

1 Introduction
Metal nanoparticles play an extremely important role in heterogeneous catalysis, where the support is often a metal oxide [1]. Traditionally, particles of a few nanometers in size are considered or have been proven to be the actors of catalysis [2,3]. An intriguing class of systems, however, which is the focus of the present brief review, is represented by very small, subnanometer clusters, i.e., aggregates composed by less than – say – 30 atoms, which are at the lowest end of the transition between molecular and metallic behavior. We propose to use for these systems the term ultrananocatalysts and ultrananocatalysis, in analogy with the use of the prefix ‘ultra’ in other fields (such as ultrathin films in surface science [4]), so as to highlight their peculiar features. We include in this class of systems also multi-component, i.e., alloyed, nanoclusters, which are especially interesting for the possibility of tuning activity and selectivity via a proper choice of the mixing elements [5–7].
Although ultrananoclusters have been known to be possible active species in heterogeneous catalysis since a long time [5], also supported by EXAFS evidence [8], and although a huge amount of work has been dedicated to the study of their properties (including chemical reactions) in the gas phase [9], only recently there has been an explosive surge of interest in supported systems. This was due to the possibility of increased control in the synthesis of model but very-well characterized heterogeneous catalytic systems by deposition of size-selected aggregates from the gas phase [10]. A further crucial step has been realized when deposition onto realistic substrates and catalysis in realistic conditions was achieved [11]. Both fundamental knowledge and a glance at possible real-world applications quickly derived from these studies [12–15]. In addition, alternative synthetic approaches, such as adduct decomposition on technologically relevant substrate (zeolites), have been investigated [16], which may be amenable to mass production (other intriguing scale-up possibilities are also being been explored [17]). This has led to a rapidly increasing wealth of information on chemical reactions occurring in different systems and varied conditions, which has been very stimulating but has also led to conflicting claims and interpretations, especially in terms of kinetic mechanisms. What in our opinion is lacking and is thus strongly needed is a coherent theoretical framework in which to organize this abundance of novel and fascinating knowledge.
The present work aims at contributing a step in this direction by discussing progress in the theoretical understanding of ultrananocatalysis by size-selected metal clusters deposited onto an oxide surface. Attention will be focused on a few key concepts, which in our opinion have a general and widespread significance. In particular, we will try to illustrate via selected examples (mostly drawn for simplicity from our own recent work) the following points:
- • the fluxional character of metal ultrananoclusters and the associated need for systematically sampling the system potential energy surface;
- • the influence of the underlying support (including possible defects) in terms of epitaxial relationships, generated electrostatic field and electronic interactions, and the possible active role of the support in the reaction mechanisms;
- • the evolution of ligand adsorption energetics, structure and dynamics as a function of coverage, including many-body and synergic interactions among ligands, and the formation of catalytically active species;
- • the robustness of the catalyst in terms of surface diffusion and cluster disaggregation.
The possibility of out-of-equilibrium effects and the theoretical methodology required for their predictive description will also be briefly discussed. In the illustrative cases hereafter discussed, the (100) surface of a simple oxide such as MgO, and the Fs-center (oxygen vacancy) on MgO(100) as a defected surface [18], will be taken as prototypical examples, due to the fact that they are extremely well characterized and at the same time interesting systems, but the considerations here proposed aim at a more general applicability. References are not exhaustive (we apologize in advance for the lack of important contributions) but try to point – to the best of our knowledge – to the first occurrence of the given concept in the field of ultrananocatalysis.
The article is organized as follows. In each of Sections 2–5 one of the four concepts outlined above is described and discussed. Out-of-equilibrium effects are mentioned in Section 6. Conclusions and perspectives are sketched in Section 7.
2 The fluxional character of metal ultrananoclusters
A system is called fluxional when it has several energetically close structures and passes from one to another of these structures fairly rapidly under relevant conditions. It is well-known that metal complexes can be highly fluxional [19] and metal clusters are no exceptions. Also for large nanoparticles “flexibility” has been advocated as a key catalytic concept [20], and this is even truer in the ultranano régime. The interaction with the oxide surface can further increase this feature. As an example, for the widely investigated Fs-center on the MgO(100) surface, it has been found that the presence of the defect profoundly alters the potential energy surface (PES) of metal atoms moving in its proximity [21]. In particular, the PES close to the defect presents a characteristically rotational invariance and, topologically, a large basin of attraction with a rather small equilibrium distance on top of the defect with respect to neighboring sites [22]. This can lead to increased fluxional behavior, as can be illustrated by considering small Ag clusters on the Fs/MgO(100) surface [23]. As shown on Fig. 1, in the gas phase small Ag clusters exhibit a crossover in terms of energetically preferred configurations from planar to compact 5-fold arrangements between size N = 6 and N = 8 (with N the number of Ag atoms in the cluster). When deposited on an Fs-center of the regular MgO(100) surface, the structural landscape of the same clusters looks rather different due to the strong interaction with the defect which forces the clusters into configurations with an appreciable stretching of Ag–Ag distances involving the Ag atom on top of the defect. The end result is that planar arrangements are energetically favored only up to size N = 4, while the compact 5-fold arrangements yield the way to compact fcc-like arrangements at size N = 10 because of the excessive strain implied by the 5-fold symmetry [23] (the actual situation is even more intricate as there is a fourth motif which is energetically favored at N = 8). In connection with this complex structural crossover, it is not surprising to find an increased fluxional character: for example, the difference between the putative global minimum and the lowest energy isomer is 0.20, 0.14, 0.12 eV for Ag6, Ag8, Ag10, respectively, in the gas phase and 0.03, 0.03, 0.11 eV for Ag6, Ag8, Ag10, respectively, in the supported case.

