

1 Introduction
Flavonoids, a wide family of natural compounds, are known to possess an anti-inflammatory effect due to their inhibitory potency towards cyclooxygenase-2 enzyme [1,2]. Besides, considering structure-activity relationship studies mainly about 1,2-diaryl substituted heterocycles, the methylsulfonyl group was found to be critical for cyclooxygenase-2 inhibitory activity and is also believed to induce cyclooxygenase-2 selectivity [3,4]. Thus, our efforts are directed towards the synthesis of flavonoid derivatives bearing this pharmacophore. 2′-Hydroxychalcones are the main precursors for the synthesis of flavonoids and the Claisen–Schmidt condensation between an acetophenone and a benzaldehyde is a key reaction for their preparation (Scheme 1) [5,6]. Therefore, we were interested in the synthesis of 2′-hydroxy-4′-methylsulfonylacetophenone (1) as a synthon for the preparation of novel flavonoid derivatives that would be potential cyclooxygenase-2 selective inhibitors.
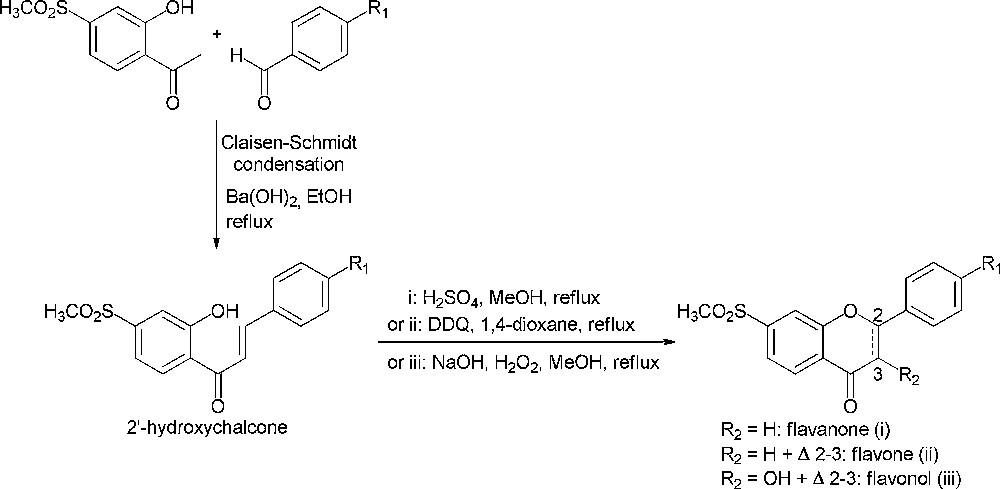
Synthetic pathways of flavonoids.
2 Results and discussions
A first strategy for the synthesis of 2′-hydroxy-4′-methylsulfonylacetophenone (1) was considered as shown in Scheme 2. This product might be synthesized from 3′-methylsulfonylacetophenone (3) through a Baeyer–Villiger oxidation followed by a Fries rearrangement from compound 2. Three synthetic pathways were envisaged in order to prepare compound 3:
- • pathway A consisted of the chlorosulfonation of acetophenone (5) followed by the conversion of the sulfonyl chloride group to methylsulfonyl group;
- • pathway B involved a Friedel–Crafts acetylation on methylsulfonylbenzene (6);
- • pathway C was directed towards the synthesis of the methyl ketone from the corresponding carboxylic acid (7) through a methylation reaction.
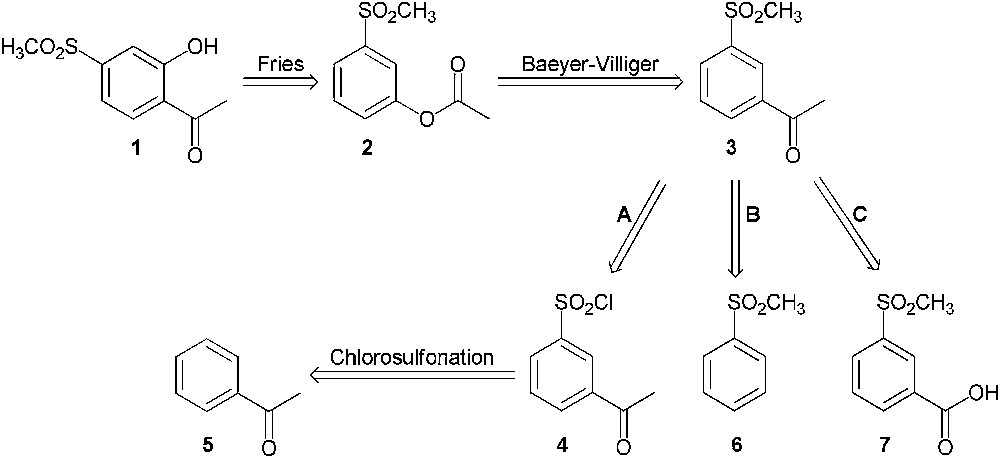
Retrosynthesis of 2′-hydroxy-4′-methylsulfonylacetophenone (1) from 3′-methylsulfonylacetophenone (3).
