting as a ballast to prevent π–π stacking in the condensed state – is indeed successful in promoting enhanced luminescence.
As a result, FONs made from dyes 3a–b show nice green emission (λem = 530 nm) whereas those made from dyes 4a–b emit red fluorescence in water (λem = 660 nm). Quite interestingly, the green emission from FONs (3a,b) and red emission from FONs (4a,b) reveal a low-medium polarity environment as compared to dyes in organic solutions; i.e., in between toluene and THF. Furthermore, in the case of FONs made from dyes 3a–b, we observe that FONs exhibit two different lifetimes, the longer and dominant one being much longer than the one measured for dyes dissolved in THF. In the case of dyes 4a–b the accessible experimental lifetime values also point to a lengthened lifetime of the emitting excited state. This clearly indicates a marked decrease of the non-radiative decay rates (as is the case for the AIEE phenomenon), a behavior which was not observed in the case of FONs made from simple dipolar push-pull chromophores also having a triphenylamine as the electron-releasing group [8,10]. This provides evidence that the molecular arrangement of bis-dipolar dyes within the nanoparticles is indeed slowing non-radiative processes that compete with fluorescence emission.
Following the observation of the role of molecular confinement in the luminescence properties of the articulated dipole dyes, we further examined the role of the alkyl appending chains. We observe that the nature of the alkyl chains (linear: nbutyl or branched: isopentyl) does not influence the photophysical properties of dyes in solution, confirming that these appending chains do not influence the electronic transition of the chromophores dissolved in an organic solvent. In contrast, we observe that the FONs made from 3a and 3b (as well as 4a and 4b to a lesser extent) exhibit different luminescence behavior, indicating that these chains do play a role in enhancing the luminescence upon molecular confinement of the dyes. The bulkier isopentyl chains do lead to larger fluorescence quantum yields (Table 2). Hence the “spatial control” of electrostatic interactions between multipolar chromophores within nanoparticles via the concomitant use of “ballast” appending moieties indeed plays a role. The lower impact on the luminescence properties of FONs made from dye 4b as compared to dye 4a further supports this statement and suggests that more efficient ballasts should be used when stronger dipolar moieties within articulated dyes are involved. As a result green-emitting FONs made from dye 3b exhibit a significant fluorescence quantum yield in pure water (23% instead of 15% for dye 3a). This underlines the appeal of controlling multipolar interactions (here by playing on both symmetry and the presence of hindering appending moieties) to generate enhanced emission in FONs made from dyes built from dipolar modules. As a result, green-emitting FONs (3b), although having a diameter of about 30 nm, show unprecedented brightness (1.6 108 M−1 cm−1) which is higher than the record brightness reported earlier for green-emitting FONs made from simpler linear dipolar chromophores [10] and showed much lower colloidal stability [8].
2.4 Two-photon absorption and brightness
Based on their design (i.e., articulated bis-dipolar dyes 3–4,a–b as chromophoric building blocks), we anticipated that FONs made from dyes 3–4,a–b could display a significant 2 PA response. Taking advantage of the fluorescence of dyes 3a–b in solution, as well as of FONs made from 3a–b and 4a–b, the 2 PA spectra could be experimentally determined by using the well-known two-photon excited fluorescence (TPEF) methodology [23]. This relies on the measurement of the excitation spectra using two-photon excitation in the red-NIR region (rather than one-photon excitation in the near UV–Visible region). Such measurements require the use of short-pulse lasers (most of the time generating ∼ 100 fs pulses) to provide sufficient peak power during the pulse duration. Relative measurements can be performed using a range of two-photon standards [23–25]. This technique also has the advantage to allow reliable measurements of the 2 PA cross-sections (σ2) by checking the quadratic dependency of the generated fluorescence signal as a function of excitation intensity (thus indicating that neither saturation nor contribution from one-photon absorption occurs). It is however limited to dyes which have a detectable fluorescence. Due to this limitation, no precise and reliable 2 PA cross-section values could be derived from TPEF measurements for dyes 4a–b in solution. In contrast, TPEF measurements could be performed with FON suspension in water thanks to their enhanced fluorescence.