(Color online.). (a) gas phase and (b) Fs-MgO-supported structural motifs of silver clusters at size 6 (first row), 8 (second row) and 10 (third row) are shown; for each cluster size, the energy differences with respect to the putative global minimum are reported in eV. In the case of Ag8, the putative global minimum in both cases belongs to a peculiar D2d symmetry group and it is not shown here.
A fluxional character of the metal cluster is considered to be beneficial for catalysis, as it may imply the possibility of novel, lower-energy-barrier mechanisms with respect to those occurring for larger clusters or extended systems (with possibly the support playing an active role, see Section 3 below), so that one can escape the boundaries imposed by the Sabatier principle and the corresponding volcano curve [24,25]. In practice, the situation at both the experimental and theoretical levels is not so simple. A first problematic issue is represented by the fact that precise knowledge on the nature of the support and its defects is required to derive an accurate modeling (different defects produce a completely different PES), and this knowledge is not easily obtained at the experimental level, so that e.g. the quantitative influence of Fs-centers on catalysis is controversial [26]. Moreover, from the purely theoretical point of view, the system's fluxional character implies the need for developing and implementing thorough and exhaustive search algorithms that are able to explore the novel paths and mechanisms that such systems exhibit. These algorithms must employ sophisticated first-principles approaches such as Density-Functional theory (DFT), because empirical potentials and force fields are usually not accurate in this size range.
The situation is therefore very complex, and one often encounters an intricate panorama, with several processes in competition: reactive dynamics of the ligands and the metal cluster, exchange with species coming from the gas phase or the support (spill-over and reverse spill-over [27]), disaggregation and/or dis-anchoring of the complex, etc., and all of these not necessarily in equilibrium, but often under kinetic conditions and interwoven as a function of experimental conditions. The most effective theoretical strategy adopted so far to tackle these complex problems consists in performing global optimization (GO) systematic searches of the DFT PES [23,28–30], followed by saddle-point search algorithms such as the nudged-elastic band (NEB) one [31] for determining reaction energy barriers and thus the rate of inter-conversion among the local minima, as exemplified in pioneering works on cluster diffusion [32,33]. Traditional alternative approaches are based on molecular dynamics simulations also amenable at the DFT level (with known problems of ergodicity and thoroughness of PES sampling), while the most recent developments rely on neighborhood exploration techniques, as will be recalled below in Section 6. It should be noted that reactant species can either come directly from the gas phase (in the so-called Eley–Ridal mechanism) or be pre-adsorbed on the metal cluster (in the so-called Langmuir–Hinshelwood mechanism) or on the oxide support (spill-over) and can then evolve back to the gas phase or to the support (inverse spill-over) after the catalytic event has taken place [34]. To simulate these phenomena at the GO level, random moves in the space of stoichiometry in addition to geometrical ones must also be considered, which require defining appropriate chemical potentials for the incoming species [35]. As a technical note, for magnetic clusters also moves in the spin space should be considered. Once low-energy structural candidates have been singled out, if thermodynamic equilibrium can be assumed, first-principles (or ab initio) thermodynamics can be employed [36]. However, this assumption is not always valid, see Section 6 below.
3 The influence of the oxide support
The influence of the support on the structure of metal clusters has been briefly recalled in Section 2. It can be simply added in passing that this influence can persist up to rather large particle sizes much beyond the ultranano régime, as demonstrated both theoretically and experimentally by the possibility of creating exotic epitaxial relationships and interface-stabilized phases [37].
The cluster electronic structure can also be significantly affected by the interaction with the support. One interesting example is the quenching or at least lowering of the spin state of the metal cluster upon adsorption [38], which is to be expected in general terms on the basis of the increased coordination (analogously to what happens at high ligand coverage, see Section 4) but is also connected with specific effects of metal/oxide interaction.
