

1 Introduction
Chemistry and the chemical industry have been shaping our lives since the beginning of the 20th century. Many positive effects of chemistry and the chemical industry are made explicit in most nowadays technologies that are pillars of the development of modern societies. However, many drawbacks still remain to achieve the expected efficiency. Waste generation and pollution caused by the low selectivity of reactions and the formation of by-products are among them. To overcome these drawbacks various solutions are proposed in the recent literature. These include the use of alternative solvents, new catalysts and alternative products with the same efficiency or even greater, but with a reduced degree of toxicity [1].
Oxidation reactions fit in a category for which the enhancement of the selectivity is still a target. Liquid phase oxidation of organic substrates, improved by solid catalysts, is considered a non-polluting and energetically favorable oxidation process on using hydrogen peroxide as an oxidizing agent. Hydrogen peroxide is one of the most active sources of oxygen, and at the same time the most acceptable oxidation agent from the point of view of environmental protection [2,3].
On the other side, apatites and apatitic materials are widely represented in nature [4]. The apatite family is formed by a large number of isomorphous compounds with the general chemical formula Me10(XO4)6Y2, where Me is a divalent cation, X, phosphorus and Y, a monovalent anion [5]. Representative members of this group are calcium phosphate fluorapatite, Ca10(PO4)6F2 and calcium hydroxyapatite (HAp) Ca10(PO4)6(OH)2. Apatites crystallize in the hexagonal system (space group P63/m), with a structure described as a compact package of Me ions and tetrahedral XO4 groups delimiting two types of unconnected channels [6]. The first one with a diameter of 2.5 Å surrounded by four Me is designated as MeI. The second type of channel has six Me, is named MeII, placed in two equilateral triangles at ¼ and ¾ and centered in a C6 axis. The diameter of this latter type of channel is 3.5 Å and is oriented along the c axis, hosting Y anions which balance the positive charge of the matrix and have great mobility [7].
One of the main characteristics of the apatite is the flexibility of its structure; all the elements can be exchanged if the charge balance is maintained. For example, it is possible to prepare a complete series of apatites by the substitution pair La3++ ↔ Ca2++ [8]. Furthermore, these compounds present a high chemical and thermal stability and a weak and retrograde solubility. These properties make apatites useful in applications like biomaterials [9], chromatography [10], sensors [11], optics, detoxification of water [12], radioactive and industrial waste immobilization [13,14] or catalysis [15]. Doping apatites with various cations enlarges the application field of these materials into luminescence [16], fuel cells [17], etc.
Hydrothermal synthesis of apatites is one of the simplest and most economical methods of preparation. Mixtures of calcium phosphates, in aqueous medium at room temperature, set and harden resulting in apatitic cements [18]. When using hydrothermal reactors, evolution from powdery to coherent materials occurs; this is due to the combined action of both physical and chemical phenomena: temperature, pressure and chemical reactions between constituents. Usually, the initial powder is pressed prior to its introduction into the autoclave, allowing the particles to approach as well as the slow diffusion of water through the pellet. In the hydrothermal process all reactions occur at temperatures above the boiling point of the liquid [19].
Benzyl alcohol is the most commonly used model compound in alcohol oxidation. Moreover, oxidation of benzyl alcohol to benzaldehyde is an important organic transformation since benzaldehyde is a very valuable chemical with applications in perfumery, agro-chemical industries, etc [20,21]. Conventional methods for performing such transformations imply the use of toxic solvents and, generally, involve the use of stoichiometric or more than stoichiometric quantities of inorganic oxidants [20]. On the other hand, oxidation of xylenes results in useful products such as terephthalic acid, mostly used as a precursor to the polyester PET.
Tungsten was reported as an active oxidizing species in a number of oxidation reactions implying hydrogen peroxide [22,23]. Venturello [24,25] reported that, under acidic conditions, aqueous hydrogen peroxide together with catalytic amounts of tungstate and phosphate ions provides a synthetically useful procedure for the highly selective oxidation or epoxidation of organic substrates. However, tungstates suffer from a rather high leaching under reaction conditions [26]. From a practical point of view, this generates some inconveniences in purification of the product and in the recycling of the catalyst. The immobilization of peroxotungstic acid on silica-grafted phosphoramides [27] or hydroxyapatite [28] led to a certain improvement of both activity and stability but leaching was not avoided. Well-defined complexes of tungsten as polyoxometalate anions [29] succeeded in this respect. However their use, for practical reasons, also requires dispersion on a high surface area support [30]. It is clear, however, from these studies the fact that association of W and P is beneficial to the activity of tungsten in liquid phase oxidation reactions.
Therefore, the aim of this work was to associate W and P through the insertion of tungsten into an apatite structure by means of a hydrothermal procedure. The scope of this association was to suppress the leaching of tungsten keeping the catalyst activity. To prove this concept the new catalysts were investigated in the oxidation of aromatic compounds (benzylic alcohol, o-xylene, m-xylene and p-xylene) using H2O2 as the oxidizing agent.
2 Materials and methods
2.1 Catalyst preparation
W-containing apatite (W/HAp) was prepared using mono-calcium phosphate (MCPM, Panreac), ®-tricalcium phosphate (®−TCP obtained from the Apatite type TCP, PROLABO Rectapur, after calcination for 3 h at 900 °C), tetracalcium phosphate (TTCP, obtained from an equimolar mixture of MCMP and CaCO3 Panreac PA, calcined for 24 h at 1350 °C, in a N2 flow) and tungstic acid (H2WO4).
The hydrothermal synthesis followed the formulation of Mejdoubi and Lacout [31]. This type of formulation using three components (TTCP, ®-TCP and MCPM) allows varying the constituent proportions, which affects the physical and chemical properties, but one always obtains hydroxyapatite according to reaction (1).
| (1) |
H2WO4 was added to the three calcium phosphates in adequate percentages to replace ∼2% of the calcium atoms in the apatite network (Table 1), and then the four components were milled together in a mortar until producing particle sizes smaller than 50 μm. A 10 mm diameter infrared die was used to pelletize 1.25 g of the mixture at 5 Tm. The pellets were introduced into a hydrothermal reactor with 100 mL of de-ionized water, and the reactor was heated in a furnace for 48 h at 200 °C (5 °C/min). Once the heating schedule was finished the reactor was allowed to cool down in air to room temperature and the obtained solid was taken away from the reactor and oven-dried at 100 °C for 30 min.