The 2 PA spectra of dyes 3a–b in solution in THF are shown in Fig. 5 and compared with their rescaled 1 PA spectrum. The comparison of the one- and two-photon absorption spectra clearly evidences a slight change in the spectral shape: a broadening of the 2 PA band is clearly observed compared to the 1 PA band. A less intense sub-band is observed at a slightly lower energy compared to the lower energy excited state attained upon one-photon excitation while a more intense sub-band is observed at higher energy (see also Table 2). These features could be related to the different symmetry-based selection rules for allowed versus forbidden transitions for 1P and 2P absorption. The less intense red-tail (hidden maximum located above 800 nm) might be ascribed to contribution of the syn conformation of dyes 3a–b in solution to the overall response (i.e., dipolar contribution) whereas the more intense band at higher energy (maximum observed at 740 nm) might be ascribed to the contribution of both anti and syn conformations of dyes 3a–b (both of them having quadrupolar contribution). The peak 2 PA response amounts to about 150 GM, which is reasonable given the small size of the chromophore and nature of electron-donating (NPh2), π-connector (Ph) and electron-withdrawing (CHO) moieties.

Comparison of one- and two-photon absorption spectra of 3a (left) and 3b (right) in THF.
Interestingly, the 2 PA spectra of the dyes when confined in FONs show similar features (Fig. 6). In addition, dyes 4a–b in FONs show much larger 2 PA responses than dyes 3a–b indicating that the stronger EWGs lead to much larger 2 PA peak response (650–720 GM) in that case. We stress that these 2 PA peak values (∼700 GM) are significant for a small chromophore suggesting that through-bond coupling (via the fluorene axis) between the two dipolar moieties is indeed operative in enhancing the 2 PA response of the dyes. Interestingly, the comparison between molecular 2 PA responses of dyes 3a–b as isolated molecules in solvents or confined within FONs yields interesting clues: the peak 2 PA responses are either maintained (3a) or only slightly reduced (3b) with respect to the corresponding 2 PA peak response of isolated chromophores in organic solutions (Table 2). Moreover, both 2 PA spectra show broadened spectrum (most probably in relation to inhomogeneous broadening) and increased 2 PA responses on the red-edge of the 2 PA band. This is an exceptional feature since previous studies conducted on push-pull dipolar (or octupolar) fluorophores showed that dipolar/multipolar interactions promoted by molecular confinement within FONs led to a significant decrease of the peak 2 PA response of the dye subunits in the NIR region as compared to isolated molecules in solution [8,10]. This reduction could be ascribed to a negative cooperative effect of dipolar interactions between chromophoric subunits [6,14], as a result of the shifted anti-parallel orientation of dipolar chromophoric subunits in the solid state [8,10]. Here the deleterious effect of the anti-parallel orientation promoted by dipolar interactions between (rod-like) dipolar subunits is annihilated thanks to the different nature (geometry, symmetry and flexibility) of the dyes that constitute the FONs. This difference clearly points to the positive effect of the “articulated dipoles” structure of dyes 3 and 4 that promotes “self-organization” within FONs that are more favorable to both luminescence and two-photon absorption characteristics of the chromophoric subunits compared to isolated molecules in an organic solvent. Another interesting aspect in this respect is that, in contrast, a decrease of the 1 PA peak response (i.e., εmax) is observed upon confinement of the dyes within FONs (Table 2). The concomitant increased transparency and enhanced two-photon absorption response of the dye subunits when self-assembled within FONs is the proof of concept that by playing on the symmetry and flexibility of articulated bis-dipolar chromophores to pilot interchromophoric interactions, desirable enhancement of both luminescence and two-photon absorption properties can be achieved also in FONs. Such a strategy was demonstrated earlier only in the case of dipolar multichromophoric architectures built for the covalent attachment of specific dipolar dyes onto a dendrimeric backbone [13,14]. We demonstrate here that such enhancement can also be achieved in much more easily accessible molecular-based nanoparticles by using articulated bis-dipolar chromophoric building blocks. This approach thus opens the gate for the dipolar-promoted aggregation-induced two-photon fluorescence enhancement phenomenon (AI2PE).