Another important effect is the transfer of electronic charge between the support and the ligand/cluster complex. It can be recalled that since early analyses [39], four main components of metal/oxide interaction have been pointed out: charge transfer and covalent bond (although a precise distinction of these two components is not easy [40]), in addition to polarization and dispersion interactions. On MgO(100) it is usually found that charge transfer is not large on the regular surface. It can however be increased when the work function of the system is reduced by creating electronic states in the band gap, as induced by defects such as an oxygen vacancy [41,42] or if the oxide is actually a medium for electron transfer from an underlying electron reservoir as it occurs for oxide ultrathin films grown on low-work-function metal or semi-conductor surfaces [14,15,43–45]. The latter possibility is particularly intriguing, as it allows one to combine ultrananoclusters with ultrathin supports, thus exploiting the novel features of both systems in catalysis [46,47] with the option of further enriching the researcher's palette by introducing an electric bias [48]. Charge transfer can be important in catalysis, as it is known from gas phase studies (usually on charged clusters) that it appreciably alters cluster reactivity: to make one of the many possible examples, a negative charge can promote O2 adsorption and dissociation on bare Ag clusters [49]. As an illustrative case, we show on Fig. 2 a comparison of plots of the spin density for different systems in which we see, e.g., that Ag on the regular MgO(100) surface exhibits weak charge transfer (also associated with weak bonding), Au on the regular surface has a less weak charge transfer (and correspondingly a bit stronger bonding [44]), Au on an Fs-center has strong bonding and a very strong charge transfer, AgPd in the gas phase contains a Pd atom whose electronic configuration has been promoted from d10 in the free atom to d9s1, i.e. to the valence state of the fully formed metallic bond [50], whereas AgPd deposited on an Fs-center of MgO(100) contains Pd with its electronic configuration switched back from d9s1 to d10. As the chosen metal systems contain an unpaired electron, the spin density gives good indications on chemical bond effects.
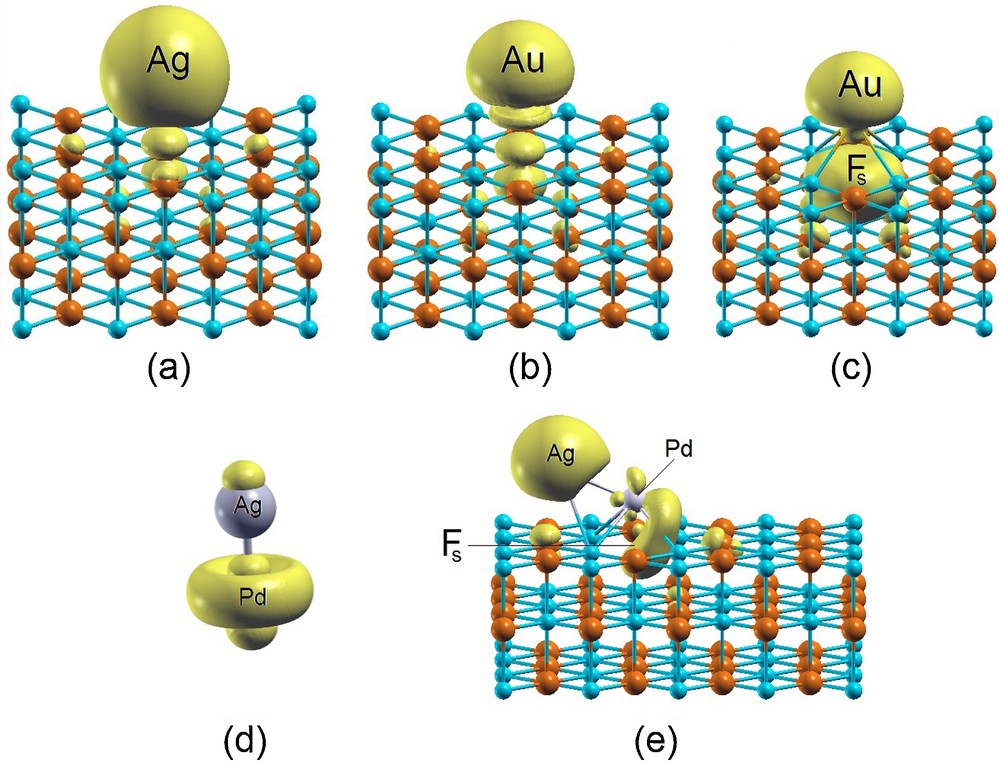
(Color online.). Spin density plots of (a) an Ag and (b) an Au adatom interacting with the regular (100) MgO surface; (c) an Au adatom interacting with the Fs-defected (100) MgO surface; (d) the gas phase PdAg dimer; (e) the PdAg dimer interacting with the Fs-defected (100) MgO surface. Isosurfaces at a value of 0.001 a.u. are plotted.