Composition of the reactant mixture.
| Compound | Weight, % |
| TTCP | 61.98 |
| ®-TCP | 23.30 |
| MCPM | 9.72 |
| H2WO4 | 5.00 |
Hydroxyapatite Ca10(PO4)6(OH)2 and Ca3(PO4)2 were purchased from Sigma–Aldrich. CaWO4 was prepared via precipitation of Na2WO4 with CaCl2 and the precipitate was further washed until it was free of chlorine. Then it was dried under vacuum at 110 °C.
2.2 Catalyst characterization
Textural characteristics (surface area and pore diameter) were determined from the adsorption–desorption isotherms of nitrogen at −196 °C using a Micromeritics ASAP 2010 Surface Area and Porosity Analyzer. Samples were degassed for 2 h at 150 °C in a vacuum.
Chemical compositions of the obtained catalysts were measured by X-ray fluorescence (XRF), employing a PANalytical AXIOS sequential spectrophotometer with a rhodium tube as a source of radiation. XRF measurements were performed onto pressed pellets (sample including 6 wt.% of wax).
X-ray diffraction (XRD) patterns were obtained in an X'Pert Pro PANalytical. Diffraction patterns were recorded using Cu Kα radiation (λ = 1.5404 Å) over a 5–90° 2θ range and a PIXcel solid state X-ray detector operating at 0.026° step size and 147 s step time. The incident beam intensity was optimized using a W/Si incident beam parabolic mirror. The software X'Pert Highscore permits obtaining a semiquantitative analysis of the phases present in the sample.
Scanning electron microscopy (SEM) observations were carried out in a JEOL JSM-5400 instrument equipped with an energy dispersive X-ray spectrometer (Oxford Link Tetra 1128-231).
Raman spectra were collected with a Horiba Jobin Yvon – Labram HR UV–Visible–NIR (200–1600 nm) Raman Microscope Spectrometer, using a laser with a wavelength of 632 nm. The spectra were collected from 10 scans at a resolution of 2 cm−1.
2.3 Catalytic tests
The catalytic tests were performed in a round bottom stirred flask with a vertical refrigerant. A thermocouple with a precision of 0.2 °C controls the reactor temperature. A heating bath using oil as thermostatic fluid ensures the reactor isothermicity. Typical catalytic tests were carried out in the presence of 50 mg catalyst with a molar substrate-to-oxidant ratio of 1/4 (1 mmol of substrate) for 4 h at 55 °C. A magnetic stirrer (4 mm) at 900 rpm homogenizes the reactant mixture. The benzyl alcohol (99.8% purity) and xylenes (99% purity) were purchased from Sigma–Aldrich.
Iodometric titrations determine the transformation degree of hydrogen peroxide. The selectivity was calculated as the ratio between the moles of the product “i”, Ni, and the total number of moles of the products, N:
| (2) |
Dehydration of the H2O2 solution (30 wt %, Consors SRL, Romania) was carried out in the presence of anhydrous MgSO4, previously calcined at 300 °C under vacuum for 24 h. Ten grams of anhydrous MgSO4 were added to a mixture of 10 mL H2O2 and 50 mL dioxane. The mixture was stirred for 2 h. After centrifugation and filtration, another 10 g of anhydrous MgSO4 were added and the solution was stirred for 1 h more. A centrifuge allows separating the MgSO4 from the solution that was finally filtered. To homogenize the substrate-hydrogen peroxide mixtures dioxane was added [32]. It is a low toxicity versatile aprotic solvent that exhibits weak interactions with the solid [33].
The H2O2 content before and after the reaction is determined by iodometry. 0.5 mL of solution was poured in an Erlenmeyer flask together with 10 mL HCl and 1 g KI. The mixture was diluted with 100–125 mL water and kept in a dark place for 15 min after that it was titrated in the presence of 14 mL Na2S2O3 (0.1N) until it turned yellow. At that point, 1 mL of starch solution was added and the solution was further titrated until the color turned from dark blue to colorless.
| (3) |
| (4) |
The initial content of hydrogen peroxide determined by iodometry was 38 wt % H2O2.
The reaction products were analyzed using an HPCL, Agilent Technologies 1260 Infinity equipped with a DAD detector, and a column Eurosphere C18 working with a flow rate of 1 mL/min, AcN: H2O = 40: 60, λ = 274.5 nm, and Vinj = 10 μL.
The leaching of tungsten was checked by analyzing samples collected at the end of the reaction using an ICP-OES-700/715 Agilent Technology equipment.
3 Results and discussion
3.1 Characterization
Table 2 presents the chemical analysis of the catalyst, expressed as weight and molar percentages.
Elemental and structural composition of the synthesized W/hydroxyapatite.
| Composition | Elemental compositiona | Structural compositionb | ||||
| Ca | P | W | Ca10(PO4)6(OH)2 | Ca3(PO4)2 | CaWO4 | |
| Weight, % | 63.20 | 30.70 | 6.13 | 83 | 11 | 6 |
| Molar, % | 60.70 | 38.03 | 1.27 |
a Determined from XRF.
b Determined from XRD.
CaWO4 had a very low surface area (2.4 m2/g), while the surface area of the W/HAp simple was much higher (18.6 m2/g). Fig. 1 shows the diffractogram of the W/HAp sample and that of a commercial pure calcium apatite. The W/Hap pattern presents diffraction lines corresponding to Ca10(PO4)6(OH)2 (01-073-0294), CaWO4 (01-077-2233) and Ca3(PO4)2 (00-009-0169), the more intense reflections corresponding to the apatitic phase, Ca10(PO4)6(OH)2. Calcium tungstate, CaWO4, and tricalcium phosphate, Ca3(PO4)2, are minor components of the W/Hap sample. A semiquantitative analysis of the sample performed with the software X'Pert Highscore is presented in Table 2. An estimate of the tungsten content of the W/HAp sample from the amount of observed crystalline phases results in a Ca:P:W atomic ratio of 62.4:37:5:0.1, which clearly evidences that tungsten is partially replacing Ca in the apatitic phase. This is also corroborated by the systematic shift of the diffraction lines of the W/Hap sample to smaller 2θ angles with respect to the pure calcium hydroxyapatite phase, for instance the diffraction line corresponding to the [002] plane appears at 25.88° for Ca10(PO4)6(OH)2 and at 25,68° for the W/HAp sample that is in accordance with the ionic radius of W6+ (0.6 Å) smaller than that of Ca2+ (1.0 Å).
