

1 Introduction
Two general types of interactions controlling the localization of the reaction site, the selectivity and the synthetic outcome are delimited within the perturbation theory of reactivity: orbital-controlled and charge-controlled interactions, depending on whether the orbital or the Coulombic contribution to the total interaction energy predominates [1,2]. While the orbital term arising from two-electron integral energy is a fundamental factor intrinsically related to the chemical structure of the reacting centers, charge interactions can be subjected to an external tuning. One of the possibilities for this is related to the media dielectric permittivity, given within Born model as:
| (1) |
To cite a few examples from such families, these are lowering the oxidation potentials of amines due to nN–σC–Si hyperconjugation [8], the cases of thio- [9], seleno- [10] and telluroethers [11] showing abnormally low oxidation potentials, the through-space interactions of two sulfur or selenium atoms in mesocyclic dichalcogenido compounds [12–18] and macrocyclic bi-, tri- and tetraselenides [19]. Through-space interaction of the 2-(2-pyridyl) ethyl pending substituent with a selenium atom was reported to remarkably facilitate electron transfer from selenides [20].
We have previously shown that similar transannular interactions take place in the cation radicals of selenoethers bearing substituents with a CO group whose oxygen can approach Se atom to a distance allowing substantial Coulombic interactions [21,22]. Usually, cation radicals resulting from the oxidation of dialkyl and alkyl arylselenides undergo monomolecular charge-controlled cleavage of bonds adjacent to selenium (hard acid center): or [23–25]; this is in contrast with the cation radicals of diaromatic selenides, for which second-order (orbital-controlled) reactions are typical [26–28], like for parent aryl thioethers [29]. In case of the selenides with transannular Se…O interactions, similar orbital-controlled bimolecular interactions were observed [21] instead of the expected first-order reactivity.
In this communication, we aim at demonstrating how orbital- and charge-controlled reactivity of cation radicals can be simply switched by the dielectric effect of the reaction media. For this, the alkyl aromatic selenoethers 1–6 were studied by cyclic voltammetry, large-scale electrolysis and IR spectroscopy in binary mixtures of various polarities. The selenide 7, in which such intramolecular interaction is absent, was considered as a test compound.
| 1 | 2 | 3 | 4 |
| PhSe(CH2)2COOMe 5 | PhSeCH2COOEt 6 | PhSeC6H12 7 |
2 Experimental
Cyclic voltammetry and large-scale electrolyses were carried out using a PAR 2273 electronic potentiostat piloted with PowerSuite software (PAR). A standard three-electrode electrochemical cell was used with a 2-mm glassy carbon (GC) working electrode. The auxiliary electrode was a GC rod and the Ag/0.1 M AgNO3 in CH3CN system served as a reference electrode. The working electrode was carefully polished with Struers 4000 paper and washed in acetonitrile before each measurement.
DFT calculations were carried out using Gaussian 03 software [30].
Aryl selenide 4 was prepared by methylation of 2-methylselenobenzoic acid [31,32] by MeI in a methanol solution of NaOH. Compounds 1–3 were prepared by electrochemical oxidation of Ph2Se2 in an acetonitrile–NaClO4 solution in the presence of cyclohexene, hex-1-ene and hept-1-ene, respectively [33]. Selenides 5 and 6 were prepared by the reaction of PhSeH with ethyl ethers of 3-chloropropionic and 2-chloroacetic acid, respectively [34]. Analytical-grade acetonitrile (AN) was purified by distillation from KMnO4–P2O5 and kept over activated 4-Å molecular sieves. Bu4NBF4 (Aldrich) was kept over P2O5 and used without any additional purification.
N-Cyclohexenylacetamide. Mp 99 °C (white needles from hexane). 1H NMR (CCl4, TMS) δ: 5.76 (s H), 3.7 (s H), 1.9 (s 3H), 1.16–1.66 (m 8H). IR (solid): 3300 cm−1; νC=O 1650 cm−1 (amide I), νN–H 1560 cm−1, δC–H 725 cm−1. Calcd, %: C 69.56, H 8.69, N 10.14; Found (C8H12O) %: C 69.72; H 8.75; N 10.05.