One less recognized effect of the oxide support is the electric field generated by the charge-separated substrate. Even on the regular and Fs-defected MgO(100) surface (see Fig. 3 for a plot of the electrostatic potential generated by the regular and the defected oxide surface) and thus for metal/oxide interactions without or with little charge transfer, this electric field can completely alter the PES of small supported clusters. For example, breaking of O2 on small Au and Ag clusters becomes thermodynamically favorable, contrary to the situation in the gas phase. It can be noted nevertheless that without charge transfer, the energy barrier to O2 dissociation can still be high, so that other mechanisms must arise for these systems to act as efficient oxidation catalysts [51]. The effect of the electrostatic field is connected with the polarization component of the metal/oxide interaction, and can be important for both small and larger clusters due to the so-called “metal-on-top” effect, i.e., the enhancement of adhesion of a given metal atom to the substrate due to the presence of other metal atoms on top of those that are directly in contact with the substrate, an effect which, e.g., favors upright with respect to epitaxial configurations of metal trimers [52].

(Color online.). Electrostatic potential generated by (a) the regular and (b) a Fs-defected (100) MgO surface are shown in a 2D map (left-side) and 3D map (right-side). In the case of the 3D map, an isosurface of −0.0005 a.u. is plotted.
Reproduced with permission from [53]. Copyright 2009 Springer.
Finally, the oxide support can take an active part in the catalytic reaction mechanism, which therefore gives rise to novel mechanisms with respect to traditional ones. Even a simple, rigid oxide such as MgO(100) can, for example, play a crucial role by coordinating ligand species such as O2 molecules as originally shown in [54]. This is exemplified on Fig. 4 for the oxidation reaction of CO to CO2 catalyzed by Ag3 supported on MgO(100), in which we see that MgO plays a crucial role in the formation of an Ag3(carbonate) complex, which then acts as the catalytically active species [51]. Clearly, these effects, which are already present in simple, rigid oxides such as MgO are amplified on reducible and fluxional oxides such as ceria or titania [55,56], in which, e.g., oxygen atoms can move from the substrate to the cluster. If some of the atoms of the support take active part to the reaction by substantially changing their geometry, the correct way of exploring these phenomena is to include these atoms into the set of active species, e.g., in the global optimization moves mentioned at the end of Section 2, or in the PES neighborhood exploration techniques mentioned in Section 6 below.

(Color online.). Mechanisms ruling the oxidation reaction of CO to CO2 catalyzed by Ag3 supported on MgO(100). The ER and LH acronyms refer to Eley–Rideal and Langmuir–Hinshelwood mechanisms, respectively.
4 Coverage effects
The evolution of ligand adsorption energetics, structure and dynamics as a function of coverage is a fascinating topic.
First of all, in general one observes a non-linear or many-body or saturation behavior, i.e., a decrease of ligand adsorption energies on a given metal cluster as a function of the number of adsorbed species. As a consequence of this saturation behavior, coverage in operating conditions depends on parameters such as temperature, so that e.g. by increasing the temperature, coverage is expected to shift at lower values [34]. In this connection, a phenomenon, which is not always underlined, is the lesser sensitivity of adsorption energies to cluster composition in the initial stages of ligand adsorption, i.e., at low coverage. At high temperature and low coverage, one can thus expect that catalysts will tend to exhibit similar activity but a low selectivity, also because of kinetic reasons: decreased Arrhenius filtering of higher energy barrier processes, see Section 6 below. One other aspect that has been investigated in previous literature on gas phase cluster but is not often recalled in the supported case is the possibility of synergic effects in ligand adsorption, i.e., the fact that the adsorption energy of a given ligand can be strongly influenced by co-adsorption of other species [57]. Although not present in all cases [51], this phenomenon can be relevant in catalysis. For example, connected with this phenomenon is the well-known influence of trace impurities, as either accelerating or poisoning species, on the catalytic activity and also for increasing or decreasing catalytic selectivity, see, e.g., [58,59] for two disparate examples. To illustrate this phenomenon, we report on Fig. 5 the energy differences obtained by adding CO2 or H2 molecules starting from a Ni3(H)(HCO2) complex deposited on the regular MgO(100) surface. It can be clearly seen in this figure that the first adsorption of CO2 or H2 lowers the energy by a quantity (0.31 or 0.41 eV for CO2 or H2, respectively) much smaller than the corresponding quantity obtained after adsorption of the other species (1.38 or 1.29 eV for CO2 or H2, respectively). In summary, it can be stated that saturation effects tend to limit adsorption energies, whereas ligand–ligand synergic effects can amplify them, especially if the two ligand species have different chemical characteristics (electronegativity), until saturation in both species is reached.