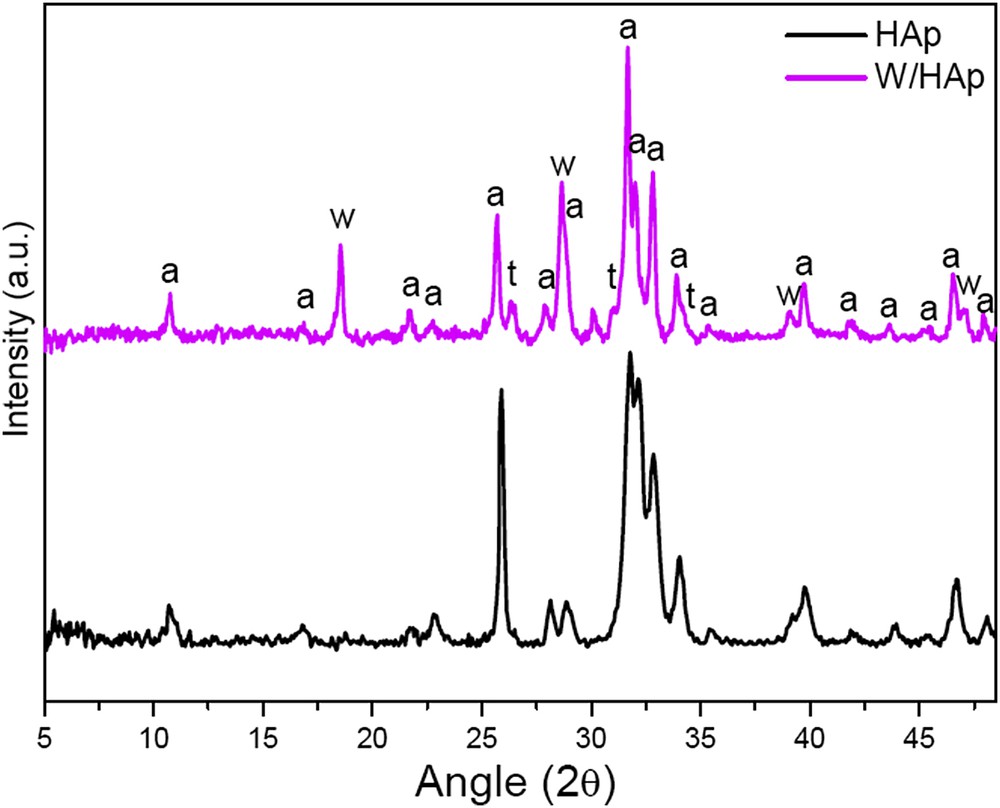
Diffractograms of the commercial apatite (HAp) and the prepared sample (W/HAp) a: Ca10(PO4)6(OH)2; w: CaWO4; t: Ca3(PO4)2.
Fig. 2 shows a representative SEM micrograph of the W/HAp sample. Three types of crystals can be distinguished. First, the needle-like crystals that are characteristic of the apatite phase. The EDX analysis of these crystals shows the presence of P and Ca, although the presence of small amounts of W, Fig. 3, cannot be disregarded in accordance with the FRX and XRD analysis.

SEM image of the prepared sample.
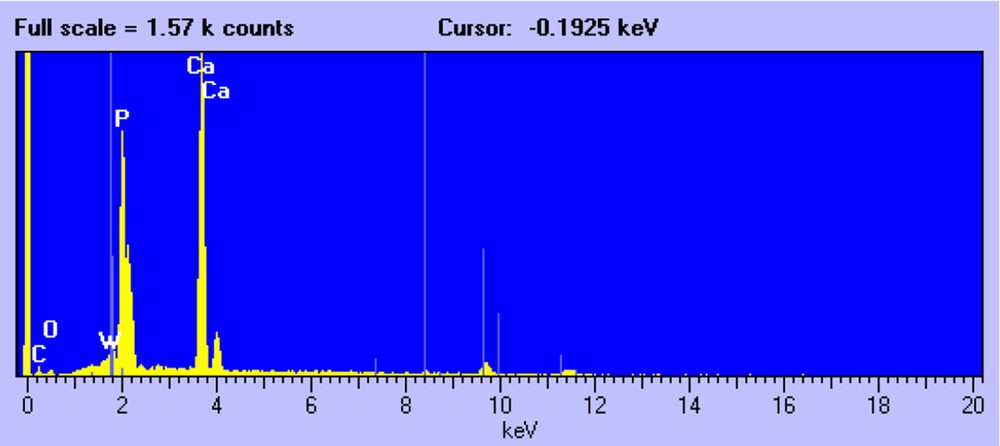
EDX analysis of apatite crystals.
The second type of crystal presents an octahedral morphology (Fig. 4). In accordance with EDX (Fig. 5) and XRD analysis they correspond to W-rich crystals, namely, CaWO4.

SEM image of W-rich crystals.
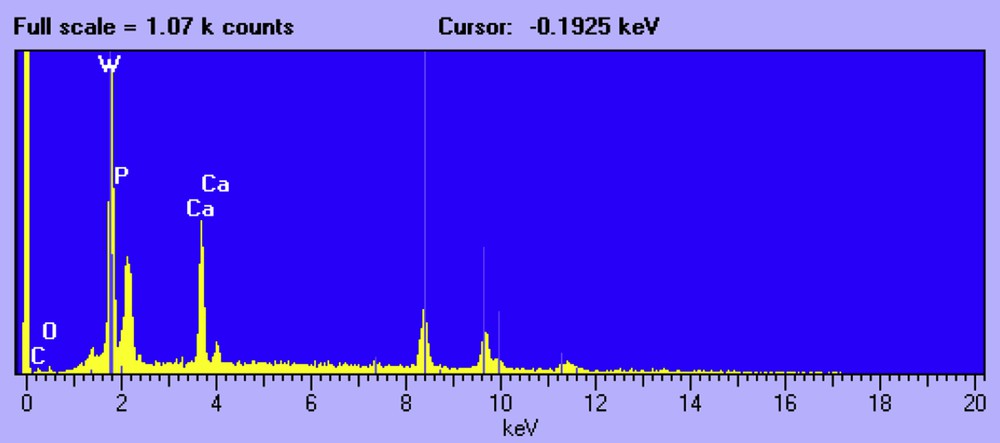
EDX analysis of W-rich crystals.
The third type of crystal, without a well-defined morphology (Fig. 6), was attributed on the basis of EDX (Fig. 7) and XRD analysis to un-reacted tricalcium phosphate Ca3(PO4)2.