1,2-Bis(acetamido)cyclohexane. Mp 268 °C (white powder from ethanol). 1H NMR (CDCl3, TMS): 6.12 (s 2H), 3.61–3.75 (m 2H), 2.1 (m 4H), 1.9 (s 6H), 1.7–1.8 (m 4H). IR: 3290 cm−1, νC=O 1645 cm−1 (amide I), νN–H 1555 cm−1, δC–H 725 cm–1. Calcd, %: C 60.58; H 9.15; N 14.13; Found (C10H18N2O2) %: C 60.73; H 9.07; N 14.21.
Diphenyldiselenid. Mp 62 °C (yellow fibers from ethanol). 1H NMR (CCl4, TMS): 7.6, 7.13 (10H). IR: νC–H 690 cm−1, νC–H 735 cm−1. Calcd, %: C 46.18; H 3.21; Found (C12H10Se2) %: C 46.23; H 3.24.
3 Results and discussion
Upon oxidation in AN/0.1 M Bu4NBF4, selenides 1–5 show a well-defined peak at Ep ≈ 1 V, followed by a less developed second oxidation step (Fig. 1). Because of adsorption of seleno ethers at the Pt interface, voltammograms of 1–5 at a glassy carbon (GC) electrode are better shaped, in spite of its higher internal capacity compared to that of a Pt electrode. The activation energy of the limiting current of 1–5 (RΔlog(ip)/Δ(1/T) = 6.2 ± 0.5 kJ mol−1) is practically the same as the temperature coefficient of viscosity of acetonitrile [35]. The oxidation currents ip vary linearly with the square root of the scan rate v1/2 and with the concentration (for C ≤ 5 × 10−3 mol L−1), which attests to a diffusion-controlled process [36]. The electron stoichiometry of the first oxidation step was determined from comparison of the oxidation currents ip of 1–5 with the one-electron current of ferrocene. Although at slow scan rates, only the anodic peak is present, at v ≥ 100 V s−1 some reversibility starts to appear, corresponding to the reduction of electrogenerated cation radicals (Fig. 2).
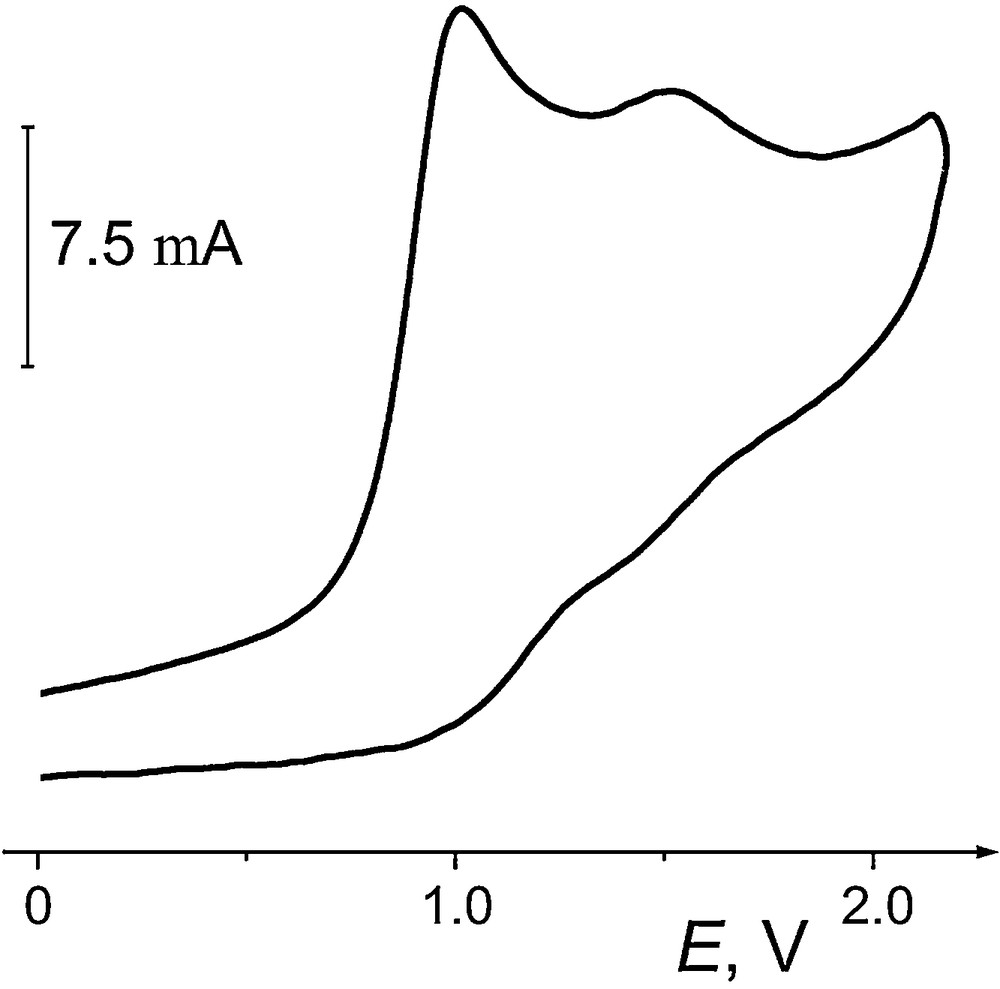
Voltammogram of oxidation of 2 (2 × 10−3 mol L−1) in AN/Bu4NBF4 at a GC microdisk electrode; scan rate v = 2 V s−1, T = 295 K.
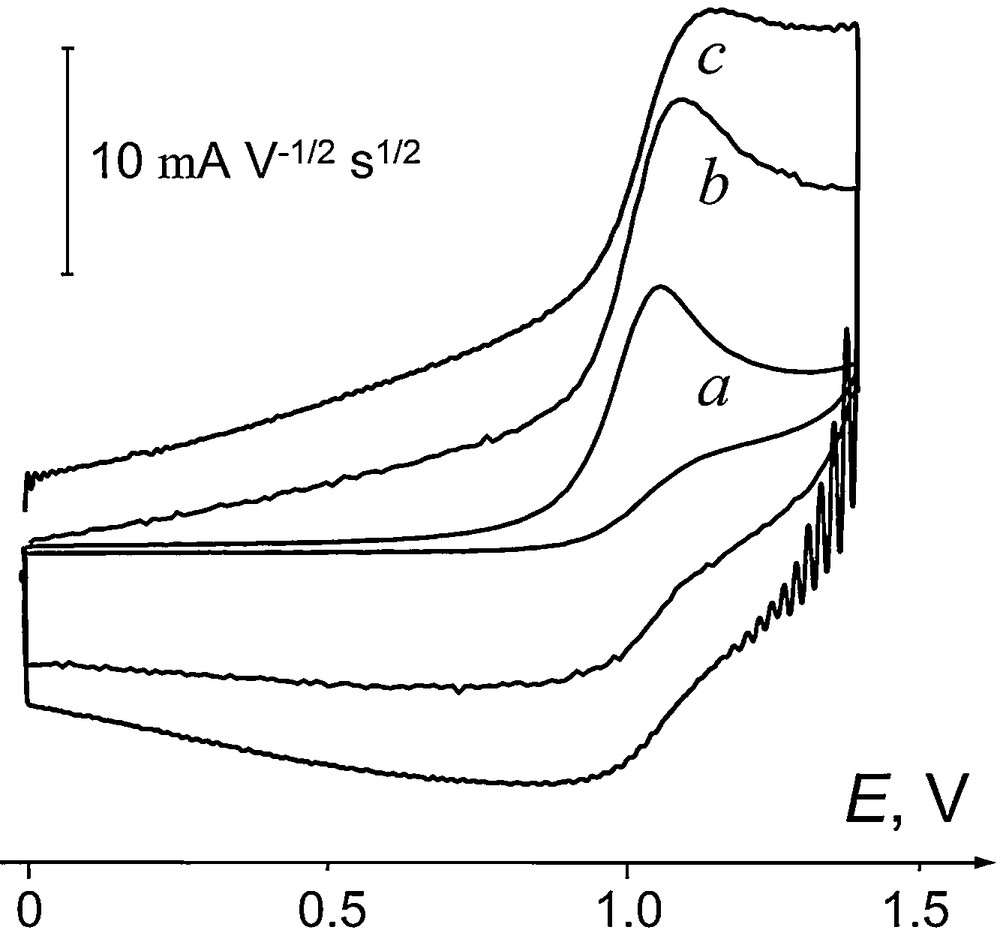
Current-normalized voltammograms of 3 (2 × 10−3 mol L−1) at different scan rates: (a) 0.5 V s−1; (b) 25 V s−1; (c) 100 V s−1.