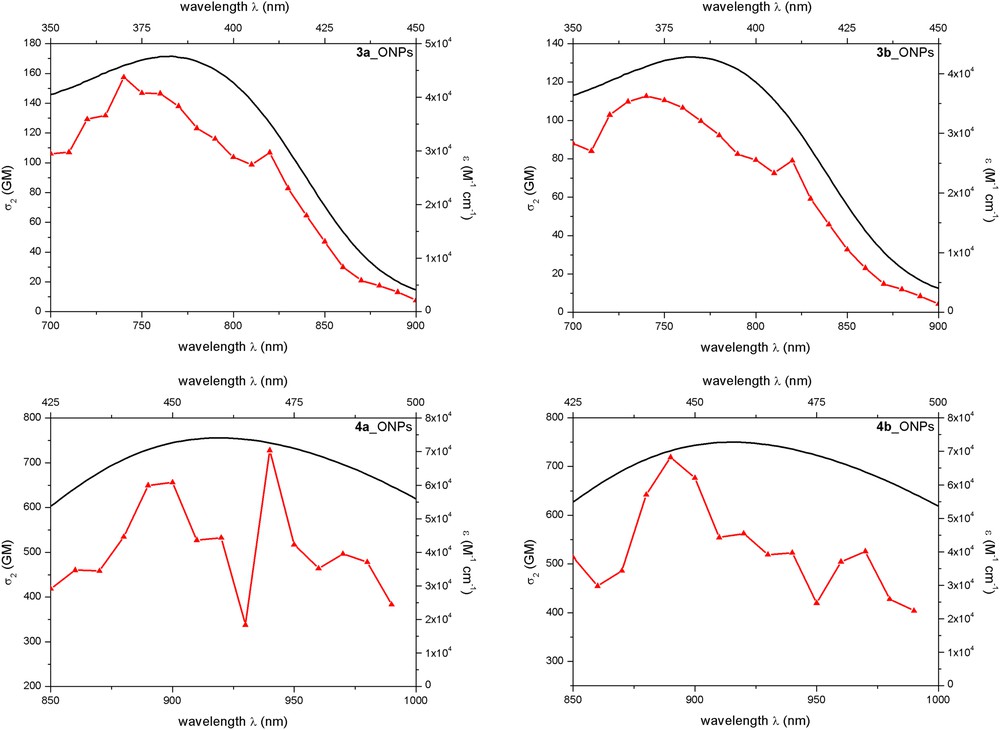
Comparison of one- and two-photon absorption spectra of top: FONs (3a) (left) and 3b (right) and bottom: FONs (4a) (left) and 4b (right) in water.
2.5 Chemical and colloidal stability of FONs in water
With the aim to use these FONs as markers and tracers, we investigated their chemical and colloidal stability by monitoring the evolution of their size as well as their absorption and emission properties over time. Even after two months, the overall size of NPs is found to remain relatively stable while no precipitation occurs. Only with a slight increase of the average diameter size (10–15%), these FONs experience very slow maturation and other growing processes. This remarkable colloidal stability can be ascribed to the highly negative ζ potential. In addition, the photophysical properties of the FONs remain almost unchanged with steady luminescence quantum yields and no modification of either absorption or emission spectra. This remarkable feature indicates that the confinement and organization of the dyes within FONs are extremely stable and the dyes do not rearrange over time thus preserving the photophysical characteristics. Furthermore, albeit chromophores 4a–b are water-sensitive (and undergo retro-Knoevenagel reactions in polar solvents like acetone or acetonitrile in the presence of trace of water) and yield back chromophores 4a–b, the retro-Knoevenagel reaction is totally suppressed in FONs made from dyes 4a–b revealing that the molecular confinement within FONs precludes reactivity between dyes on the surface and water molecules from the bath. As such FONs made from dyes 3a–4b show chemical and colloidal stability reminiscent of those of inorganic nanoparticles emphasizing their potential as tracers or markers for future applications in imaging.
