1 Introduction
The development of new cyclization reactions has been greatly advanced by the use metal-catalyzed processes called cascade or domino reaction [1]. These processes allow a succession of events that are triggered by well-placed functionalities in the structure of the starting material. In this direction, a useful transformation that has been extensively used is the intramolecular carbopalladation of alkynes [2] ended, for example, by a cross-coupling reaction such as Stille [3], Heck–Mizoroki [4], Suzuki–Miyaura [5], or Sonogashira reaction [5], a pericyclic reaction such as electrocyclization [6], or a CO insertion [7]. CH activation has been also used after cyclocarbopalladations to end cascade reactions, C(sp2)-H activation was the most common one [8], C(sp3)-H activation is lightly described in the literature [9]. In this context, to develop novel cyclocarbopalladation/CH activation cascade reactions, we have designed starting materials of types 1 and 3 (Scheme 1). Depending on the starting material, C(sp2)-H or C(sp3)-H activation should finish the cascade process leading to naphthalene derivatives (type 2) and to original polycycles containing seven-membered rings (type 4).
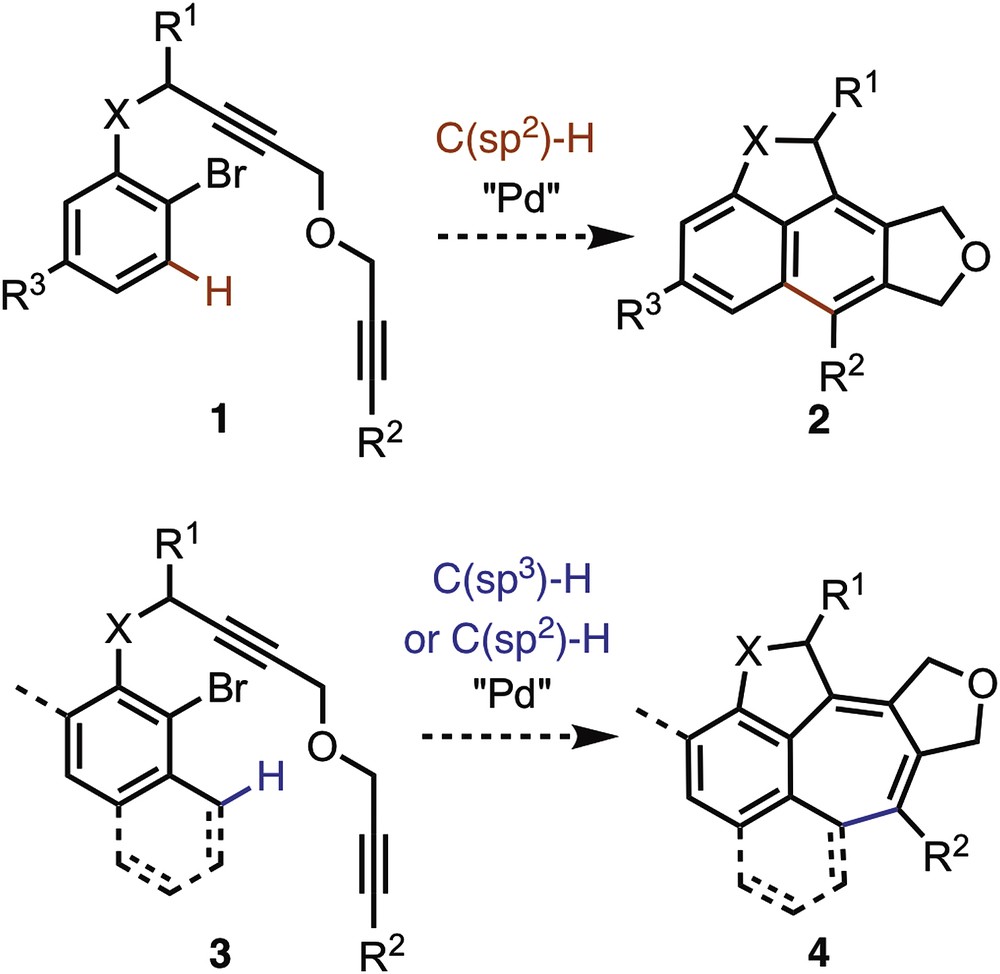
Synthesis of polycycles containing five- and seven-membered rings.