Considering the pathway A, the synthesis of 3′-chlorosulfonylacetophenone (4) was envisaged through the action of chlorosulfonic acid upon acetophenone (5). This acid is considered as the reagent of choice for the direct conversion of aromatic compounds into their sulfonyl chlorides [7–9]. However, several studies, which investigated the chlorosulfonation of acetophenone, showed contradictory results despite similar reaction conditions (Scheme 3). Thus, the work of Shingare and Ingle was the only one to describe the synthesis of the expected 3′-chlorosulfonylacetophenone (4) through this reaction [10]. Otherwise, Riesz and Frankfurter reported that this reaction yielded 5-acetylbenzene-1,3-disulfonyl dichloride (8) [11,12], even if, a few years later, Weston and Suter had shown that the resulting product was not compound 8, but 2-(2-(chlorosulfonyl)acetyl)benzene-1-sulfonyl chloride (9) [12]. Woodruff also described the synthesis of compound 9 and showed that the chlorosulfonation of acetophenone, depending on the conditions, was able as well to give the α, meta-sulfonation product (10) [13,14]. Finally, Chapman claimed the formation of another new compound identified as 3-chlorobenzothiophen-1,1-dioxide-2-sulfonyl chloride (11) [15].
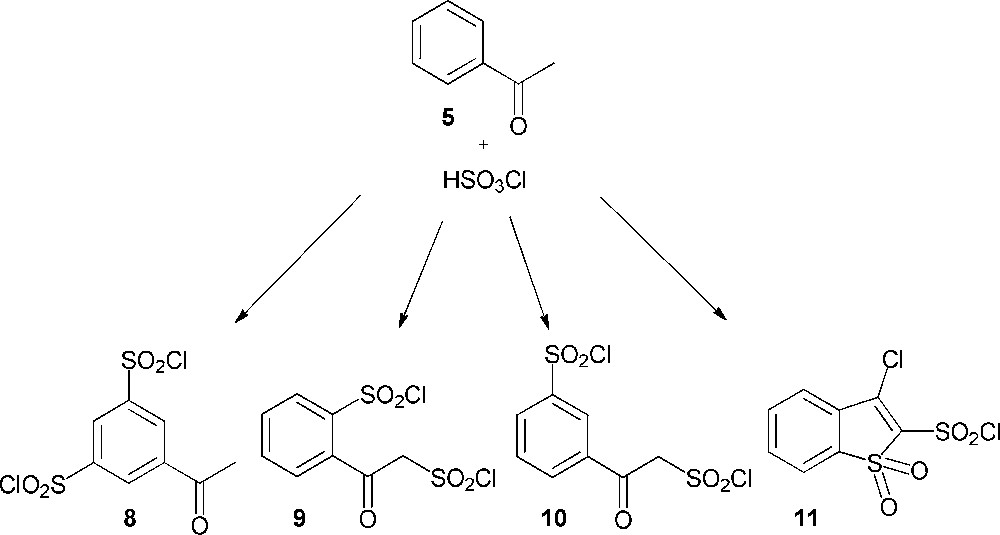
Reaction of acetophenone (5) with chlorosulfonic acid.
Our investigations dealing with the chlorosulfonation of acetophenone (5) were performed by varying the reaction conditions, but did not allow us to obtain the desired product. In most cases, no evolution of the reaction medium was noticed; in a few cases, even when using the procedure described by Shingare and Ingle [10], acetophenone (5) was partly transformed into benzothiophen-3(2H)-one-1,1-dioxide (12), which had never been described in the previous reports (Scheme 4). As seen for the action of chlorosulfonic acid on ethyl phenyl ketone, the reaction probably involves an initial chlorosulfonation of the enolic form of acetophenone (5) followed by a rearrangement to form the intermediate 2-oxo-2-phenylethanesulfonyl chloride. Subsequent electrophilic aromatic substitution might afford the compound 12 (Scheme 5) [16].
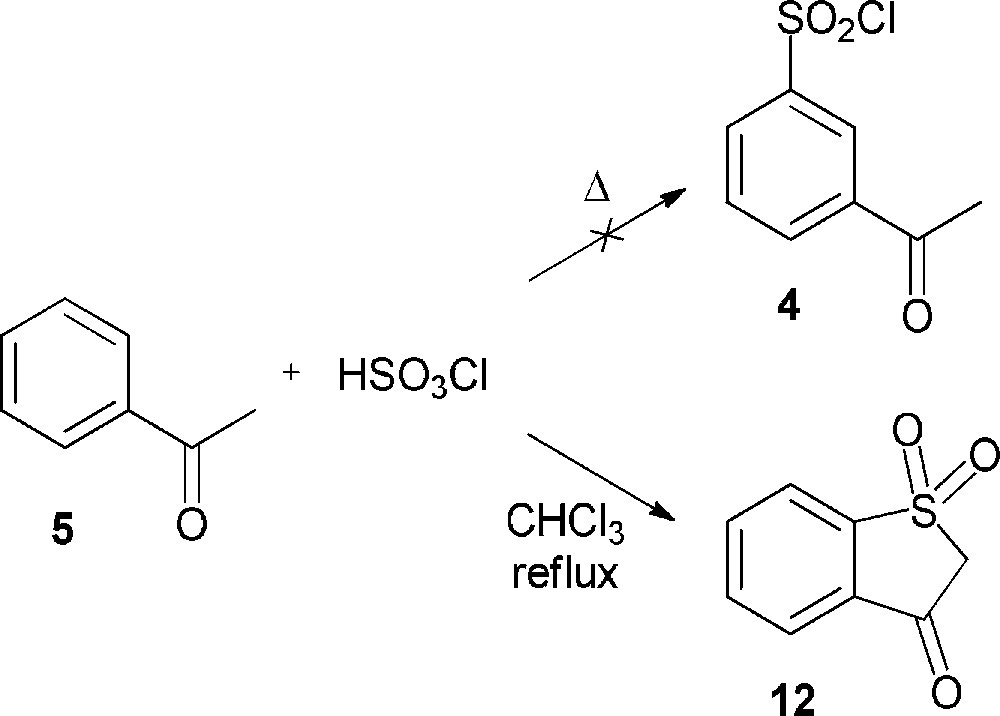
Formation of benzothiophen-3(2H)-one-1,1-dioxide (12) through chlorosulfonation of acetophenone (5).