Characteristic slopes ΔEp/Δlog(v), determined from cyclic voltammetry at various scan rates (Table 1), are consistent with those obtained earlier using a rotating disk electrode, ΔEp/Δlog(ω) [21], both indicating the bimolecular character of the reaction of electrogenerated cation radicals 1+•–5+•. The process at Ep1 thus corresponds to an EC scheme with electrochemically reversible electron transfer followed by fast second-order bulk reaction of the primary products [36]. Such reactivity is typical for thioethers [37] or diarylselenides [26,28,38,39], but is quite unusual for selenides, for which monomolecular cleavages usually occur. This reveals radical side of reactivity of the cation radicals 1+•–5+•, which became possible because the charge localization on Se in these species is insufficient for inducing SeC or (Se)CH bond cleavage directly, as in dialkyl and alkyl arylselenides [25]. Remarkably, the cation radicals of 6 and 7 both react in a monomolecular process (Table 1), in accordance with the general trend for selenoethers [38].
Parameters of the electrochemical oxidation of aryl alkyl selenides ArSeAlk at a GC electrode in AN and is mixtures with THF and EC (0.1 M Et4NBF4).
| Cpd | Ep, Va | Ep–Ep/2, mV | n | ΔEp/Δlog(v), mV | ΔE1/2/Δlog(ω) | ||
| THF-ANb | AN | EC-ANb | |||||
| 1 | 0.965 | 62 | 1.3 | 21 | 21 | 30 | 19c |
| 2 | 0.972 | 60 | 1.2 | 22 | 20 | 30 | 20c |
| 3 | 1.125 | 64 | 1.1 | 23 | 20 | 32 | 20c |
| 4 | 1.090 | 67 | 1.2 | 20 | 22 | 30 | 20c |
| 5 | 1.065 | 62 | 1.2 | 22 | 21 | 31 | |
| 6 | 1.235 | 105 | 2.0 | 20 | 29 | 31 | 30c |
| 7 | 1.110 | 95 | 2.0 | 32 | 31 | 32 | 31d |
The chemical nature and the size of Se atom [40] imply that the positive charge in the cation radicals of selenoethers, even those of diarylselenides [39], is mostly localized on Se, so that this dichotomy directly stems from transannular stabilization of the charge on Se by the oxygen of internal carboxy or acetamide groups in 1+•–5+•.
Supposing equation (1) to hold for cation radicals 1+•–5+•, we considered their reactivity in media of different polarities, using binary mixtures of acetonitrile (ɛ = 36) with THF (ɛ = 7.4) and ethylene carbonate (EC, ɛ = 90 at 313 K [35]). The available polarities in this medium cover the 8 < ɛ < 90 range. The complete account of all solute–solvent interactions is quite complex [3] and the “polarity” of a solvent can be described through a number of parameters [41,42]. In this context, certainly not the most selective [43], though having a simple physical sense, the solvent dielectric permittivity ɛ is a convenient characteristic of the polarity of the used medium.
The evolution of the slope of the characteristic function Ep–log(v) for the oxidation of selenides 1–5 with the dielectric permittivity of the binary mixture is shown in Fig. 3. All selenides, for which transannular Se…O interaction is important, exhibit a change of this parameter from 20 mV to 30 mV upon increasing the medium's polarity, attesting to a progressive evolution of the potential-determining reaction of the cations radicals 1+•–5+• from disproportionation (kinetic order α = 2) to a first-order reaction (α = 1). The test compound 7, in which such interactions are absent, shows no dependence of the nature of the follow-up reaction on solvent polarity. Selenide 6, whose cation radicals exhibit monomolecular reaction in AN (Table 1), also shows the dichotomy of reactivity (Fig. 3), as observed for 1–5; however, this change occurs in the media less polar than AN, at ɛ ≅ 16.