2 Carbopalladations/C(sp2)-H activation cascade reactions
We started our investigations with the diyne 1a to yield naphthalene derivatives. First, we decided to start our investigations with conditions that are classically used in our laboratory for such type of cascades, Pd(PPh3)4/K2CO3/PhH, at 100 °C under microwave irradiations (Table 1, entry 1). We were pleased to observe the formation of compound 5, which represents the desilylated derivative of the desired product, in 33% yield. The protodesilylation is probably because of the proton source coming from the in situ formation of protonated base during the cascade process [10]. It is important to note that even if the desilylated compound 5 is obtained, the silyl group on the starting material is necessary to avoid degradation of the reaction mixture [11]. With an organic base, such as Et3N and iPr2NH, the reaction was complete and a separable mixture of 2a and 5 was obtained (entries 2 and 3). Yields were around 25% and 65% for 2a and 5, respectively. By decreasing the amount of base (1 equiv instead of 5), the yield of 5 was improved (80%, entry 4). Finally, the use of less amount of catalyst (5 mol % instead of 20 mol %), gave similar result in terms of yield and ratio between 2a and 5 (entry 5). When the palladium diacetate/triphenylphosphine system was used, the quantity of compound 5 formed decreased and a large quantity of the starting material was recovered (entry 6). Eventually, the use of Pd(PPh3)4 (5 mol %) and diisopropylamine in benzene under microwave irradiation proved to be the conditions of choice to obtain 5 in good yield (72%, entry 5, Table 1).
Optimization of the cascade reaction.
| Entry | Catalyst (mol %) | Base (equiv) | Time | 1a (%) | 2a (%) | 5 (%) |
| 1 | Pd(PPh3)4 (20) | K2CO3 (5) | 2 h | 34 | – | 33 |
| 2 | Pd(PPh3)4 (20) | Et3N (5) | 30 min | – | 24 | 65 |
| 3 | Pd(PPh3)4 (20) | iPr2NH (5) | 30 min | – | 25 | 64 |
| 4 | Pd(PPh3)4 (20) | iPr2NH (1) | 30 min | – | 15 | 80 |
| 5 | Pd(PPh3)4 (5) | iPr2NH (1) | 30 min | – | 19 | 72 |
| 6 | Pd(OAc)2 (5)/PPh3 (10) | iPr2NH (1) | 30 min | 66 | 8 | 20 |
Concerning the formation of compound 5, we propose the following mechanism (Scheme 2): after oxidative addition of the active palladium species into the CBr bond of 1a, a 5-exo-dig cyclocarbopalladation gives rise to intermediate 7 which undergoes a 5-exo-dig cyclocarbopalladation giving 8. A C(sp2)-H activation takes place to provide 9 followed by a reductive elimination leading to silylated compound 2a. Finally, compound 5 was obtained by protodesilylation of 2a.

Proposed catalytic cycle for the formation of 2a and 5.
A structural confirmation was obtained by an X-ray crystallographic analysis of compounds 2a and 5 (Fig. 1).