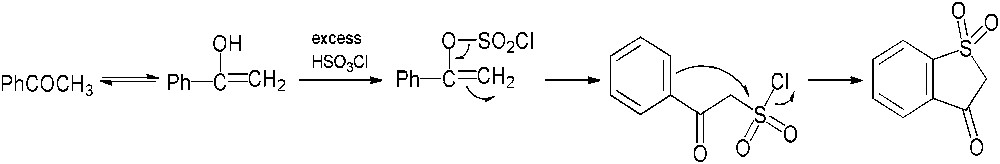
Proposed mechanism for the formation of benzothiophen-3(2H)-one-1,1-dioxide (12).
At the same time, the Friedel–Crafts acetylation on methylsulfonylbenzene (6) with acetyl chloride in the presence of aluminium trichloride was undertaken (pathway B, Scheme 2). Different reaction parameters were explored but unfortunately, no conversion of the starting material was observed.
The difficulty of these meta electrophilic substitutions prompted us to direct our efforts towards the transformation of a pertinent meta-substituted methylsulfonylbenzene. 3-Methylsulfonylbenzoic acid (7) was selected to envisage its methylation using methyllithium (pathway C, Scheme 2). For this reaction, the literature describes the use of two equivalents of the organometallic reagent; one equivalent acts as a base by deprotonation of the carboxylic acid while the second one acts as a nucleophilic agent on the carbonyl group of the lithium carboxylate [17]. Thus, the methylation was performed in a successful way to afford compound 3 (Scheme 6). However, in order to optimize the synthesis yield, reaction conditions had to be changed. The results of our investigations are shown in Table 1. In our work, the optimal yield (35%) was obtained when the synthesis was performed with 4 equiv. of methyllithium, by adding the starting materials at 0 °C, then stirring the reaction medium at room temperature. On the other hand, by introducing the reactants at –78 °C, the reaction yield decreased from 35% to 22% due to the formation of a side product (compound 13, 10% yield) (Scheme 6). The excess of methyllithium should deprotonate the methylsulfonyl moiety of compound 3; then, the attack of the formed carbanion on the carbonyl group of the undeprotonated 3′-methylsulfonylacetophenone (3) leads to compound 13 as a by-product.
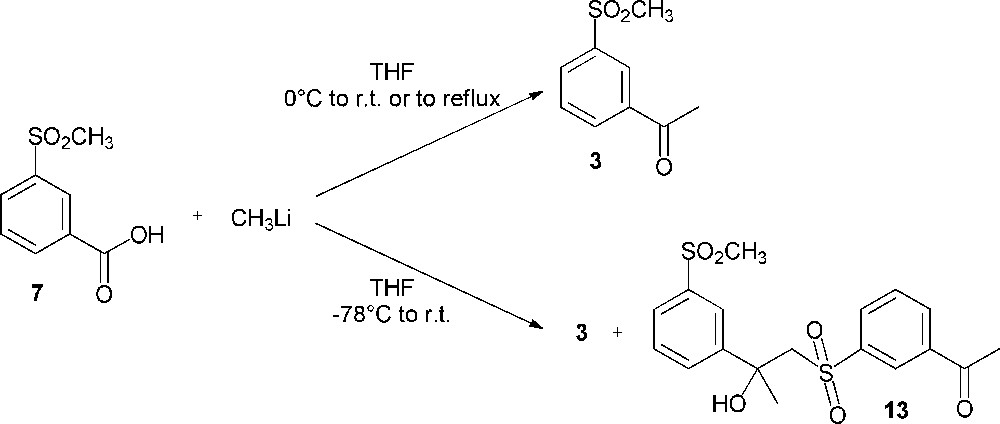
Reaction of 3-methylsulfonylbenzoic acid (7) with methyllithium.