Dependence of the ΔEp/Δlog(ν) value on the molar fraction of acetonitrile in binary solvents THF-AN and AN-EC. (8) 1; (7) 2; (–) 3; (,) 4; (4) 6; (∀) 7. GC electrode, T = 298 K.
The solvatochromism of electronic (UV/Vis) or molecular (IR) modes is often considered for studying specific interactions of substrates with the solvent [44]; obviously, intramolecular interactions show similar trends of optical properties. However, UV/Vis spectroscopy is precluded for the case of selenides 1–5 because of strong aromatic absorption of the PhSe moiety. In place, solution IR spectroscopy was used [42], because THF, AN, and EC are all optically transparent in the window of characteristic amide I and II vibrations [45]. Since it is difficult to separate specific solute–solvent interactions from purely dielectric effects [46], a simple model was adopted. In binary solvents, given a large excess of solvent compared to the selenide (at a substrate concentration of 1 mmol L−1, for each molecule of the selenide there are approximately 104–102 molecules of solvent, depending on whether it is pure or added as a co-solvent at ≥ 1%), solvent–solute interactions (solvation of CO and NH groups and Se) in the whole range of co-solvent concentrations are supposed to be constant, since only few solvent molecules can coordinate at once with the substrate. With this assumption (corroborated by the continuity of the Ep vs. log(v) graph upon switching THF-AN to EC-AN mixture), any change of vibrational modes can be considered as arising from dielectric-sensitive intramolecular interactions. At 1 mmol L−1, solubility changes and aggregation phenomena are neglected. For this model of solvent polarity, the dielectric function (ɛ–1)/(2ɛ + 1), developed for a diatomic oscillator in a dielectric continuum with macroscopic dielectric constant ɛ, was used (Fig. 4) [47]. Dielectric permittivities of solvents in binary mixtures being additive, the resulting ɛ has been calculated using the simplified Shakhparonov equation [48]:
| (2) |

Dielectric function (ɛ–1)/(2ɛ + 1) for the binary mixtures THF-AN and EC-AN with different compositions.
Corresponding shifts of CO and NH vibrations with solvent polarity are shown in Fig. 5. In IR spectra of neat compounds, the amide I band of the stretching vibrations of CO group in N-(2-(phenylselenyl)hexyl)acetamide 2 is shifted to 1625 cm−1 compared to N-hexyl acetamide 7 in which Se…O interactions are absent, vC=O = 1645 cm−1. In dilute acetonitrile solutions, this vibration is freed from solid-state associations and appears at higher frequency (1670 cm−1), and is progressively shifted to higher frequencies with a change of polarity of the solvent (Fig. 5). For amide II band vibration vN–H, an analogous trend was found, which was even more pronounced. The influence of Se on amide II absorption (1530 cm−1 in acetonitrile) is complex, resulting from the complex nature of this vibration due to the interaction between CN stretching and NH bending modes of the CNH fragment. The fact that vN–H vibration mode is more sensitive to solvent polarity than vC=O attests to the fact that Se…O interaction prevails over Se…H(N) in solution for neutral selenides. This feature reflects the fact that Se…O interaction in a non-oxidized molecule 2 has a repulsive and destabilizing character, contrary to stabilizing Se(+)…O interaction between positively charged Se and electron-rich carbonyl oxygen in the cation radical (Scheme 1). Induced by electron withdrawal, the geometry change in 2+• occurs due to free rotation about Csp3-N σ-bond, driven by the formation of a 2c–3e system similar to what was realized in di- and tri-seleno-substituted derivatives upon one-electron oxidation [17,18].