3 Conclusion
In conclusion, the bottom-up approach for the design of ultra-bright FONs for bioimaging purposes based on the replacement of linear push-pull chromophores by articulated bis-dipolar dyes proved successful. Luminescence enhancement was observed in FONs compared to dyes dissolved in THF in opposition to what was reported earlier for FONs made from more classical push-pull chromophores [8–10]. Although such a phenomenon is well known for typical AIEE systems (i.e., hindered conjugated systems which undergo planarization upon aggregation), planarization of the conjugated system is not involved in the present case, as indicated by the elongated fluorescence lifetimes. This unveils different mechanisms for fluorescence enhancement based on the restriction of dipole–dipole interactions. Moreover, due to the specific nature of the bis-dipolar dyes, cooperative enhancement of the 2 PA response and/or broadening of the 2 PA spectra is achieved while a marked decrease of the peak 2 PA response of molecular subunits of FONs made from linear push-pull chromophores was observed earlier [8]. As a result, the combined effect of molecular confinement of articulated bis-dipolar dyes on luminescence and 2 PA leads to FONs with giant two-photon brightness. Indeed green-emitting FONs (3b) exhibit giant two-photon brightness of 430 000 GM while red-emitting FONs (4b) show a record 2P brightness among red-emitting nanoparticles (320 000 GM). In addition, these nanoparticles show exceptional chemical, structural and colloidal stability, retaining their size and luminescence properties over long periods of time. This provides evidence that the articulated bis-dipolar scheme has also a positive effect on the surface properties (as attested by the highly negative ζ potential) and on the nanostructuration of the FONs. As such these novel molecular-based nanoparticles hold major promise as ultra-bright nanotools (markers and tracers) for applications in (bio)imaging.
4 Experimental section
4.1 Synthesis
4.1.1 General methods
Commercially available reagents (purchased from Aldrich and Alfa) were used without further purification. Dry solvents were distilled from the appropriate drying reagents immediately before use. All air- or water-sensitive reactions were carried out under argon. Reactions were monitored by thin-layer chromatography carried out on silica gel precoated aluminium sheets (60F-254). Column chromatography was performed using Fluka silica gel Si 60 (40–63 μm, 230–400 mesh). Melting points were measured on Stuart SMP 10. 1H and 13C spectra were recorded on a Bruker Advance I 300 spectrometer at 300 MHz and 75 MHz, respectively, or on a Bruker Advance II 400 spectrometer at 400 MHz and 100 MHz, respectively. Chemical shifts are given in parts per million with respect to the solvent residual peak and the coupling constant (J) is given in Hz. HRMS spectra were recorded by CESAMO (Bordeaux, France) on a JEOL AccuTOF GCv mass spectrometer using an FD emitter with an emitting voltage of 10 kV. Infrared spectra were recorded on a Nicolet iS10 FT IR spectrometer in the range 4000–1000 cm−1. Elemental analyses were carried out at ICSN (CNRS Gif/Yvette, France).
4.1.2 9H-Fluorene-2,7-diamine, 9,9-dibutyl-N2, N2, N7, N7-tetraphenyl (2a)
To a mixture of diiodofluorene, (1.32 g, 2.5 mmol), diphenylamine (928 mg, 5.5 mmol), dry potassium carbonate (2.76 g, 10 mmol) and 18-crown-6 (13 mg, 0.5 mmol) in 1,2-dichlorobenzene under argon, copper powder (32 mg, 0.5 mmol) was added and the suspension was heated to 200 °C for two days. After filtration using Celite and evaporation to dryness, the residue was chromatographed on a silica column eluting with a petroleum ether/CH2Cl2 (90/10) mixture. The expected product was obtained as a white powder (53% yield, 812 mg). Mp 205 °C, 1H NMR (DMSO-d6, 300 MHz) δ 7.65 (d, J = 8.3 Hz, 2H), 7.29 (m, 8H), 7.04 (m, 14H), 6.64 (dd, J = 8.3, 1.9 Hz, 2H), 1.77 (m, 4H), 1.03 (m, 4H), 0.68 (t, 6H, J = 7.2 Hz), 0.60 (m, 4H). 13C NMR (CDCl3, 100 MHz) δ 152.27, 148.30, 146.71, 136.50, 129.36, 123.88, 123.67, 122.62, 119.98, 119.71, 55.15, 40.00, 26.34, 23.20, 14.18. Anal. Calcd for C45H44N2: C, 88.19; H, 7.24; N, 4.57. Found: C, 87.78; H, 7.20; N, 4.79.