Conversion of 3-methylsulfonylbenzoic acid (7) to 3′-methylsulfonylacetophenone (3).
| Entry | CH3Li (equiv.) | Reaction conditions | Yield (%) (compound 3) |
| 1 | 2.2 | 0 °C to rt | 6 |
| 2 | 3.0 | 0 °C to rt | 8 |
| 3 | 3.0 | 0 °C to reflux | 13 |
| 4 | 4.0 | 0 °C to reflux | 20 |
| 5 | 4.0 | 0 °C to rt | 35 |
| 6 | 4.0 | –78 °C to rt | 22 |
| 7 | 5.0 | 0 °C to reflux | 12 |
Thus, the synthesis of the expected 3′-methylsulfonylacetophenone (3) has been achieved through the methylation of the corresponding benzoic acid (7) and a Baeyer–Villiger oxidation followed by a Fries rearrangement, which should be the next step to further obtain 2′-hydroxy-4′-methylsulfonylacetophenone (1).
The Baeyer–Villiger oxidation of 3′-methylsulfonylacetophenone (3) was carried out using meta-chloroperoxybenzoic acid (m-CPBA) as the oxidizing agent in the presence of a catalytic amount of ceric ammonium nitrate (CAN) in methylene chloride (Scheme 7) [18]. Unfortunately, the expected 3-(methylsulfonyl)phenyl acetate (2) was obtained in a low yield; indeed, an appreciable amount of the starting 3′-methylsulfonylacetophenone (3) was not transformed, while two side products were also recovered: the starting material was mono- and di-chlorinated (compounds 14 and 15 respectively) in α to the carbonyl group.
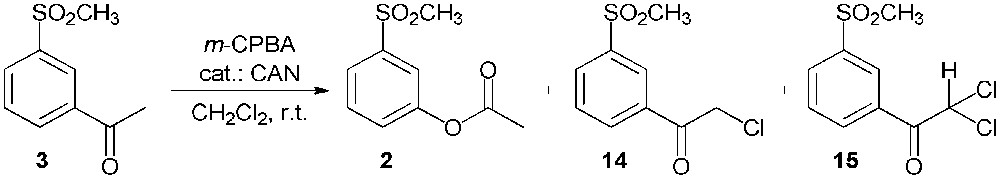
Baeyer–Villiger oxidation of 3′-methylsulfonylacetophenone (3).
Due to the low yield of the Baeyer–Villiger oxidation, the Fries rearrangement was not undertaken and a second synthetic strategy had to be considered (Scheme 8). This latter started from 3-mercaptophenol (16) from which the thioether 17 was selectively formed through a methylation reaction [19]. A subsequent Friedel–Crafts acetylation provided the desired intermediate (18) [20,21]. The site of acetylation was first assigned by comparison of proton nuclear magnetic resonance spectra of products 17 and 18. The hydroxyl group signal was shifted from 9.47 ppm for 3-(methylthio)phenol (17) to 12.53 ppm for compound 18. This deshielding effect is due to the intramolecular hydrogen bond between the oxygen atom of the carbonyl and the phenolic proton [22]. This attractive interaction could not occur if the acetyl was introduced at the para position relative to the hydroxyl function. In addition, a long range (4J) correlation between the phenolic proton and the carbonyl carbon was observed on the heteronuclear multiple bond correlation spectrum and further confirmed the structure of the acetophenone 18. Compounds 19, 20 and 21 were also isolated as the minor side products. Finally, 2′-hydroxy-4′-methylsulfonylacetophenone (1) was obtained through an oxidation of the corresponding sulphide (18) using oxone® [23]. This second strategy is an alternative synthetic route to compounds 1 and 18, which have already been prepared from 4′-fluoro-2′-methoxyacetophenone [24]. Compound 1 has also been used for the synthesis of a chalcone derivative designed as an antioxidant and an anti-microbial agent [25].
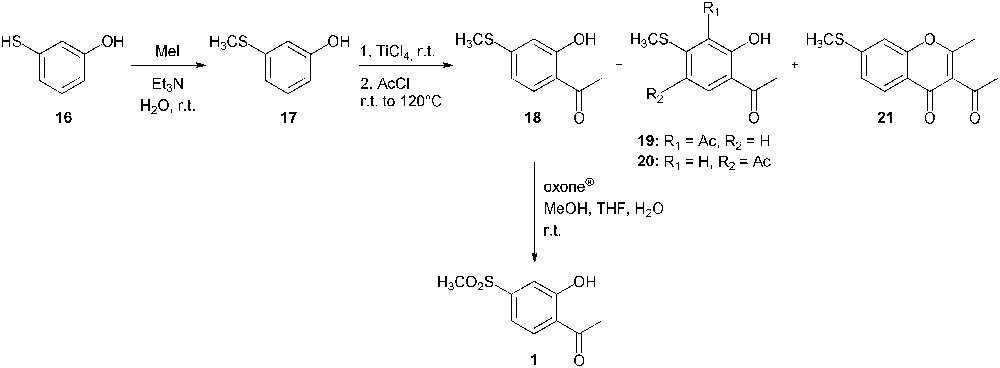
Synthesis of 2′-hydroxy-4′-methylsulfonylacetophenone (1) from 3-mercaptophenol (16).
3 Conclusion
In summary, different synthetic routes to 2′-hydroxy-4′-methylsulfonylacetophenone were described.