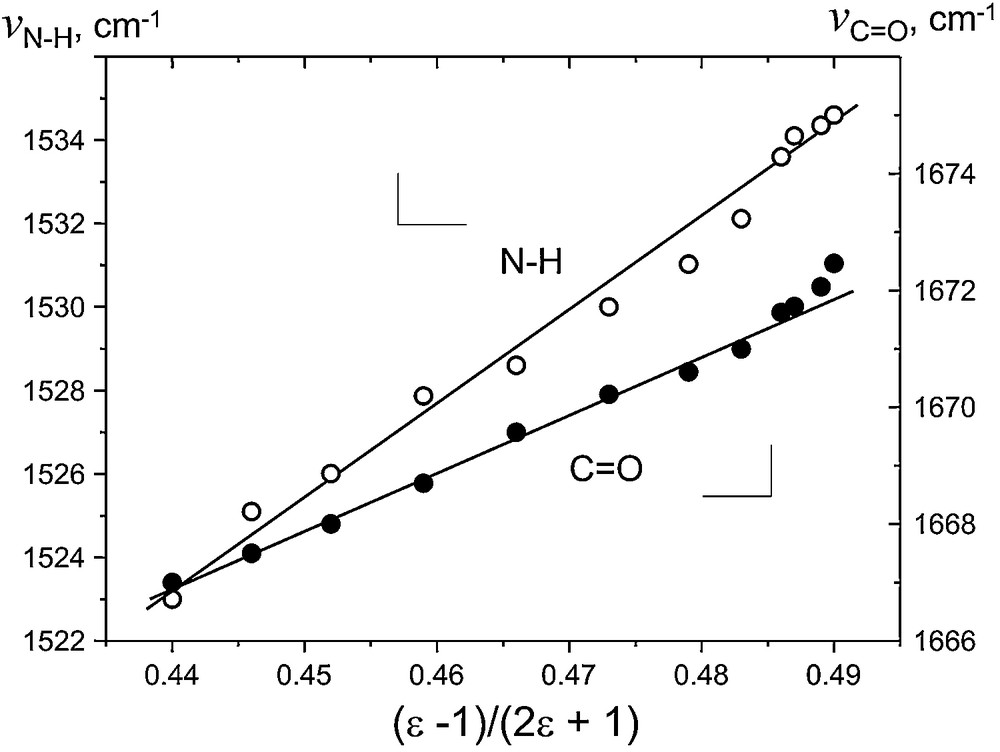
Solvatochromic shifts of vC=O and vN–H vibration frequencies in 2 with solvent dielectric parameter (ɛ–1)/(2ɛ + 1).

Through-space interactions in 2 and 2+•.
The low stability of cation radicals 1+•–5+• does not allow direct measurement of the vC=O shift caused by the Se(+)…O(C) interaction. In order to support the above model, the geometries of selenides 1–5 and of their cation radicals were optimized at the DFT B3LYP/6-311+G* level for media with different dielectric permittivities, using the isodensity polarized continuum model (IPCM) implemented in Gaussian 03 [49]. Thus obtained atomic charges at Se and carbonyl oxygen in these species in AN are collected in Table 2.
Distances Se…O and CO, Mulliken (M) and NBO (NBO) charges in selenides 1–7 and their cation radicals (CR) in CH3CNa.
| Cpd | lSe–O, Å | lC=O, Å | qSe | qO | ||||||
| Neutr. | CR | Neutr. | CR | Neutr. | CRM | CRNBO | Neutr. | CRM | CRNBO | |
| 1 | 3.892 | 2.441 | 1.230 | 1.251 | 0.123 | 0.740 | 1.108 | –0.690 | –0.697 | –0.745 |
| 2 | 3.891 | 2.449 | 1.230 | 1.251 | 0.123 | 0.739 | 1.108 | –0.690 | –0.697 | –0.744 |
| 3 | 3.882 | 2.482 | 1.230 | 1.251 | 0.135 | 0.758 | 1.118 | –0.690 | –0.699 | –0.747 |
| 4 | 3.071 | 2.526 | 1.210 | 1.226 | 0.144 | 0.773 | 1.137 | –0.654 | –0.678 | –0.693 |
| 5 | 3.377 | 2.555 | 1.208 | 1.224 | 0.130 | 0.765 | 1.132 | –0.656 | –0.658 | –0.697 |
| 6 | 3.353 | 2.808 | 1.211 | 1.205b | 0.204 | 0.805 | 1.162 | –0.709 | –0.711 | –0.615 |
| 7 | 0.111 | 0.742 |
a From DFT B3LYP/6–311+G* using IPCM solvation model [44].
b Se…O(Csp3) coordination instead of Se…O = C.