SEM image of tricalcium phosphate crystals.
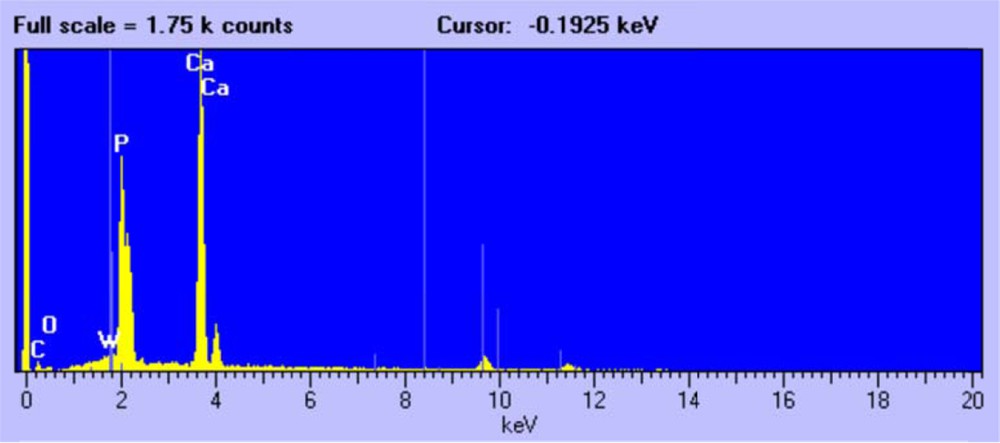
EDX analysis of tricalcium phosphate crystals.
Raman spectra provided additional structural information and evidence on the formation of the different phases. Raman spectra of investigated W/HAp display lines that are characteristic of apatites [34] (Fig. 8A). Several absorptions are assigned to P–O bonds in the PO4 group. The absorptions at 1079 and 1051 cm−1 are assigned to the triply degenerate asymmetric stretching mode (υ3) of the P–O bond, the strong line at 966 cm−1 to totally symmetric stretching mode (υ1) of the P−O bond, the line at 594 cm−1 to the triply degenerate bending mode (υ4) of the O−P−O bond and that at 436 cm−1 assigned to the doubly degenerate bending mode (υ2) of the O−P−O bond. These spectra also indicate the presence of tungsten. In the isopolytungstate species, the W exhibits a distorted octahedral geometry with generally more WO2(t) groups, where t stands for a terminal oxygen atom. According to Payen and co-workers the symmetric and antisymmetric stretching modes of these groups are observed at 960–980 and 900–920 cm−1, respectively [35,36]. However, the line assigned to symmetric stretching mode overlaps the signal of the strong P–O bond and it is very difficult to be decomposed. In addition to this line, the line at 337 cm−1 is typically assigned to the vibrations of W-O bonds in tungstates [36] including CaWO4 [37]. These results are in good agreement with the XRD results.
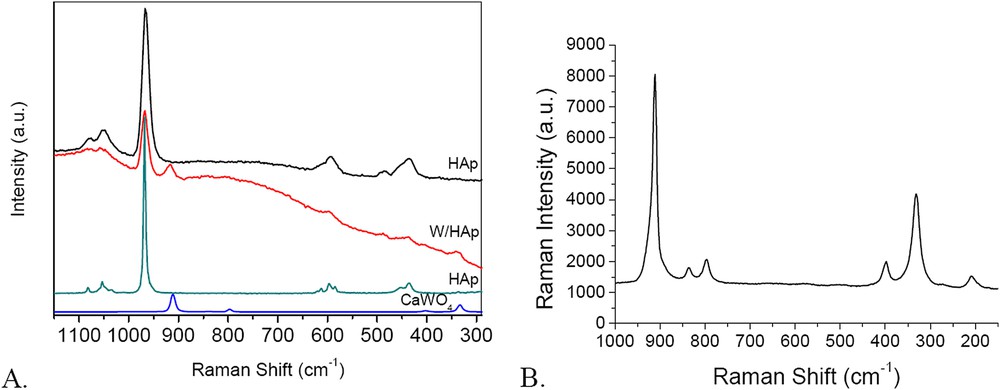
Raman spectra of the W/HAp sample in comparison with commercial HAp and CaWO4 (A); Raman spectra of the CaWO4 (B).
Fig. 8B presents the Raman spectrum of CaWO4 used as the reference in this study and prepared by precipitation of Na2WO4 with CaCl2. Six Raman phonon modes located 915, 839, 799, 398, 332, and 210 cm−1, respectively, were observed in this spectrum. The line at 915 cm−1 has the assignment discussed above. Those at 839 and 799 cm−1 correspond to Bg and Eg modes that degenerated from A1(υ1) and F2(υ3) vibration modes, while those at about 398 and 332 cm−1 correspond to Bg and (Ag + Bg) vibrations which are attributed to the degeneration of E(υ2) vibrations of tungstate groups. The line at about 210 cm−1 belongs to the external mode of CaWO4 [37].
3.2 Catalytic tests
For the sake of comparison, blank experiments were made using a commercial hydroxyapatite. No conversion was observed using benzyl alcohol or xylenes. However, apatite decomposed over 20% of hydrogen peroxide in a non-selective way. Similar results were obtained for Ca3(PO4)2.
Tests with the pure CaWO4 phase, also found in the new W/HAp catalyst, indicated a catalytic activity that is in agreement with the previous report of Noyori et al. on the role of tungsten in liquid phase oxidation [22]. Thus for the oxidation of benzyl alcohol 18.8% conversion was achieved after 4 h reaction time. Time evolution of the conversion and reaction products is presented in Fig. 9. In the first two hours the oxidation occurred only with the selective formation of benzyl aldehyde. After three hours the conversion reached a plateau and the resulting benzaldehyde underwent further oxidation to benzoic acid. Moreover, the aromatic cycle starts to oxidize giving rise to three more products which are clearly observed after four hours. Thus, the selectivity decreased and up to 5 products were identified, Scheme 1.