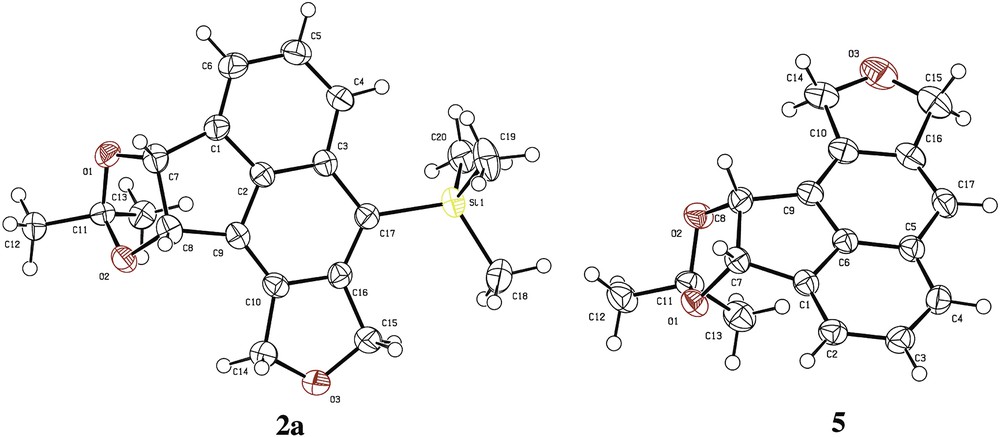
X-ray crystal analysis of the compounds 2a and 5.
The scope and limitations of this reaction were next investigated. As summarized in Scheme 3, substrates 1a–f were treated under the optimized conditions to provide the naphthalene derivatives 2a–f and 5 in 6–72% yields. With starting material 1b, bearing a triethylsilyl group, the optimized conditions also induce a desilylation of the final product leading to a separable mixture of 2b and 5 in, respectively, 12% and 54% yield. The use of propargyl alcohol derivative 1c gave the naphthalene derivative 2c in only poor yield. Almost the same result was observed with the ethyl or the phenyl starting materials 1d–f.

Scope and limitations.
The optimized conditions were applied on phenol derivative 10, which was synthesized in three steps from the 2-bromophenol (Scheme 4). Only degradation of the starting material was observed. With the system Pd(OAc)2/P(OPh)3/Cs2CO3 in 1,4-dioxane, compound 10 underwent the desired cascade reaction ended by a protodesilylation to afford compound 11 in 65% yield.

Synthesis of naphthalene derivative 11.
To have an access to more challenging polycycles containing seven-membered rings, a similar starting material 3a was designed in three steps in 69% yield starting from 1-bromonaphthalen-2-ol. Of note, because of the naphthalene moiety, the last step of the cascade reaction should as well be a C(sp2)-H activation leading to the formation of a seven-membered ring. Degradation occurred with the use of Pd(PPh3)4 in PhH/iPr2NH (Table 2, entry 1). The catalytic system Pd(OAc)2/P(OPh)3/Cs2CO3 in 1,4-dioxane was the best catalytic system to obtain compound 12 (entries 2–5). Yields were quite similar in toluene or 1,4-dioxane and under microwave irradiation or with an oil bath (15–25%). In one case, it was possible to isolate the desilylated compound 13 (entry 4, 10% yield). The proposed mechanism is similar: a twofold 5-exo-dig cyclocarbopalladation ended by a C(sp2)-H activation. Yields are modest but during this process three carbon–carbon bonds are formed in a single step, and the resulting compounds are original pentacycles containing seven-membered rings.
Optimization of the cascade reaction from 3a.
| Entry | Catalyst | Base/additive | Solvent | Heating | Time | 3a (%) | 12 (%) | 13 (%) |
| 1 | Pd(PPh3)4 (5) | iPr2NHa | PhH | MW | 1.5 hb | – | 9 | – |
| 2 | Pd(OAc)2 (15)/P(OPh)3 (30) | Cs2CO3 (5) | 1,4-Dioxane | MW | 30 min | 24 | 15 | – |
| 3 | Pd(OAc)2 (15)/P(OPh)3 (30) | Cs2CO3 (5) | 1,4-Dioxane | Δ | 30 min | 12 | 25 | – |
| 4 | Pd(OAc)2 (15)/P(OPh)3 (30) | Cs2CO3 (5) | Toluene | Δ | 30 min | 9 | 19 | 10 |
| 5 | Pd(OAc)2 (15)/P(OPh)3 (30) | Cs2CO3 (5) | Toluene | MW | 30 min | 5 | 22 | – |
a As cosolvent.