4.1.3 9H-Fluorene-2,7-diamine, 9,9-di(3-methylbutyl)-N2, N2, N7, N7-tetraphenyl (2b)
The same procedure was applied for compound 2b starting from the corresponding substituted diiodo fluorene derivative (1b). 2b was obtained in 45% yield as a white solid after column chromatography (silica, eluent: Petroleum ether-CH2Cl2, 90:10). It could be recrystallized from heptane. Mp 183 °C; 1H NMR (DMSO-d6, 300 MHz) δ 7.64 (d, J = 8.3 Hz, 2H), 7.29 (m, 8H), 7.08 (d, J = 1.9 Hz, 2H), 7.03(m, 12H), 6.93 (dd, J = 8.3, 1.9 Hz, 2H), 1.79 (m, 4H), 0.87 (m, 2H), 0.67 (d, J = 6.6 Hz, 12H), 0.47 (m,4H). 13C NMR (CDCl3, 100 MHz) δ 152.15, 148.23, 146.69, 136.54, 129.36, 123.95, 123.61, 122.58, 120.00, 119.67, 54.99, 38.17, 33.05, 28.46, 22.80.
4.1.4 Benzaldehyde, 4, 4′-[(9,9-dibutyl-9H-fluorene-2,7-diyl)bis(phenylimino)]bis (3a)
POCl3 (0.6 ml) was added dropwise to a solution of 2a (918 mg) in dry DMF (7.5 ml). The mixture was heated at 90 °C for 16 h. After cooling, the reaction mixture was poured on ice and carefully neutralized with a saturated NaHCO3 solution, and then the precipitate was filtered, rinsed with water and dried. The residue was chromatographed on a silica column using a CH2Cl2/MeOH (98/2) mixture. After eluting a small fraction of the monoaldehyde, the dialdehyde was obtained in 74% yield (741 mg). Mp 186 °C. IR (cm−1) 1687 νCO. 1H NMR (CDCl3, 300 MHz) δ 9.87 (s, 2H), 7.73 (d, J = 8.8 Hz, 4H), 7.64 (d, J = 7.9 Hz, 2H), 7.39 (m, 4H), 7.18 (m, 14H), 1.86 (m, 4H), 1.12 (m, 4H), 0.76 (t, J = 7.4 Hz, 6H), 0.70 (m, 4H). 13C NMR (CDCl3, 75 MHz) δ 190.42, 153.39, 152.60, 146.31, 145.22, 137.67, 131.34, 129.72, 129.24, 125.99, 125.28, 125.04, 120.95, 120.60, 119.61, 55.27, 39.69, 26.19, 22.94, 13.98. Anal. Calcd for C47H44N2O2: C, 84.40; H, 6.63; N, 4.19. Found: C, 84.34; H, 6.66; N, 4,00. HRMS (FD) calcd. for C47H44N2O2: 668.3403. Found: 668.3406.
4.1.5 Benzaldehyde, 4, 4′-[(9,9-bis(3-methyl)butyl-9H-fluorene-2,7-diyl)bis(phenylimino)]bis (3b)
The same procedure was applied starting from derivative 2b and compound 3b was obtained in 67% yield as a yellow solid after column chromatography (silica, eluent : CH2Cl2/MeOH, 98:2). Mp 195 °C. IR (cm−1) 1687 νCO. 1H NMR (CDCl3, 300 MHz) δ 9.86 (s, 2H), 7.73 (d, J = 8.9 Hz, 4H), 7.64 (d, J = 7.9 Hz, 2H), 7.39 (m, 4H), 7.18 (m, 14H), 1.87(m, 4H), 1.32 (m, 2H), 0.74 (d, J = 6.6 Hz, 12H), 0.58 (m, 4H). 13C NMR (CDCl3, 75 MHz) δ 190.40, 153.39, 152.51, 146.31, 145.24, 137.77, 131.34, 129.74, 129.22, 121.01, 125.36, 125.04, 120.91, 120.66, 119.53, 55.10, 37.84, 32.94, 28.16, 22.56. Anal. Calcd for C49H48N2O2: C, 84.45; H, 6.94; N, 4.02. Found: C, 84.18; H, 7.03; N, 3.85. HRMS (FD) calcd. for C49H48N2O2: 696.3716. Found: 696.3717.