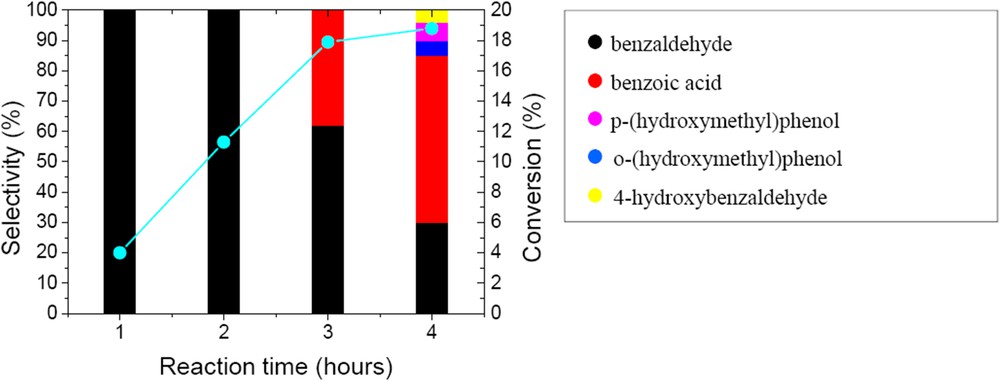
Time evolution of the conversion and selectivity of the prepared CaWO4 in the oxidation of benzyl alcohol (50 mg catalyst, substrate-to-oxidant molar ratio of 1/4 (1 mmol substrate), 4 h, 55 °C and 900 rpm).
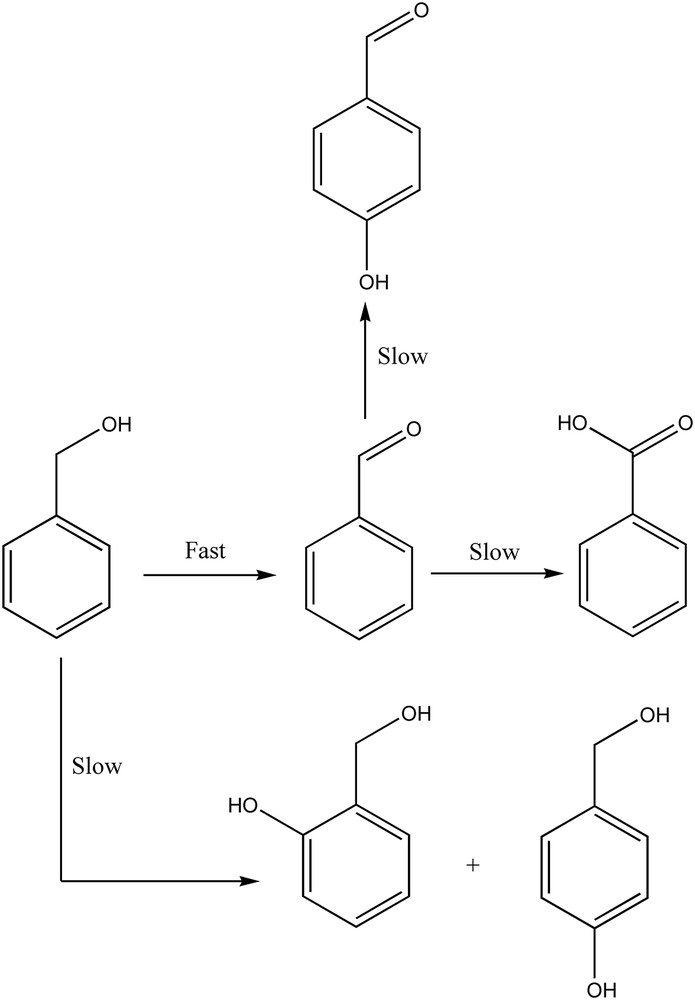
Evolution of the reaction products in the oxidation of benzyl alcohol on the W-apatite catalyst.
The advantage of using dioxane is its (http://en.wikipedia.org/wiki/miscible) miscibility with water that generates an azeotrope with a boiling point of around 90 °C [38]. Also, it has weak interactions with the catalyst affording the access of the reactants to the active sites [33]. In this way its use in mixtures with hydrogen peroxide has two advantages: (i) an advanced solubilization of the substrate, and (ii) “solubilization” of the water produced in the reaction, thus diminishing its competition with the substrate for the catalyst active sites.
Xylenes are also oxidized on CaWO4 catalysts. 22.4% conversion in the oxidation of p-xylene was achieved after 4 h reaction time. The evolution of the conversion and of the product distribution with time is presented in Fig. 10. The evolution of both conversion and reaction products differ from that observed in the case of the oxidation of benzyl alcohol. The p-xylene conversion increased continuously with time, while the selectivity changed from around 90% in p-tholylmethanol after 1 h to around 70% in 4-methylbenzaldehyde after 4 h reaction time. Scheme 2 depicts the evolution of the reaction products.
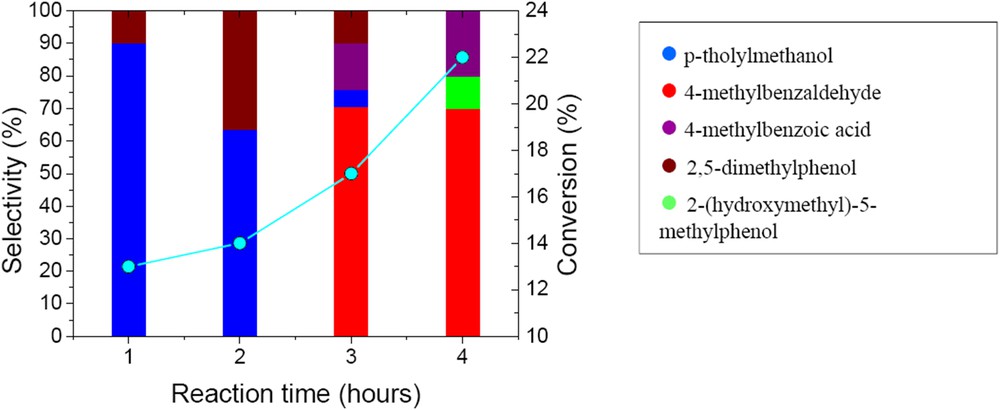
Time evolution of the conversion and selectivity of the prepared CaWO4 in the oxidation of p-xylene (50 mg catalyst, substrate-to-oxidant molar ratio of 1/4 (1 mmol substrate), 4 h, 55 °C and 900 rpm).