Partitioning of charges in the Mulliken scheme is not distance selective, so it only shows their evolution upon electron withdrawal. Distance-dependent CHelpG calculations [50] would supposedly provide a better account of charge distribution in these species, but proper open-shell treatment involving Se atom is still a very delicate task. As a compromise, NBO charges can be used for estimating Se…O interaction energy in a first approach (Fig. 6).
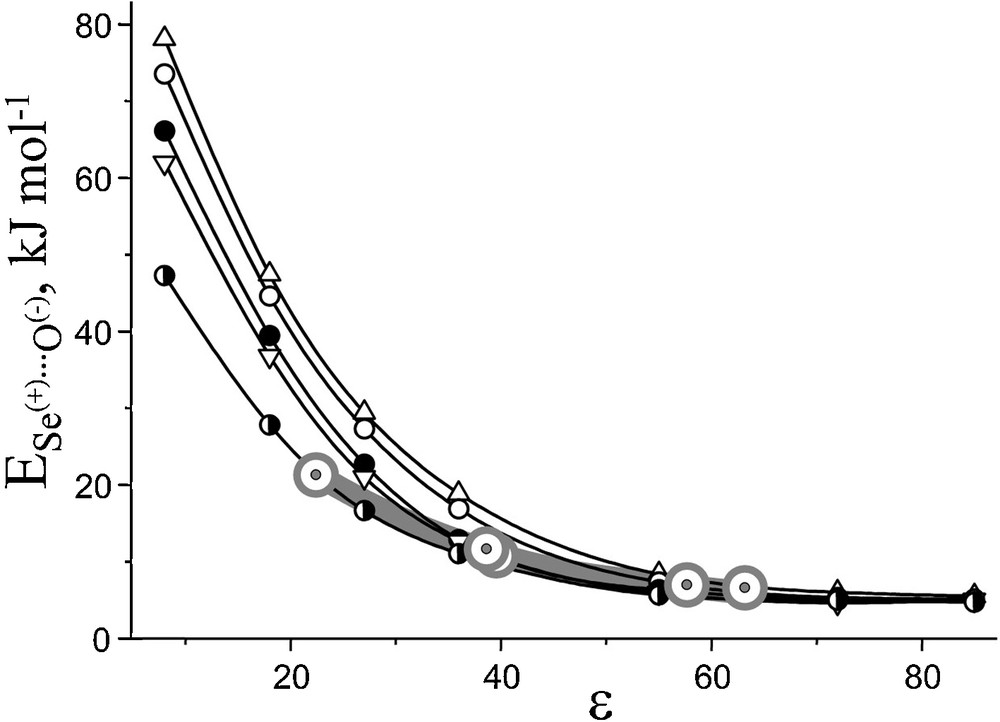
Correlation of Se…O interaction energy with solvent polarity for cation radicals: (8) 1+•; (–) 3+•; (,) 4+•; (X) 5+•; (4) 6+•. From DFT B3LYP/6-311+G*. Dichotomy points from Fig. 2 (ɛ at α = 1.5) are marked with grey circles.
Depending on the structure of the selenide, Se and O atoms can form six- to four-membered spatial rings (Fig. 7) providing their maximal approach. Only cation radical 4+• is planar and rigid, while in 1+•, 2+•, 5+• and 6+•, a free rotation about SeC and CC bonds is possible, which enables minimizing the Se…O distance. While these atoms approach, dative interaction from n-electrons of the carbonyl oxygen to positively charged Se provokes the concomitant elongation of the CO bond in 1+•–5+• (Table 2). Interestingly, CO bond becomes longer in 6+• compared to 6, but this is due to a different orientation of the carboxyethyl group: contrary to 1+•–5+•, the Se…O coordination in 6+• occurs at the ester oxygen (Fig. 7).
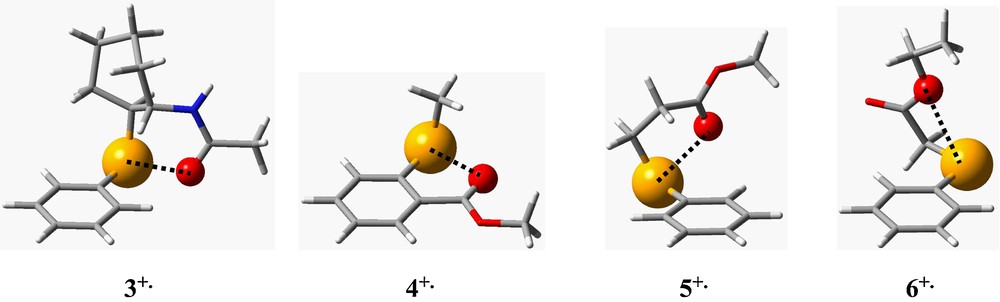
Optimized geometries of cation radicals 3+•–6+• and transannular distances Se…O. Van der Waals radii for Se and O atoms are used.