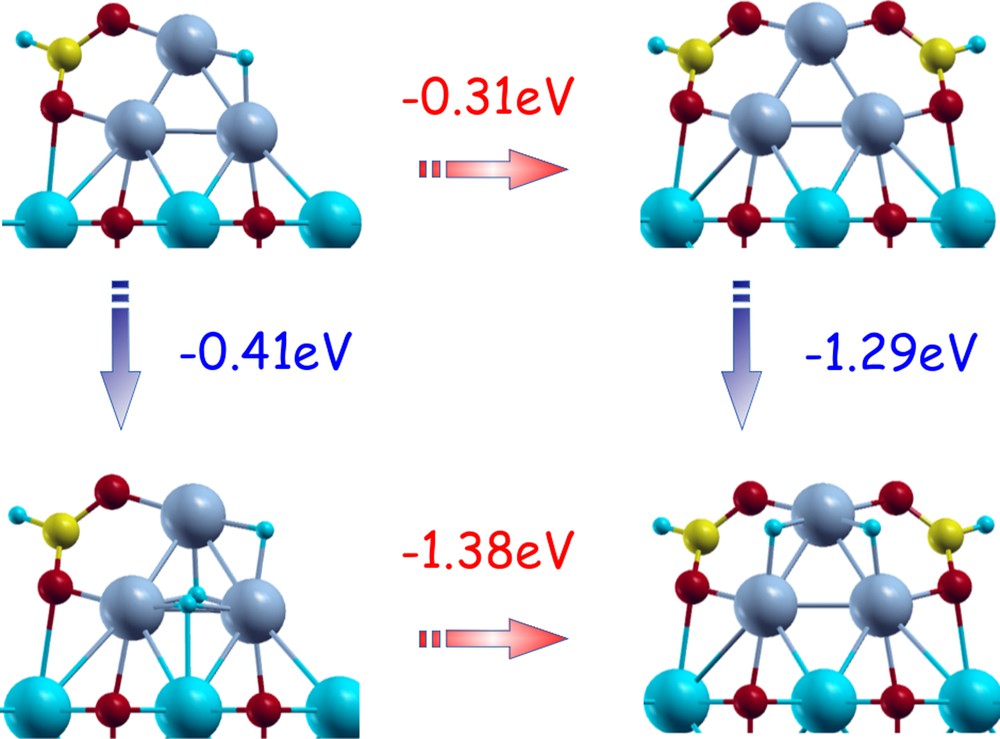
(Color online.). Energetics of sequential adsorption of CO2 or H2 molecules onto a Ni3(H)(HCO2) complex deposited on the regular MgO(100) surface. The energy needed for adding CO2 is given in red, that for adding H2 is given in blue. For each stoichiometry, the global minimum configurations are considered. Energies in eV. For interpretation of references to color, see the online version of this article.
Second, ligand adsorption can modify the structural framework of the metal ultrananocluster [60]. What happens is that the cluster possesses several isomers in a narrow interval of energy, and some of the higher energy ones can be more apt to ligand adsorption so that a crossover between these configurations and the global minimum one is realized by increasing ligand coverage. From a technical point of view, it can be noted parenthetically that high ligand coverage and the resulting high coordination often correspond to quenching of the electronic spin of the metal cluster, which can be beneficial in terms of the stability of convergence of the self-consistent Kohn–Sham DFT equations.
The concept of the evolution of ligand adsorption structure as a function of coverage can be further illustrated and enriched in the case of ethylene adsorption on gas phase Pt2 and Pt3 clusters, see Fig. 6 [61]. Here, we can see in the example of Pt2 that depending on ethylene coverage, one has two rather different régimes: a saturation one and an unsaturation one, with an interesting energetic switch of the adsorption mode from a di-σ mode characteristic of low coverage to a π-mode characteristic of high coverage. This is in keeping with experimental data: a dependence on the adsorption mode upon coverage has been seen experimentally on extended surfaces [62]. This switching of adsorption mode can have a great influence on the catalytic properties of the system. As an example, in the same figure, it is shown that on Pt3 in the unsaturation or low coverage régime, the oxidative insertion of Pt into the CH bond can be activated, whereas this process is not favorable in the saturation régime [61]. In passing, it can be noted that the interaction with ligand species such as a carboncarbon double bond can lead to a quenching of spin on metal atoms and, e.g., back-promotion of Pt from the d9s1 valence state to the atomic state d10 [61,63].