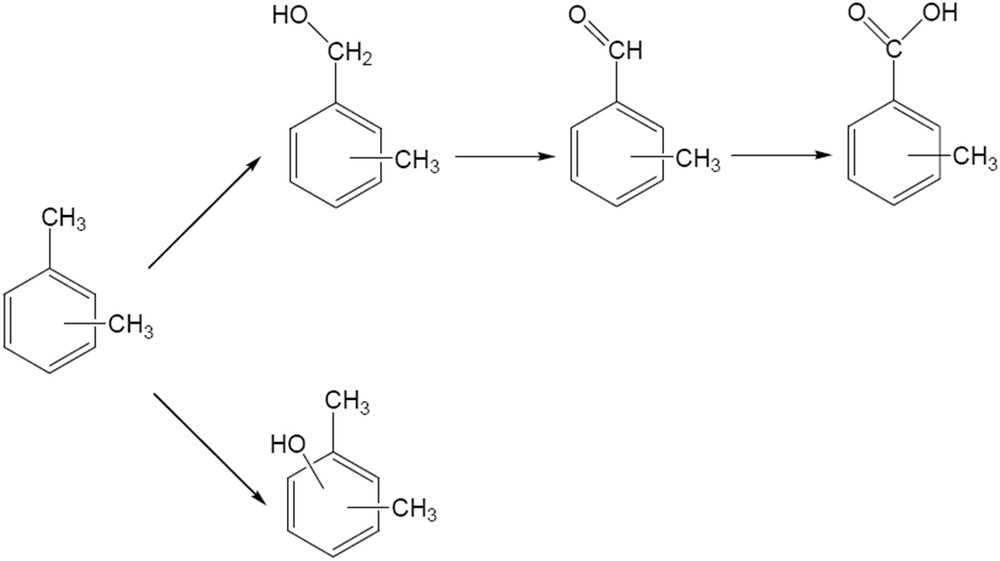
Evolution of the reaction products in the oxidation of xylenes on the W-apatite catalyst.
A significant leaching of tungsten was detected by ICP-OES for both the oxidation of benzyl alcohol and xylenes on CaWO4 catalysts. Tungsten concentration was in the range of 600–900 ppm for the oxidation of benzyl alcohol, while for the oxidation of xylenes it was in the 500–850 ppm range.
The efficiency of the H2O2 decomposition is a very important parameter in liquid phase hydrogen peroxide oxidation since H2O2 is still expensive enough. Obviously, the efficiency of the hydrogen peroxide decomposition is referred to as the ratio of the number of moles of H2O2 consumed in the oxidation of the reactants and intermediate products to the total number of moles of H2O2 transformed during the oxidation process. This ratio expresses the percentage of the H2O2 molecules effectively used for the oxidation of the substrate. Fig. 11a shows the evolution with time of the number of moles of H2O2 consumed per gram of substrate and reaction products as a function of time during the oxidation of both benzyl alcohol and p-xylene. A rather fast H2O2 decomposition is observed in the first hour of the reaction while the substrate conversion was only of around 4% for benzyl alcohol and around 13% for p-xylene. This demonstrates a non-selective decomposition of H2O2 to water and molecular oxygen. Although not identified by analytical measurements it may also correspond to a certain leaching of tungsten. The efficiency of the H2O2 decomposition presented in Fig. 11b confirms the fact that on pure CaWO4, H2O2 is decomposed non-selectively in a large extent.
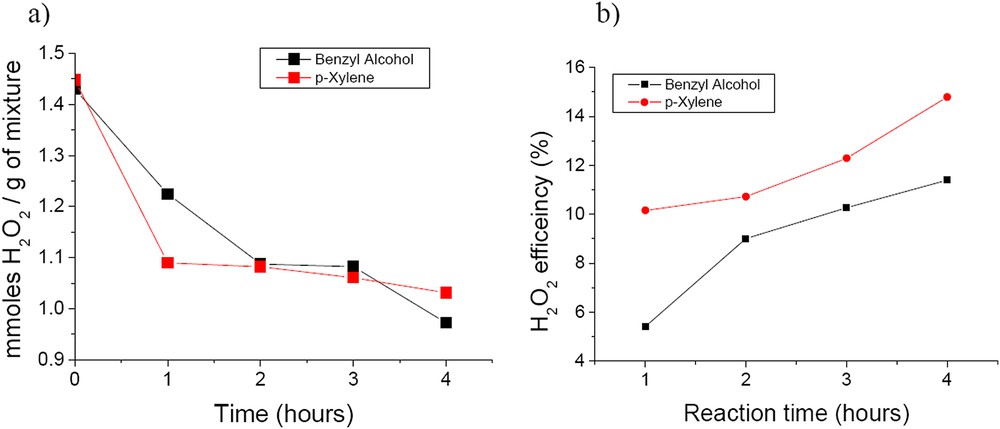
a) Time evolution of H2O2 decomposition and b) efficiency using the prepared CaWO4.
Fig. 12 presents the conversion of benzyl alcohol and xylenes on the W/HAp catalyst. On this catalyst 27.6% conversion in the oxidation of benzyl alcohol was achieved after 4 h reaction time with a selectivity of 100% for benzyl aldehyde. It is noteworthy that the content in tungsten in W/Hap was 6.1 wt.% compared to 63.8 wt.% in CaWO4. Based on these it may be estimated that the W/HAp catalyst was 15.6 times more active than the CaWO4 one.
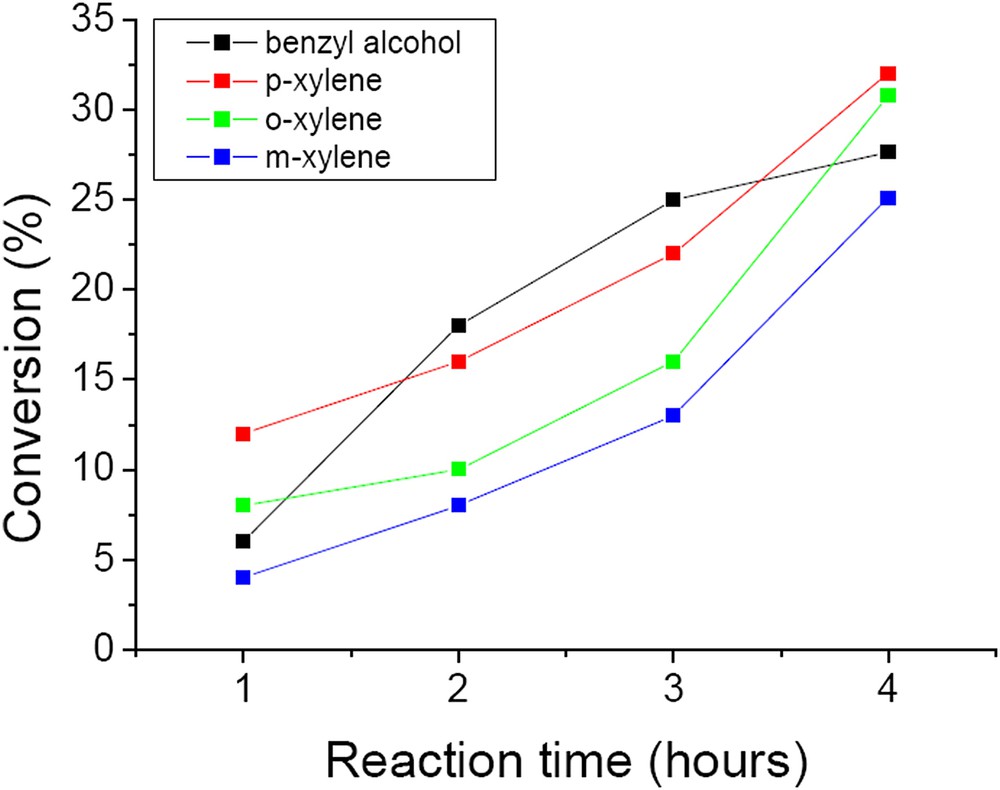
Conversion of benzyl alcohol and xylene substrates on the W/HAp catalyst (50 mg catalyst, substrate-to-oxidant molar ratio of 1/4 (1 mmol substrate), 4 h, 55 °C and 900 rpm).