From this consideration, it is clear that taking into account solely the electrostatic interaction between two point charges, one neglects ring strain energy and orbital overlap contribution [51], since interatomic distance Se…O (2.44–2.80 Å) becomes comparable to that of a stretched SeO chemical bond (gas phase SeO length is 1.779 Å [52]). In addition, polarization of non-valent core electrons of bulky interacting atoms is totally omitted in this model.
In order to check the synthetic consequences of solvent-induced dichotomy, large-scale electrooxidation of 2 was carried out in binary solvents corresponding to the zones of first- (ɛ = 36) and second-order (ɛ = 68) reactivity of its cation radicals. The oxidation was carried out at a GC anode at the potential set at 0.9 V (at ca. 80% of the ip). The electrolyses were stopped when the current dropped to ca. 5% of the initial value, which was checked to correspond to 2.3 and 1.2 F/mol in polar and non-polar media, respectively, in good agreement with the results of cyclic voltammetry. In acetonitrile, N-cyclohexenyl acetamide was found to be the main oxidation product. Its formation involves β-elimination of the intermediate selenoxyde [53] resulting from the reaction of a selenium-centered dication with traces of water. The eliminated phenylselenenic acid was transformed into diphenyldiselenide during the work-up of the reaction mixture. In a more polar AN-EC (1:8) mixture, bifunctional 1,2-bis(acetamido)cyclohexane was formed as the main product. A direct cleavage of Se-Csp3 bond and second electron transfer gave PhSe+ and a carbocation, whose reaction with acetonitrile finally led to the di-acetamido product. Similarly, Ph2Se2 was the final product carrying eliminated PhSe moiety (Scheme 2).
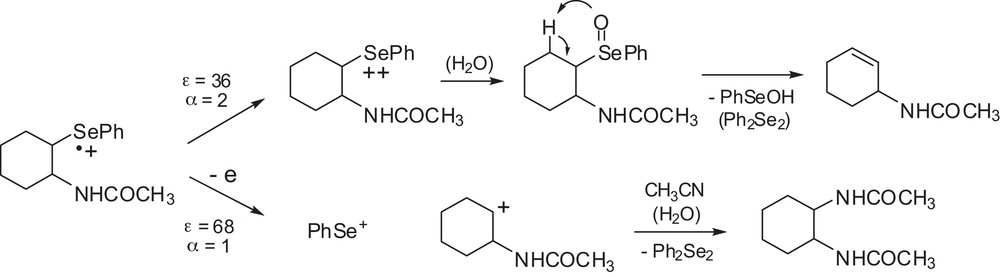
Electrooxidation of 3 in media of different polarities.
4 Conclusion
The electrooxidation of the family of alkyl arylselenides, whose cation radicals show Se…O transannular interactions, was studied by cyclic voltammetry and the related methods and supported by DFT calculations. Using binary mixtures of acetonitrile with aprotic solvents of lower and higher polarity allowed one to demonstrate the alteration of the reaction of electrogenerated cation radicals from bimolecular (orbital-controlled) to monomolecular (charge-controlled); this dichotomy has distinct synthetic consequences, as was shown by large-scale electrolysis. Whatever the exact mechanism of this phenomenon, a simple parameter linked to solvent polarity, dielectric permittivity, provides a rather good account for the observed behavior.
The only variable parameter in the present study was solvent polarity. It is clear that such an approach takes into account only intramolecular phenomena, leaving aside intermolecular interactions (e.g., with an external nucleophile or with the anions of supporting electrolyte), which also must play an important role in the electrochemical reactivity of cation radicals of alkyl arylselenides.
Disclosure of interest
The authors declare that they have no conflicts of interest concerning this article.