b Then 12 h at 100 °C, Δ
3 Carbopalladations/C(sp3)-H activation cascade reactions
Following these investigations, we were interested in an access to other polycycles containing a seven-membered rings. To this purpose, we have designed other compounds of type 3 (Scheme 1) bearing a methyl group in the ortho position to the bromine. One key point of this method is the final C(sp3)-H activation. Because of structural similitudes between 3b and the nonmethylated analogue 1a, the previously reported “optimal” conditions (Table 1) have been used first: Pd(PPh3)4 (5 mol %) and diisopropylamine in benzene under microwave irradiation (Table 3). No conversion was observed after 3 h (entry 1). With the same conditions in an oil bath instead of microwave irradiation and after 18 h, we observed the formation of compound 14 in 25% yield (entry 2). It seems that during the process the desired compound 4b undergoes a protodesilylation leading to compound 14. With iPr2NH as solvent, a separable mixture of 4b and 14 was observed in, respectively, 12% and 19% yield (entry 3).
Optimization of the cascade reaction from 3b.
| Entry | Solvent | Heating | Time | 3b (%) | 4b (%) | 14 (%) |
| 1 | PhH | MW | 3 h | 100 | – | – |
| 2 | PhH | Δ | 18 h | 20 | Traces | 25 |
| 3 | iPr2NH | Δ | 22 h | – | 12 | 19 |
Concerning the formation of these two compounds, we propose the following mechanism (Scheme 5): after oxidative addition of the active palladium species into the CBr bond of 3b, a 5-exo-dig cyclocarbopalladation gives rise to intermediate 16, which undergoes a 5-exo-dig cyclocarbopalladation giving 17. A C(sp3)-H activation takes place to provide 18 followed by a reductive elimination leading to silylated compound 4b. Compound 14 is obtained by protodesilylation.

Proposed catalytic cycle for the formation of 4b and 14.
To the best of our knowledge, this represents the first example of a cascade reaction using carbopalladations/C(sp3)-H activation allowing the formation of seven-membered rings in a polycyclic scaffold. However, because of modest yields in the seven-membered formation, we decided to design other substrates.
Compound 3c was synthesized in five steps and 14% yield starting from the commercially available 2,5-dimethylphenol. With compound 3c in hand, we tried diverse reaction conditions (Table 4). With Pd(PPh3)4 in PhH/iPr2NH, mainly degradation occurred, no desired compound 4c was observed but only 7% of compound 19 (entry 1). After the twofold 5-exo-dig cyclocarbopalladation and the C(sp3)-H activation, the desired compound 4c undergoes a 1,5-prototropy giving compound 19. This compound shows a similar skeleton to the natural product malibatol A, which exhibits cytotoxic and human immunodeficiency virus (HIV)-inhibitory activity [12].
Optimization of the cascade reaction from 3c.
| Entry | Catalyst | Base/additive | Solvent | Time | 3c (%) | 19 (%) |
| 1 | Pd(PPh3)4 (5) | iPr2NHa | PhH | 30 minb | 19 | 7 |
| 2 | Pd(OAc)2 (10)/P(OPh)3 (20) | Cs2CO3 (5) | 1,4-Dioxane | 1.5 h | – | 40 |
| 3 | Pd(OAc)2 (10)/P(OPh)3 (20) | Cs2CO3 (1, 3) | 1,4-Dioxane | 1.5 h | 14 | 30 |
| 4 | Pd(OAc)2 (10)/P(OPh)3 (20) | Cs2CO3 (5) | Toluene | 1.5 h | – | 36 |
| 5 | Pd(OAc)2 (10)/P(OPh)3 (20) | Cs2CO3 (5)/nBu4NCl(1) | Toluene | 1.5 h | – | 11 |
| 6 | Pd(OAc)2 (10)/PPh3 (20) | Cs2CO3 (5) | Toluene | 30 min | 11 | 28 |
a As cosolvent.
b Then 1.5 h at 130 °C.