Oxidation of xylenes on these catalysts depends on the nature of the isomer. Thus, p- and o-xylenes led to higher conversions than m-xylene (p-xylene (32%) > o-xylene (30.8%) > m-xylene (25%)). The selectivity in this reaction also depends on the substrate. After 4 h, the selectivity for 3-methylbenzaldehyde was 93.8% for m-xylene, with m-tolylmethanol and 2,6-dimethylphenol as by-products. The oxidation of p-xylene oxidation resulted in 45.5% p-tholylmethanol, 40.1% p-tolualdehyde and 14.4% p-toluic acid. The oxidation of the second methyl group (4-carboxybenzaldehyde or therephthalic acid) was not observed in this reaction. It is very probable that the remaining methyl groups were deactivated by the electron-withdrawing effect of the carboxyl group. Oxidation of o-xylene led to 100% o-tolualdehyde. The pathways of xylene oxidation are presented in Scheme 2.
Leaching measurements confirmed the protection effect of the phosphate groups. Irrespective of the fact that the substrate has been oxidized or the reaction conditions the leaching was smaller than 12 ppm, accounting for the reduced fraction of tungsten present in the form of CaWO4 since metal leaching is extremely reduced in apatitic structures [13].
Hydrogen peroxide efficiency was also substantially much higher for the W/HAp than that of the CaWO4 sample. In all cases, the hydrogen peroxide efficiency for the W/HAp catalyst was 22–24% and followed the conversion order (Fig. 13). These values are higher than those for pure CaWO4 but still below 25%. The observed efficiency is rather high for a tungsten based catalyst although smaller than those previously reported for titania [3,27,28].
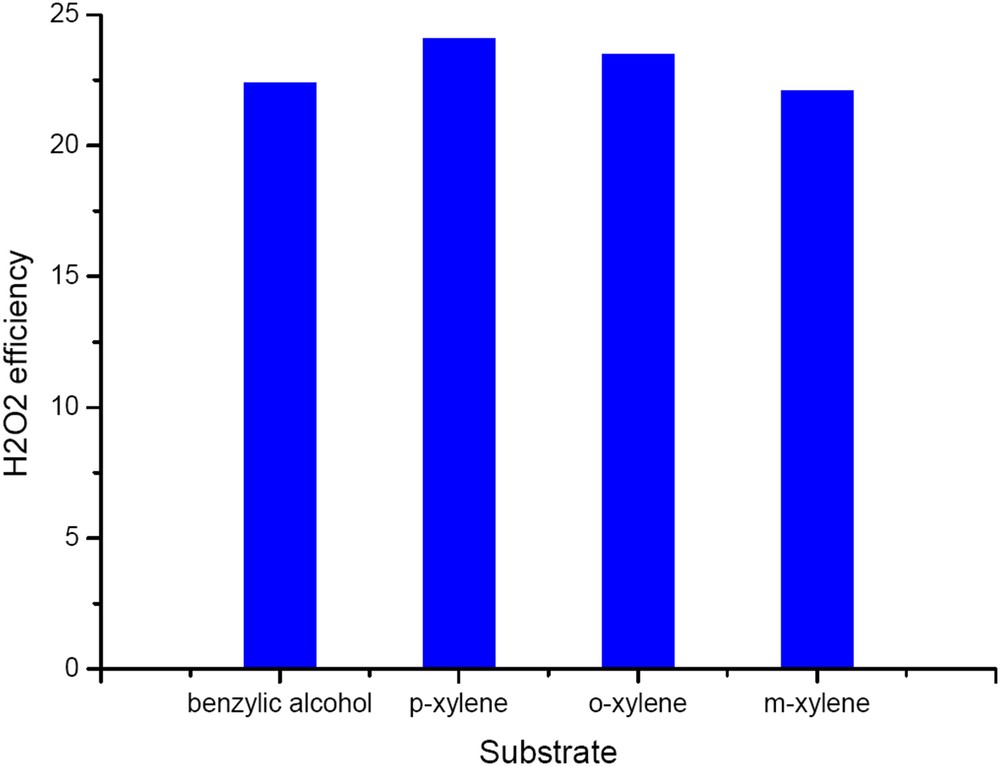
Hydrogen peroxide efficiency on the W-apatite catalyst.
According to the model we proposed in Scheme 3, the increased values of hydrogen peroxide efficiency and also of the conversions could be associated to the existence of tungsten cations in lattice sites of the hydroxyapatite structure and/or to the interaction between Ca10(PO4)6(OH)2 and CaWO4. Contrarily, on Ca3(PO4)2, as mentioned above, hydrogen peroxide nonselectively decomposed to water and oxygen.