The first strategy via 3′-methylsulfonylacetophenone did not allow the preparation of the expected 2′-hydroxy-4′-methylsulfonylacetophenone. Indeed, in contrast to the previous reports, the chlorosulfonation of acetophenone produced benzothiophen-3(2H)-one-1,1-dioxide. In the same way, the Friedel–Crafts acetylation of methylsulfonylbenzene failed. Then, even if 3′-methylsulfonylacetophenone was obtained from the corresponding benzoic acid, the low yield of the Baeyer–Villiger oxidation led us to consider another approach.
Thus, the methylation of 3-mercaptophenol followed by a Friedel–Crafts acetylation provided 2′-hydroxy-4′-methylthioacetophenone, which was then oxidized using oxone®. Therefore, 2′-hydroxy-4′-methylsulfonylacetophenone was synthesized in a three-step pathway. This compound is now considered as a key synthon for the synthesis of new flavonoid derivatives designed as potential cyclooxygenase-2 inhibitors.
4 Experimental
4.1 General
Chemical reagents and solvents were purchased from Alfa Aesar (Schiltigheim, France), Fisher Scientific (Illkirch, France), Sigma–Aldrich (Saint-Quentin-Fallavier, France) or Carlo Erba Réactifs–SDS (Val-de-Reuil, France) and were used without further purification. TLC was performed using Merck silica gel 60 F254 plates. Merck silica gel 60 H was used for preparative TLC. The spots were visualized with a UV lamp. Melting points were determined with a Wagner & Munz Kofler bench WME. NMR spectra were recorded with a Bruker DPX-400 spectrometer (1H: 400 MHz; 13C: 100 MHz) using Me4Si as an internal standard. IR spectra (solutions in CH2Cl2) were recorded with a Mattson Satellite FT–IR spectrometer and only major peaks are reported. UV spectra (solutions in EtOH) were recorded with a Shimadzu UV-2401PC spectrophotometer. Mass spectra were acquired at the “Centre régional de mesures physiques de l’Ouest” (Rennes, France) using a Waters Q-TOF 2 spectrometer.
4.2 Syntheses
4.2.1 Benzothiophen-3(2H)-one-1,1-dioxide (12)
Acetophenone (5) (0.20 mL, 1.71 mmol) was added dropwise to an ice-cooled solution of HSO3Cl (1.10 mL, 17.11 mmol) in CHCl3 (5 mL). The reaction mixture was stirred at room temperature for 3 h then refluxed during 4 h. A biphasic mixture was formed and stirred at room temperature overnight. After addition of H2O and concentrated NaOH, the product was extracted with CHCl3. Combined organic layers were dried (Na2SO4), filtered and concentrated under reduced pressure. The residue (0.098 g) was purified by preparative TLC on silica gel (eluent: 7:3 hexane/EtOAc) to afford yellowish crystals (yield 14%, 0.041 g); Mp: 131 °C; IR (CH2Cl2, cm−1): 523, 768, 1145, 1199, 1312, 1724; UV (EtOH, nm): λmax = 208, 244; 1H NMR (400 MHz, CDCl3): δ 4.11 (s, 2H, CH2), 7.84 (br ddd, J = 1.2, 7.3, 7.8 Hz, 1H, H-6), 7.96 (td, J = 1.1, 8.0 Hz, 1H, H-7), 8.00–8.05 (m, 2H, H-5, H-8); 13C NMR (100 MHz, CDCl3): δ 57.5 (C-2), 122.0 (C-8), 124.9 (C-5), 133.3 (C-4), 134.4 (C-6), 137.4 (C-7), 148.0 (C-9), 187.0 (C-3); HRMS (ESI+): m/z calculated for C8H6O3NaS: 204.9935, found: 204.9937.
4.2.2 3′-methylsulfonylacetophenone (3) and 1-(3′-(2′′-hydroxy-2′′-(3′′′-(methylsulfonyl)phenyl)propylsulfonyl)phenyl)ethanone (13)
To a solution of 3-methylsulfonyl benzoic acid (7) (0.200 g, 1 mmol) in anhydrous THF (15 mL) was added dropwise MeLi (1.6 M in Et2O, 2.50 mL, 4 mmol) at –78 °C under N2. The reaction mixture was then allowed to warm to room temperature, stirred during 2 h 25 min and treated with saturated aqueous NH4Cl. EtOAc was added and the organic layer was separated. The aqueous layer was extracted with EtOAc. Combined organic layers were dried (Na2SO4), filtered and concentrated under reduced pressure. The residue (0.149 g) was purified by preparative TLC on silica gel (eluent: 8:2 CH2Cl2–EtOAc) to provide:
3′-methylsulfonylacetophenone (3) as yellowish crystals (yield 22%, 0.044 g); Mp: 106 °C; IR (CH2Cl2, cm−1): 539, 1149, 1262, 1300, 1316, 1693; UV (EtOH, nm): λmax = 205, 236; 1H NMR (400 MHz, CDCl3): δ 2.68 (s, 3 H, COCH3), 3.10 (s, 3 H, SO2CH3), 7.72 (br t, J = 7.8 Hz, 1 H, H-5′), 8.15 (ddd, J = 1.2, 1.6, 7.8 Hz, 1 H, H-4′), 8.24 (dt, J = 1.3, 7.8 Hz, 1 H, H-6′), 8.50 (br t, J = 1.6 Hz, 1 H, H-2′); 13C NMR (100 MHz, CDCl3): δ 26.9 (COCH3), 44.6 (SO2CH3), 127.4 (C-2′), 130.2 (C-5′), 131.6 (C-4′), 133.3 (C-6′), 138.3 (C-1′), 141.7 (C-3′), 196.3 (CO); HRMS (ESI+): m/z calculated for C9H10O3NaS: 221.0248, found: 221.0246.