Compound 19 was obtained in better yield with Pd(OAc)2/P(OPh)3/Cs2CO3 in 1,4-dioxane (40% yield, entry 2). By decreasing the amount of base (entry 3) in toluene instead of 1,4-dioxane (entry 4), with nBu4NCl as additive (entry 5) or with PPh3 instead of P(OPh)3 (entry 6), the reaction gave compound 19 in lower yield.
A new starting material 3d was synthesized from 2-bromo-3-methylbenzoic acid in six steps and in 45% yield. This compound could undergo a new cascade reaction: a 6-exo-dig carbopalladation, followed by a 5-exo-dig cyclocarbopalladation and ended by a C(sp3)-H activation. After a long optimization of the reaction conditions, with the screening of the different ligands (XPhos, dppp, PPh3, and P(OPh)3), palladium (Pd(OAc)2, PdI2, Herrmann–Beller catalyst, and Pd(PPh3)4), solvent (1,4-dioxane, toluene, DMF, DCM, and CH3CN), base (Cs2CO3, K2CO3, CsOPiv, and Hünig base), and temperature (85, 100, 130 °C, oil bath, or under microwave irradiation), the best found condition was Pd(PPh3)4, Cs2CO3 in 1,4-dioxane. However, only 10% of the desired compound 21 was formed (Scheme 6). During this optimization, either degradation or starting material is often observed. In some case the intermediate 20 was formed, this later arises from the 6-exo-dig cyclocarbopalladation followed by a demetalation. It seems that this substrate is not suitable to undergo the cascade reaction completely.

Synthesis of seven-membered ring starting from 3d.
4 Conclusions
In summary, different types of polycyclic molecules were synthesized by the palladium-catalyzed cascade reaction. The cascade cyclocarbopalladations followed by a C(sp2)-H activation allowed the synthesis of eight fused naphthalenes in 6–72% yield. With cyclocarbopalladations ended by a C(sp2 or sp3)-H, we were able to synthesize six polycycles containing one seven-membered ring in 10–40% yield, leading to original scaffolds by using an elegant pathways to get them. During these cascades, the palladium is moving several times along the carbon structure forming three new CC bonds and three new cycles. Work is currently in progress to increase yields and to design new starting materials that should lead to original scaffolds containing seven-membered ring.
5 Experimental section
5.1 General information
All reagents, chemicals, and dry solvents were purchased from commercial sources and used without purification. Reactions were monitored by thin-layer silica gel chromatography (TLC) using Merck silica gel 60 F254 on aluminum sheets. TLC plates were visualized under UV light and revealed with acidic p-anisaldehyde or KMnO4 stain. Crude products were purified by flash column chromatography on Merck silica gel Si 60 (40–63 μm). All NMR spectra were recorded in CDCl3, C6D6, or CD2Cl2 on a Bruker Avance III 400 MHz BBFO+ probe spectrometer for 1H NMR and 100 MHz for 13C NMR, and a Bruker Avance 300 MHz dual probe spectrometer for 1H NMR. Proton chemical shifts are reported in parts per million (ppm) (δ), relatively to residual solvent. Multiplicities are reported as follows: singlet (s), doublet (d), doublet of doublet (dd), triplet (t), quartet (q), broad singlet (br s), multiplet (m). Coupling constant (J) values are given in hertz (Hz). Carbon chemical shifts are reported in ppm with the respective solvent resonance as the internal standard. 1H NMR and 13C NMR signals were assigned mostly on the basis of distortionless enhanced polarization transfer (DEPT) and 2D-NMR (correlation spectroscopy [COSY], heteronuclear multiple-bond correlation spectroscopy [HMBC], and heteronuclear multiple-quantum correlation [HMQC]) experiments. High resolution mass spectrometry (HRMS) was performed using an Agilent 1200 rapid resolution liquide chromatographie (RRLC) high performance liquid chromatography (HPLC) chain and an Agilent 6520 accurate mass quadrupole time-of-flight (QToF). Microwave irradiation was carried out with a microwave reactor from Biotage using pressurized vials. Infrared spectra (IR) were recorded on a FT IR Thermo Nicollet ATR 380, diamond spectrometer. Microwave irradiations have been performed using a Biotage Smith Creator apparatus.