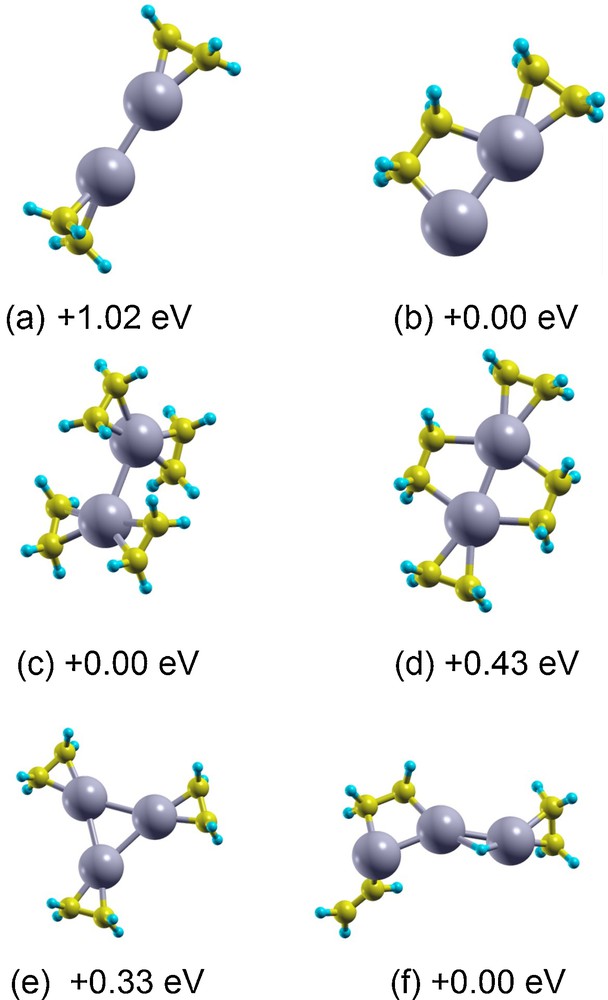
(Color online.). (a–d) Pt2 dimer in the case of absorption of 2–4 ethylene molecules: a: Pt2eth2 in di-σ mode; b: Pt2eth2 in a mixed di-σ mode and π-mode; c: Pt2eth4 in full π-mode; d: Pt2eth4 in a mixed di-σ mode and π-mode. Pt3eth3 molecule in the case of (e) pure π-mode and (f) formation of a hydride complex. Energy differences in eV with respect to the putative global minimum for each species are reported.
In conclusion, especially at low temperature, one can expect (and should investigate theoretically) that more than one species of the same reactant can be adsorbed on the metal ultrananocluster, as has been proven to be true, even for extended systems [64]. It can be noted in passing that under this point of view, catalysis by metal oxide or hydride clusters can be simply seen as an O2 or H2 high-coverage variant of the present subject. Multiple ligand adsorption corresponds to the formation of ligands + metal–cluster complexes that then act as the real catalytically active species, as in the example of CO oxidation by Ag3/MgO(100) recalled in Section 3 in which an Ag3(carbonate) adduct is predicted to be the real catalyst in operating conditions, see Fig. 4 [51]. These catalytic complexes are the analogous of metal complexes in homogeneous catalysis, with the difference that in the heterogeneous case these species are not preformed, but are formed in situ with a stoichiometry depending on the experimental conditions (in passing, we believe it would be worthwhile developing this parallel in more depth). In connection with Section 2, it should be underlined that the fluxional character beneficial for catalytic activity pertains to the catalytic complex and not to the bare metal cluster, as often implied for simplicity in current literature. These two quantities do not coincide in general, as the presence of the reactants can appreciably modify the PES, and thus the fluxional character, of the catalytic system. In our experience, we have observed for example that an excessively fluxional metal cluster by reacting with ligands can give rise to a rigid adduct, thus eventually being less catalytically active than another cluster which is less fluxional but keeps this character after ligand adsorption (and may be additionally more robust, which is the subject of the next section).
To summarize and close the cycle initiated in Section 2, the formation of catalytic complexes entails the need of singling out the correct mechanism of the process under the given operating conditions. Once this is done, one can search for appropriate descriptors and then construct a volcano curve according to the Sabatier principle. Such descriptors will not necessarily be the same and/or with a peak located at the same position for all possible mechanisms, even though accidental degeneracies/coincidences may occur, explaining the paradox of correct claims based on oversimplified mechanisms.
5 Catalyst stability
For small supported metal clusters, due to their fluxional character and incompletely developed metallic bond, the robustness of the catalyst is something that should always be tested. The two major phenomena here are: surface diffusion and cluster disaggregation.
Surface diffusion is essential in determining the growth of metal clusters via epitaxial aggregation of deposited adatoms, and presents interesting aspects because diffusion of small clusters has been shown to occur through an unexpected variety of possible mechanisms [32,33]. Diffusion is therefore essential for cluster growth, but—once a metal cluster is formed with the right size—it can be detrimental to catalysis, as it can lead to sintering into larger, and possibly less active, aggregates [65–67]. Metal atom diffusion into the substrate also leads to loss of catalytically active components and should be taken into account as a possibility. In this connection, it can be noted that not much investigated so far (a rare exception is [68]) is the diffusion of the composite ligand + metal aggregate (e.g., the catalytic complex of the previous section), although it can be expected to be equally important and possibly as facile as diffusion of the bare metal cluster. A trapping center such as a defect is therefore needed to anchor the clusters to the surface and hinder the achievement of the thermodynamically stable but catalytically less interesting crystalline phase.