1-(3′-(2′′-hydroxy-2′-(3′′′-(methylsulfonyl)phenyl)propylsulfonyl)phenyl)ethanone (13) as a colourless paste (yield 10%, 0.041 g); IR (CH2Cl2, cm−1): 535, 1145, 1262, 1300, 1689, 3492; UV (EtOH, nm): λmax = 204; 1H NMR (400 MHz, DMSO-d6): δ 1.61 (s, 3 H, C(OH)CH3), 2.62 (s, 3 H, COCH3), 3.15 (s, 3 H, SO2CH3), 3.98 (d, J = 15.0 Hz, 1 H, H-1′′), 4.10 (d, J = 15.0 Hz, 1 H, H-1“), 5.71 (s, 1 H, OH), 7.44 (br t, J = 7.8 Hz, 1 H, H-5′′′), 7.64 (br t, J = 7.8 Hz, 1 H, H-5′), 7.66–7.72 (m, 2 H, H-4′′′, H-6′′′), 7.91–7.92 (m, 2 H, H-4′, H-2′′′), 8.12 (br s, 1 H, H-2′), 8.16 (br d, J = 7.8 Hz, 1 H, H-6′); 13C NMR (100 MHz, DMSO-d6): δ 26.8 (C-2), 30.4 (C-3′′), 43.5 (SO2CH3), 65.5 (C-1′′), 71.6 (C-2′′), 123.4 (C-2′′′), 125.1 (C-6′′′), 127.1 (C-2′), 128.7 (C-5′′′), 129.5 (C-5′), 130.4 (C-4′′′), 131.9 (C-4′), 132.7 (C-6′), 136.9 (C-1′), 140.3 (C-3′′′), 141.7 (C-3′), 147.7 (C-1′′′), 196.7 (C-1); HRMS (ESI+): m/z calculated for C18H20O6NaS2: 419.0599, found: 419.0596.
4.2.3 3-(methylsulfonyl)phenyl acetate (2), 2-chloro-1-(3-(methylsulfonyl)phenyl)ethanone (14) and 2,2-dichloro-1-(3-(methylsulfonyl)phenyl)ethanone (15)
CAN (0.056 g, 0.1 mmol) and m-CPBA (0.602 g, 2.5 mmol) were added to a solution of 3-methylsulfonylacetophenone (3) (0.200 g, 1 mmol) in CH2Cl2 (10 mL). The reaction mixture was kept at room temperature during 45 days then worked up by pouring into H2O. The products were extracted with Et2O. The organic layers were combined then successively washed with aqueous solutions of KI, Na2S2O3 and NaHCO3. The organic extract was dried (Na2SO4), filtered and evaporated under reduced pressure. The residue (0.196 g) was purified twice by preparative TLC on silica gel (eluents: 9.5:0.5 CH2Cl2/EtOAc then 8:2 Et2O/hexane) to give:
- • 3-(methylsulfonyl)phenyl acetate (2) as a colourless oil (yield 8%, 0.018 g); IR (CH2Cl2, cm−1): 531, 760, 1145, 1196, 1300, 1771; UV (EtOH, nm): λmax = 204; 1H NMR (400 MHz, CDCl3): δ 2.34 (s, 3 H, OCOCH3), 3.07 (s, 3 H, SO2CH3), 7.40 (br dd, J = 1.4, 8.1 Hz, 1 H, H-6), 7.60 (br t, J = 8.0 Hz, 1 H, H-5), 7.71 (br t, J = 1.8 Hz, 1 H, H-2), 7.82 (br d, J = 7.8 Hz, 1 H, H-4); 13C NMR (100 MHz, CDCl3): δ 21.0 (OCOCH3), 44.5 (SO2CH3), 121.0 (C-2), 124.7 (C-4), 127.2 (C-6), 130.5 (C-5), 141.9 (C-3), 151.0 (C-1), 168.8 (CO); HRMS (ESI+): m/z calculated for C9H10O4NaS: 237.0198, found: 237.0198;
- • 2-chloro-1-(3-(methylsulfonyl)phenyl)ethanone (14) as a colourless oil (yield 3%, 0.011 g); IR (CH2Cl2, cm−1): 539, 1145, 1300, 1708; UV (EtOH, nm): λmax = 206, 238; 1H NMR (400 MHz, CDCl3): δ 3.11 (s, 3 H, SO2CH3), 4.72 (s, 2 H, CH2), 7.76 (br t, J = 7.8 Hz, 1 H, H-5), 8.20 (br d, J = 7.8 Hz, 1 H, H-4), 8.26 (br d, J = 7.8 Hz, 1 H, H-6), 8.51 (br s, 1 H, H-2); 13C NMR (100 MHz, CDCl3): δ 44.4 (SO2CH3), 45.4 (CH2), 127.6 (C-2), 130.3 (C-5), 132.2 (C-4), 133.5 (C-6), 135.2 (C-1), 141.8 (C-3), 189.7 (CO); HRMS (ESI+): m/z calculated for C9H10O3ClS: 233.0039, found: 233.0039;