5.2 Experimental procedures
5.2.1 General procedure A: protection of diols in acetonide
A solution of diol (1 equiv), 2,2-dimethoxypropane (5 equiv) and p-TsOH (0.1 equiv) in acetone (0.19 M) was stirred at room temperature for 1 h. The mixture was quenched by addition of brine and extracted with Et2O. Combined organic layers were washed with water, brine, dried over Na2SO4, filtered, and concentrated under reduced pressure. Crude product was purified by flash column chromatography over silica gel to afford the corresponding acetonide.
5.2.2 General procedure B: deprotection of alkyne
A solution of silylated alkyne (1 equiv) and K2CO3 (1 equiv) in MeOH (0.2 M) was stirred at room temperature for 1 h. The mixture was quenched by addition of a saturated solution of NH4Cl and extracted with EtOAc. Combined organic layers were washed with water, brine, dried over Na2SO4, filtered, and concentrated under reduced pressure. Crude product was purified by flash column chromatography over silica gel to afford the corresponding terminal alkyne.
5.2.3 General procedure C: preparation of propargyl alcohol
A solution of terminal alkyne (1 equiv) and EtMgBr (1 M in THF, 2.5 equiv) in anhydrous THF (0.09 M) was heated at 50 °C and stirred for 30 min. Paraformaldehyde (2.5 equiv) was added and the suspension was stirred at 50 °C for 3.5 h. The mixture was quenched by addition of a saturated solution of NH4Cl and extracted with Et2O. Combined organic layers were washed with water, brine, dried over Na2SO4, filtered, and concentrated under reduced pressure. Crude product was purified by flash column chromatography over silica gel to afford the corresponding propargyl alcohol.
5.2.4 General procedure D: biphasic Williamson ether synthesis
To a solution of propargyl alcohol (1 equiv) in CH2Cl2 (0.18 M) were added propargyl bromide (80% in toluene, 3.5 equiv), n-Bu4NHSO4 (0.1 equiv), and NaOH (0.5 M, 50% aq). The reaction mixture was stirred at room temperature for 2 h and extracted with CH2Cl2. Combined organic layers were washed with water, brine, dried over Na2SO4, filtered, and concentrated under reduced pressure. Crude product was purified by flash column chromatography over silica gel to afford the corresponding propargyl ether.
5.2.5 General procedure E: silylation of alkyne
A solution of propargyl ether (1 equiv) and EtMgBr (1 M in THF, 2.5 equiv) in anhydrous THF (0.1 M) was heated at 50 °C and stirred for 1 h. Trimethylsilyl chloride (freshly distilled, 2.5 equiv) was added and the suspension was stirred at 50 °C. The mixture was quenched by addition of a saturated solution of NaHCO3 and extracted with Et2O. Combined organic layers were washed with water, brine, dried over Na2SO4, filtered, and concentrated under reduced pressure. Crude product was purified by flash column chromatography over silica gel to afford the corresponding silylated compound.
5.2.6 General procedure F: palladocatalyzed cascade reactions 5-exo-dig/CH activation
A solution of cascade reactions' substrate (1 equiv) and diisopropylamine (1.3 equiv) in benzene (0.1 M) was degassed with argon. Pd(PPh3)4 (0.05 equiv) was added. The reaction mixture was degassed again and heated at 100 °C under microwave irradiation. The mixture was concentrated in vacuo and purified by flash column chromatography over silica gel to afford the corresponding polycyclic compound.