4.1.6 Propanedinitrile,2,2′[[(9,9-dibutyl-9H-fluorene-2,7-diyl)bis(phenylimino)]4,1-phenylenemethylidyne] bis (4a)
A solution of 3a (67 mg, 0.1 mmol), malononitrile (8 mg) and β-alanine (1 mg) in absolute ethanol (2 ml) was heated to reflux for 24 h. The bis Knoevenagel adduct precipitate and water (1 ml) was added to the cool mixture in order to ensure complete precipitation. The red-orange powder was collected by filtration, rinsed with water and dried. It could be purified by column chromatography on silica (eluent: CH2Cl2). Yield 91%. Mp 218 °C. IR (cm−1) 2215 νCN. 1H NMR (CDCl3, 400 MHz) δ 7.77 (d, J = 8.8 Hz, 4H), 7.66 (d, J = 8.8 Hz, 2H), 7.55 (s, 2H) 7.41 (m, 4H), 7.25 (m, 6H), 7.16 (m, 4H), 7.04 (d, J = 8.8 Hz, 4H) 1.85 (m, 4H), 1.09 (m, 4H), 0.72 (t, J = 7.7 Hz, 6H), 0.66 (m, 4H). 13C NMR (CDCl3, 100 MHz) δ 157.98, 153.60, 153.06, 145.51, 144.63, 138.44, 133.18, 130.13, 126.55, 126.24, 125.81, 123.23, 121.45, 121.13, 119.14, 115.41, 114.28, 75.90, 55.58, 39.77, 26.37, 23.05, 14.13. Anal. Calcd for C53H44N6: C, 83.22; H, 5.80; N, 10.99. Found: C, 82.47; H, 5.82; N, 10.75. HRMS (FD) calcd. for C53H44N6: 764.3627. Found: 764.3604.
4.1.7 Propanedinitrile,2,2′[[(9,9-bis(3-methyl)butyl-9H-fluorene-2,7-diyl)bis(phenylimino)]4,1-phenylenemethylidyne]bis-(4b)
The same procedure was applied starting from 3b. 4b could be purified by column chromatography on silica (eluent: CH2Cl2). Yield 76%. Mp 239 °C. IR (cm−1) 2220 νCN. 1H NMR (CDCl3, 400 MHz) δ 7.77 (d, J = 8.8 Hz, 4H), 7.67 (d, J = 8.8 Hz, 2H), 7.55 (s, 2H), 7.41 (m, 4H), 7.24 (m, 6H), 7.16 (m, 4H), 7.04 (d, J = 8.8 Hz, 4H) 1.86 (m, 4H), 1.29 (m, 2H), 0.71 (d, J = 7.6 Hz, 12H), 0.53 (m, 4H). 13C NMR (CDCl3, 100 MHz) δ 157.96, 153.60, 152.93, 145.49, 144.64, 138.53, 133.19, 133.19, 126.59, 126.27, 125.85, 123.21, 121.37, 121.17, 119.07, 115.42, 114.27, 75.90, 55.41, 37.93, 33.13, 28.31, 22.71. Anal. Calcd for C55H48N6: C, 83.30; H, 6.10; N, 10.60. Found: C, 83.14; H, 6.09; N, 10.56. HRMS (FD) calcd. for C55H48N6: 792.3940. Found: 792.3943.
4.2 Synthesis and characterization of fluorescent organic nanoparticles (FONs)
Nanoparticles of chromophores 3a, 3b, 4a and 4b were prepared using the reprecipitation method at room temperature. FON solutions were obtained via dropwise addition of 100 μL of a 1 mM spectroscopic grade THF stock solution of compounds 3a, 3b, 4a or 4b to 9.9 ml of deionized water under magnetic stirring. A fluorescent limpid solution is readily obtained as the addition is completed.