- • 2,2-dichloro-1-(3-(methylsulfonyl)phenyl)ethanone (15) as a colourless oil (yield 4%, 0.007 g); IR (CH2Cl2, cm−1): 535, 1149, 1300, 1716; UV (EtOH, nm): λmax = 203; 1H NMR (400 MHz, CDCl3): δ 3.12 (s, 3 H, SO2CH3), 6.63 (s, 1 H, CHCl2), 7.78 (br t, J = 7.8 Hz, 1 H, H-5), 8.23 (ddd, J = 1.2, 1.7, 7.9 Hz, 1 H, H-4), 8.42 (ddd, J = 1.2, 1.6, 7.9 Hz, 1 H, H-6), 8.66 (br t, J = 1.6 Hz, 1 H, H-2); 13C NMR (100 MHz, CDCl3): δ 44.4 (SO2CH3), 67.7 (CHCl2), 128.8 (C-2), 130.2 (C-5), 132.3 (C-1), 132.7 (C-4), 134.6 (C-6), 141.9 (C-3), 184.4 (CO); HRMS (ESI+): m/z calculated for C9H9O3Cl2S: 266.9649, found: 266.9655;
- • 3′-methylsulfonylacetophenone (3) as yellowish crystals (yield 25%, 0.050 g).
4.2.4 3-(methylthio)phenol (17)
MeI (0.50 mL, 7.93 mmol) and Et3N (1.20 mL, 8.46 mmol) were added to a solution of 3-mercaptophenol (16) (0.61 mL, 7.93 mmol) in H2O (5 mL). The resulting mixture was stirred at room temperature for 4 h 20 min then the product was extracted with EtOAc. Combined organic layers were dried (Na2SO4), filtered and concentrated under reduced pressure to afford a brown liquid (yield 96%, 1.066 g); IR (CH2Cl2, cm−1): 687, 776, 1215, 1440, 1475, 1584, 3363; UV (EtOH, nm): λmax = 215, 254; 1H NMR (400 MHz, DMSO-d6): δ 2.41 (s, 3 H, SCH3), 6.53 (br dd, J = 1.8, 8.0 Hz, 1 H, H-6), 6.63 (br t, J = 1.8 Hz, 1 H, H-2), 6.66 (br d, J = 7.8 Hz, 1 H, H-4), 7.09 (br t, J = 7.9 Hz, 1 H, H-5), 9.47 (s, 1 H, OH); 13C NMR (100 MHz, DMSO-d6): δ 14.5 (SCH3), 112.0 (C-6), 112.4 (C-2), 116.5 (C-4), 129.7 (C-5), 139.1 (C-3), 157.7 (C-1); HRMS (ESI+): m/z calculated for C7H9OS: 141.0374, found: 141.0373.
4.2.5 2′-hydroxy-4′-methylthioacetophenone (18), 3′-acetyl-2′-hydroxy-4′-methylthioacetophenone (19), 5′-acetyl-2′-hydroxy-4′-methylthioacetophenone (20) and 3-acetyl-2-methyl-7-(methylthio)-4H-chromene-4-one (21)
TiCl4 (0.17 mL, 1.57 mmol) was slowly added to 3-(methylthio)phenol (17) (0.200 g, 1.43 mmol) placed in a flask flushed with N2. The reaction mixture was kept at room temperature during 2 h and AcCl (0.17 mL, 2.14 mmol) was added. The resulting thick solution was kept at room temperature for 15 min then brought to 120 °C and left at this temperature for an additional hour. The reaction mixture was cooled to room temperature, diluted with CH2Cl2 and quenched with H2O. The products were extracted with CH2Cl2. Combined organic layers were dried (Na2SO4), filtered, and concentrated under reduced pressure. The residue (0.233 g) was purified by preparative TLC on silica gel (eluent: 8:2 hexane-EtOAc) to provide:
2′-hydroxy-4′-methylthioacetophenone (18) as yellowish crystals (yield 53%, 0.137 g); Mp: 85 °C; IR (CH2Cl2, cm−1): 803, 838, 1219, 1347, 1549, 1631; UV (EtOH, nm): λmax = 215, 248, 307; 1H NMR (400 MHz, CDCl3): δ 2.49 (s, 3 H, SCH3), 2.58 (s, 3 H, COCH3), 6.72–6.74 (m, 2 H, H-3′, H-5′), 7.57 (d, J = 8.2 Hz, 1 H, H-6′), 12.53 (s, 1 H, OH); 13C NMR (100 MHz, CDCl3): δ 14.5 (SCH3), 26.3 (COCH3), 112.9 (C-3′), 116.3 (C-5′), 116.4 (C-1′), 130.5 (C-6′), 150.3 (C-4′), 162.8 (C-2′), 203.3 (CO); HRMS (ESI+): m/z calculated for C9H10O2NaS: 205.0299, found: 205.0301.