A second important phenomenon is cluster disaggregation. This has been investigated in detail for the gas phase [69], but it is less known for supported metal clusters, except for a few instances. As an example, in the case of Ag clusters supported on a double vacancy defect of MgO(100), disaggregation has been shown to preferentially occur via emission of an Ag2 dimer [30]. Even less investigated is disaggregation induced by ligand binding to the metal cluster. Strong ligands such as CO can in fact weaken the metal/metal interaction and trigger cluster breaking. For example, the combined bonding of CO and O2 onto small Au clusters has been shown to disrupt AuAu bonds and favor cluster breaking [51]: see a pictorial illustration of this phenomenon on Fig. 7. The predicted instability of Au clusters under realistic conditions of CO oxidation is in keeping with experimental observations [70]. Again, cluster disaggregation leading to Ostwald ripening into larger aggregates can be detrimental to the catalytic activity [65–67], even though under appropriate conditions it can be hypothesized that a dynamic equilibrium and a steady state is reached between aggregation and disaggregation [71].
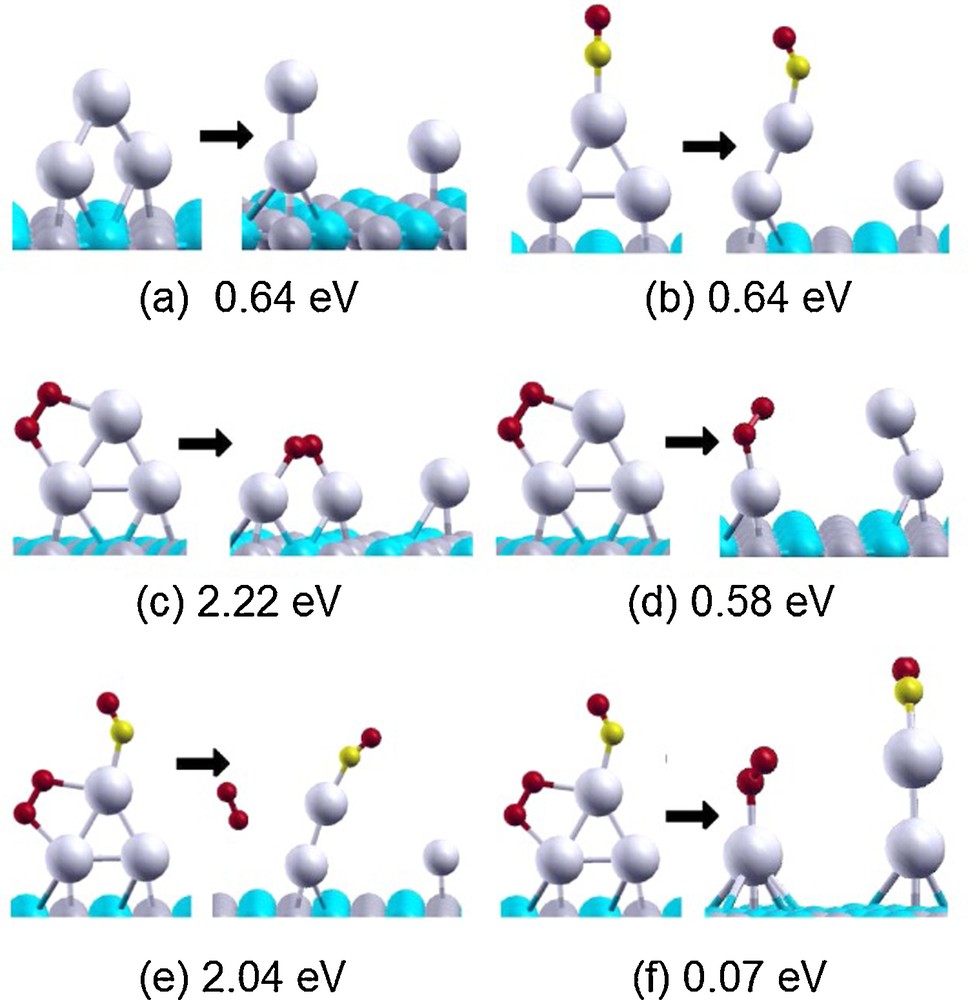
(Color online.). Breaking of Au3OnCOm complexes. Minimum energies required to break the complexes are reported in eV.
Partially adapted with permission from [51]. Copyright 2012 The American Chemical Society.
To summarize: ultrananoclusters can be more fluxional and thus more interesting, but also more fragile and more prone to instability than larger systems. A compromise between these two characteristics must be found to exploit their novel features in practical devices.