TEM was carried out using a HITACHI H7650. The copper grid coated with a carbon membrane was pretreated using the Glow discharge technique to yield a positively charged hydrophilic carbon surface to allow stronger interactions between the sample and the grid itself and thus easier imaging. One droplet of the dilute aqueous suspension was deposited on the grid and the excess liquid was eliminated with a paper. A staining procedure using uranyl acetate was then used to enhance the contrast. Taking advantage of the unimodal size distribution, the average number of dye subunits per FONs could be calculated using the radius values measured from TEM and assuming a density of 1, which is reasonable for such organic molecules.
Dynamic Light Scattering (DLS) was performed using a Zetasizer Nano SZ-100Z Horiba instrument operating at 173°. The hydrodynamic diameters Dh were calculated from diffusion coefficients using the Stokes–Einstein equation. All correlogram analyses were performed using the software supplied by the manufacturer. Zeta-potential analysis was performed with the SZ-100Z Horiba instrument. Several measurements were realized for each sample according to a predefined operating procedure.
4.3 Photophysical and two-photon absorption studies
All photophysical studies have been performed with freshly prepared air-equilibrated solutions at room temperature (298 K). UV/Vis absorption spectra were recorded on a Jasco V-670 spectrophotometer. Steady-state and time-resolved fluorescence measurements were carried out on a Fluorolog spectrofluorometer. Fully corrected emission spectra were obtained under excitation at the wavelength of the absorption maximum. Fluorescence quantum yields of dilute dye solutions were measured according to literature procedures using fluorescein in 0.1 M aqueous NaOH (Φ = 0.90) or Nile blue in ethanol (Φ = 0.27) depending on the emission range. The fluorescence quantum yield of the FON suspension in water was determined using a center-mount sample holder. Fluorescence decays were measured in a time-correlated single photon counting (TCSPC) configuration, under excitation from selected nanoLED (370 or 455 nm). The instrument response was determined by measuring the light scattered by a Ludox suspension. The lifetime values were obtained from the reconvolution fit analysis of the decay profiles; the quality of the fits was judged by the reduced χ2 value (χ2 <1.1). The reported lifetimes are within ±0.1 ns
Two-photon absorption cross-sections (σ2) were determined from the two-photon excited fluorescence (TPEF) cross-sections (σ2Φ) and the fluorescence emission quantum yield (Φ). TPEF cross-sections were measured relative to fluorescein in 0.01 M aqueous NaOH for 715–980 nm [23,24], using the well-established method described by Xu and Webb [23] and the appropriate solvent-related refractive index corrections [26]. The quadratic dependence of the fluorescence intensity on the excitation power was checked for each sample and all wavelengths. Measurements were conducted using an excitation source delivering fs pulses. This allows avoiding excited-state absorption during the pulse duration, a phenomenon which has been shown to lead to overestimated 2 PA cross-sectional values. To span the 680–1080 nm range, a Nd:YLF-pumped Ti:sapphire oscillator was used. The excitation was focused into the cuvette through a microscope objective (10X, NA 0.25). The fluorescence was detected in epifluorescence mode via a dichroic mirror (Chroma 675dcxru) and a barrier filter (Chroma e650sp-2p) by using a compact CCD spectrometer module BWTek BTC112E. Total fluorescence intensities were obtained by integrating the corrected emission. The experimental uncertainty of the action cross-sectional values determined by this method has been estimated to be ±10%.
Acknowledgments
MBD gratefully acknowledges the Conseil Régional d’Aquitaine for financial support (Chair of Excellence to MBD and fellowship to JD). TEM microscopy was done in the Bordeaux Imaging Center (UMS 3420 CNRS – University of Bordeaux/Inserm US4). This work was partially supported by the European Commission and received funding from the People Programme (Marie Curie Actions) of the European Union's Seventh Framework Programme FP7/2007-2013/under REA grant agreement n°607721.