A mixture of 3′-acetyl-2′-hydroxy-4′-methylthioacetophenone (19) and 5′-acetyl-2′-hydroxy-4′-methylthioacetophenone (20) as a dark paste (yield 2% 0.006 g); IR (CH2Cl2, cm−1): 990, 1258, 1351, 1557, 1635, 2924; UV (EtOH, nm): λmax = 203, 261, 317; compound 19: 1H NMR (400 MHz, CDCl3): δ 2.47 (s, 3 H, SCH3), 2.61 (s, 3 H, 3′C–CO–CH3), 2.62 (s, 3 H, 1′C–CO–CH3), 6.80 (d, J = 8.6 Hz, 1 H, H-5′), 7.70 (d, J = 8.6 Hz, 1 H, H-6′), 13.06 (s, 1 H, OH); compound 20: 1H NMR (400 MHz, CDCl3): δ 2.43 (s, 3 H, SCH3), 2.63 (s, 3 H, 5′C(CO)CH3), 2.67 (s, 3 H, 1′C(CO)CH3), 6.83 (s, 1 H, H-3′), 8.26 (s, 1 H, H-6′), 12.78 (s, 1 H, OH); compound 19: 13C NMR (100 MHz, CDCl3): δ 15.8 (SCH3), 26.5 (1′C–CO–CH3), 31.9 (3′C–CO–CH3), 114.9 (C-5′), 116.4 (C-1′), 127.5 (C-3′), 131.9 (C-6′), 149.4 (C-4′), 160.7 (C-2′), 201.5 (3′C–CO–CH3), 203.6 (1′C–CO–CH3); compound 20: 13C NMR (100 MHz, CDCl3): δ 16.1 (SCH3), 26.2 (1′C(CO)CH3), 27.5 (5′C(CO)CH3), 113.4 (C-3′), 114.9 (C-1′), 125.5 (C-5′), 134.9 (C-6′), 154.7 (C-4′), 164.6 (C-2′), 196.2 (5′C(CO)CH3), 203.0 (1′C(CO)CH3); HRMS (ESI+): m/z calculated for C11H12O3NaS: 247.0405, found: 247.0404.
3-acetyl-2-methyl-7-(methylthio)-4H-chromene-4-one (21) as a dark paste (yield 2%, 0.008 g); IR (CH2Cl2, cm−1): 1425, 1549, 1619, 1635, 1701; UV (EtOH, nm): λmax = 203, 262, 319; 1H NMR (400 MHz, CDCl3): δ 2.50 (s, 3 H, CH3), 2.56 (s, 3 H, SCH3), 2.63 (s, 3 H, COCH3), 7.16 (d, J = 1.6 Hz, 1 H, H-8), 7.24 (dd, J = 1.7, 8.5 Hz, 1 H, H-6), 8.05 (d, J = 8.4 Hz, 1 H, H-5); 13C NMR (100 MHz, CDCl3): δ 15.0 (SCH3), 20.0 (CH3), 32.5 (COCH3), 112.7 (C-8), 120.6 (C-4a), 123.4 (C-6), 124.0 (C-3), 126.1 (C-5), 148.2 (C-7), 156.0 (C-8a), 168.3 (C-2), 175.6 (C-4), 200.8 (CO); HRMS (ESI+): m/z calculated for C13H12O3NaS: 271.0405, found: 271.0407.
4.2.6 2′-hydroxy-4′-methylsulfonylacetophenone (1)
Oxone® (2.350 g, 3.82 mmol) was added to 2′-hydroxy-4′-methylthioacetophenone (18) (0.620 g, 3.41 mmol) previously dissolved in a mixture of MeOH (9 mL), THF (9 mL) and H2O (9 mL). The resulting mixture was stirred at room temperature for 1 h 25 min then the product was extracted with EtOAc. Combined organic layers were dried (Na2SO4), filtered and concentrated under reduced pressure to afford yellowish crystals (yield 91%, 0.663 g); Mp: 141 °C; IR (CH2Cl2, cm−1): 772, 1149, 1203, 1285, 1308, 1650; UV (EtOH, nm): λmax = 213, 249; 1H NMR (400 MHz, CDCl3): δ 2.71 (s, 3 H, COCH3), 3.07 (s, 3 H, SO2CH3), 7.46 (dd, J = 1.7, 8.3 Hz, 1 H, H-5′), 7.56 (d, J = 1.6 Hz, 1 H, H-3′), 7.95 (d, J = 8.3 Hz, 1 H, H-6′), 12.34 (s, 1 H, OH); 13C NMR (100 MHz, CDCl3): δ 27.3 (COCH3), 44.2 (SO2CH3), 117.0 (C-3′), 118.2 (C-5′), 122.8 (C-1′), 132.1 (C-6′), 147.0 (C-4′), 162.7 (C-2′), 204.4 (CO); HRMS (ESI+): m/z calculated for C9H10O4NaS: 237.0197, found: 237.0205.
Acknowledgments
The authors gratefully thank the “Région Limousin” for the PhD grant to Rokhaya Gueye, the Organic and Therapeutic Chemistry Laboratory of Limoges School of Pharmacy for its support and the “CRMPO” (Rennes, France) for recording the high-resolution mass spectra.