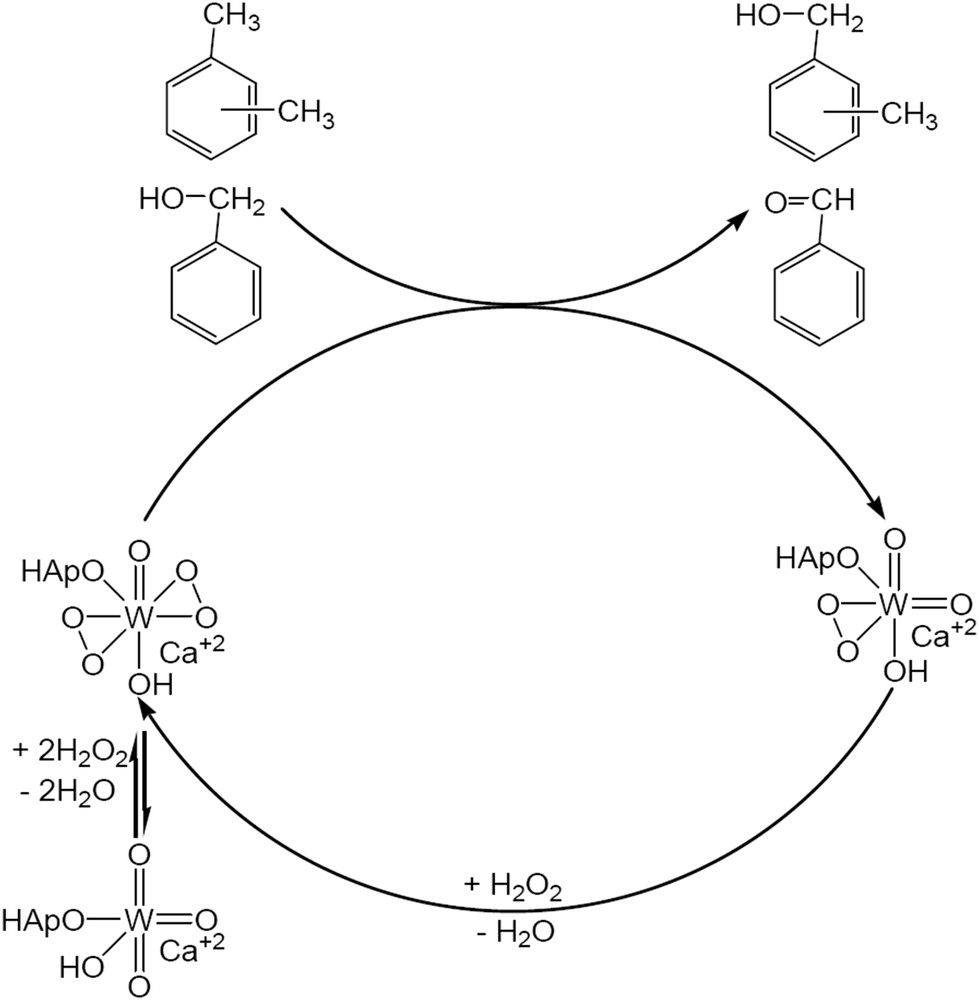
Oxidation of xylene and benzyl alcohol on Ca10(PO4)6(OH)2 – CaWO4 catalysts.
Previous reports state the role of phosphorus in accelerating the epoxidation of olefins [22]. It was considered that a phosphonate or a phosphate anion coordinating the W center could increase the electrophilic nature of the peroxo moiety of the tungstate anion. The model presented in Scheme 3 is in line with these findings. It can also explain the increase of the activity of the W/hydroxyapatite compared with that of the pure CaWO4. Besides this, a secondary effect of the texture cannot be neglected. CaWO4 exhibits a very low surface area (2.4 m2/g) while for W/HAp the surface area was much higher (18.6 m2/g). This makes the 6.1 wt.% W more accessible in W/HAp than the 63.8 wt.% W in CaWO4.
After the reaction the catalysts were separated by centrifugation and recycled in a new catalytic run for six successive cycles. For the W/HAp catalyst, after six cycles the loss of activity was less than 15% compared to the fresh catalyst with no effect on the selectivity. Combining the weight percentages of the different elements and the phase percentages determined by XRD, Table 2, the apatite phase may be formulated as Ca9,58W0,14(PO4)6(OH)2 accounting this phase for around 66% of the total amount of tungsten present in the W/HAp catalyst. Thus, the observed leaching should be ascribed to the dissolution of the CaWO4 phase that, as demonstrated above, shows low activity and a poor selectivity. This demonstrates the robustness of the new catalyst. Further work is in progress to increase the H2O2 selectivity.
A comparison with literature reports on liquid phase oxidation on apatites or tungsten related catalysts reveals the advantages of the W/HAp catalyst described in this study. Comparable conversions but a more severe leaching were observed for 25 wt.% 12-tungstophosphoric acid deposited on different mesoporous molecular sieves (19.1 wt.% W) [39]. Moreover, homogeneous catalysts based on tungsten also required higher tungsten loadings and have the typical disadvantage of catalyst recovery [40]. It is also worth noting that this catalyst has a behavior comparable to HAp containing ∼5 wt.% Ru and Ru co-doped with Co, Ni, Pb or Fe using molecular oxygen as the oxidant [41]. The oxidation of benzyl alcohol at 60 °C on these catalysts presented a TOF of about 11 h−1 for the sample containing only Ru, and 14, 23 and 38 h−1 for the sample co-doped with Fe, Ni, and Ni and Pb, respectively. These values are comparable with the one obtained in the present study, i.e. 23 h−1. Other apatite reported systems required tens of hours to reach significant conversion in the oxidation of alcohols [42].
4 Conclusions
In conclusion, the new W/HAp catalyst prepared using the described hydrothermal approach is composed from a mixture of W-hydroxyapatite, as a major component, calcium phosphate and CaWO4, as minor components. The tungsten environment generates a co-factor effect. This favors both an activity increase and a stabilization of the catalyst against leaching. The catalyst was investigated in the liquid phase oxidation of benzyl alcohol and xylenes using hydrogen peroxide as the oxidant. For comparison, calcium phosphate, hydroxyapatite and CaWO4 were tested in the same reaction. Calcium phosphate and hydroxyapatite appeared to be inactive and decomposed non-selectively hydrogen peroxide. A moderate activity but low hydrogen peroxide efficiency was observed for the CaWO4 phase. In contrast to these solids the W/HAp catalyst shows a reasonable activity and a better hydrogen peroxide efficiency in the oxidation of benzyl alcohol and xylenes. This new W/HAp catalyst showed, after six cycles, losses of the activity below 15% compared to the fresh catalyst with no effect on the selectivity.
Acknowledgments
Financial support for this work has been obtained from the Spanish Ministry of Economy and Competitiveness (ENE2012-37431-C03) and the Andalucía Government (Junta de Andalucía TEP8196), both co-financed by FEDER funds from the European Union. Also, Romanian team acknowledges UEFISCDI for the project Idei/275/2011.