6 Out-of-equilibrium effects
As a final topic, the possibility of out-of-equilibrium effects and the theoretical methodology required for their predictive description are briefly discussed.
It was mentioned in Sections 2 and 4 that ultrananoclusters or their catalytic complexes can be fluxional species, exhibiting several energetically close local minima rapidly interconverting among each other. It was also recalled that the most effective theoretical strategy adopted so far to tackle the issue of their description consists in performing global optimization (GO) sampling, followed by saddle-point search for determining reaction energy barriers. This strategy however does not work in all cases. Suppose in particular to consider kinetics-driven catalytic processes. In such out-of-equilibrium cases, the system does not follow the path, which minimizes its energy (or free energy), but the path, which presents lower (and accessible in the given experimental conditions) energy (or free energy) barriers. Let us consider, for example, the partial oxidation of propylene to propylene oxide or acrolein, as experimentally realized, e.g., via catalysis by Ag3 supported on an ultrathin amorphous alumina substrate [14]. If one runs a GO algorithm on a system constituted by propylene and O2, one invariably finds that the algorithm will produce CO2 and H2O as the thermodynamic lowest energy state, and it is extremely difficult if not impossible to try to bias the system to avoid this end result. What happens in reality is that the catalytic system during its evolution encounters one or more branching points in which the thermodynamically favored product is not produced because it presents a higher energy barrier with respect to other, kinetically favored products. This is also intimately connected with the topic of catalytic selectivity, which is especially important in modern catalysis studies, as in the case recalled above of partial oxidation of propylene with the formation of two different products: propylene oxide and acrolein. The theoretical approaches needed to tackle this class of problems must therefore rely on very thorough and accurate PES neighborhood exploration techniques, such as, e.g., those proposed in [72], which are computationally expensive but promise to be able to capture the subtle interplay of several competing reaction paths and to identify the branching points that determine the catalyst's selectivity toward kinetics-favored products. For example, in the case of propylene oxidation by Ag3 on MgO(100), it has been suggested that replacing one of the Ag atoms with an Au atom should favor upright configurations and thus selectivity towards propylene oxide rather than acrolein, without compromising the cluster stability, a suggestion still waiting to be tested experimentally. A discussion of transition search algorithms and/or in combination with databases and system comparison approaches analogous to the techniques used in GO [73,74] will be the subject of a future report.
7 Conclusions
In summary, in the present brief article we tried to present and illustrate with simple examples some of the concepts that in our opinion play a key role in heterogeneous catalysis by very small, subnanometer metal clusters (both pure and alloyed), or—as we propose to name it—in heterogeneous ultrananocatalysis. Fluxional character, support and coverage effects, and catalyst stability, in addition to out-of-equilibrium phenomena, have been suggested as basic concepts to be taken into account in the study of this fascinating and rapidly developing field. Attention has been focused on an oxide support, but the concepts here discussed should be applicable in a much broader context. One fundamental idea hopefully emerging from this presentation is that the formation and evolution under the given conditions of a metal/oxide/ligand hybrid or composite system, which exhibits characteristics often strongly different from the individual components is the core of ultrananocatalysis. The consequent possibility of novel reaction mechanisms that can overcome current volcano curve limitations, the atom-economic use of metal elements (especially appealing for precious metals), and the non-scalable dependence of catalytic properties on cluster size and composition lending itself to surgical investigations, all make that ‘ultranano’ represents one of the most promising fields in catalysis.
Even from this quick and necessarily incomplete overview should emerge the complex, multidimensional character of the problem, which limits the validity of simple extrapolations and one-dimensional diagrams. As outlined above, the challenge for theoretical and computational approaches is indeed to be able to accurately describe the intricate interplay of several competing processes: reactive dynamics of the ligands and the metal cluster, exchange with species coming from the gas phase or the support (spill-over and reverse spill-over), disaggregation and/or dis-anchoring of the catalytic complex, etc., and all of these not necessarily in equilibrium but often under kinetic conditions and interwoven as a function of experimental conditions.
In our opinion, from a computational point of view, the development and continuous improvement of systematic sampling techniques (clearly relying on sufficiently predictive theoretical approaches) represent a proper answer to this challenge [72], together with the parallel development of experimental characterization techniques able to furnish precise information on the energetics, structure and dynamics of catalytic actors and events in situ and under realistic conditions. This however does not preclude – on the contrary it must stimulate – the search for descriptors [25,75,76], which – although in a limited range of validity and for a given and well-defined mechanism – can be derived to orient oneself in this complex and multidimensional panorama and achieve the design of novel and improved catalytic systems.
Acknowledgements
Financial support from the European Research Council-Advanced Grants SEPON project No: 227457 is gratefully acknowledged.