5.2.6.1 (((4S*,5R*)-5-(2-Bromophenyl)-2,2-dimethyl-1,3-dioxolan-4-yl)ethynyl)trimethylsilane (23)
Compound 23 (white solid) was prepared following the general procedure A starting from diol 22 [13] (1.995 g, 6.37 mmol).
Rf = 0.74 (Heptane/EtOAc 8:2). 1H NMR (400 MHz, CDCl3) δ (ppm) = 7.66 (dd, 3J = 7.6 Hz, 4J = 0.8 Hz, 1H, H-6), 7.51 (dd, 3J = 7.6 Hz, 4J = 1.2 Hz, 1H, H-3), 7.34 (br t, 3J = 7.6 Hz, 1H, H-5), 7.17 (td, 3J = 7.6 Hz, 4J = 0.8 Hz, 1H, H-4), 5.49 (d, 3J = 6 Hz, 1H, H-7), 5.27 (d, 3J = 6 Hz, 1H, H-8), 1.71 (s, 3H, H-12 or 12′), 1.49 (s, 3H, H-12 or 12′), −0.08 (s, 9H, TMS). 13C NMR (100 MHz, CDCl3) δ (ppm) = 136.8 (C-1), 132.1 (C-3), 129.3 (C-4), 128.8 (C-6), 127.1 (C-5), 122.1 (C-2), 110.6 (C-11), 101.4 (C-9), 93.6 (C-10), 79.8 (C-7), 69.7 (C-8), 27.5 (C-12 or 12′), 26.2 (C-12 or 12′), −0.5 (TMS). IR (CDCl3) ν (cm−1) = 2989, 2957, 1246, 1208, 1164, 1061, 1044, 1025, 841, 758. HRMS (ESI, 120 eV) calculated for C16H21BrO2Si [M] 352.04942, found 352.04991 (Diff.: −1.39 ppm). Mp = 65 °C.
5.2.6.2 (4R*,5S*)-4-(2-Bromophenyl)-5-ethynyl-2,2-dimethyl-1,3-dioxolane (24)
Terminal alkyne 24 (colorless oil) was prepared following the general procedure B starting from silylated alkyne 23 (1 g, 2.83 mmol).
Rf = 0.46 (Heptane/EtOAc 95:5). 1H NMR (400 MHz, CDCl3) δ (ppm) = 7.70 (dd, 3J = 8 Hz, 4J = 1.6 Hz, 1H, H-6), 7.53 (dd, 3J = 8 Hz, 4J = 0.8 Hz, 1H, H-3), 7.36 (td, 3J = 8 Hz, 4J = 0.8 Hz, 1H, H-5), 7.19 (td, 3J = 8 Hz, 4J = 1.6 Hz, 1H, H-4), 5.48 (d, 3J = 6 Hz, 1H, H-7), 5.33 (dd, 3J = 6 Hz, 4J = 2 Hz, 1H, H-8), 2.20 (d, 4J = 2 Hz, 1H, H-10), 1.71 (s, 3H, H-12 or 12′), 1.50 (s, 3H, H-12 or 12′). 13C NMR (100 MHz, CDCl3) δ (ppm) = 136.2 (C-1), 132.2 (C-3), 129.5 (C-4), 128.7 (C-6), 127.3 (C-5), 121.9 (C-2), 110.6 (C-11), 79.8 (C-9), 79.6 (C-7), 76.4 (C-10), 69.1 (C-8), 27.6 (C-12 or 12′), 26.3 (C-12 or 12′). IR (CDCl3) ν (cm−1) = 3246, 2993, 2936, 2877, 2117, 1567, 1231, 1161, 1073, 1056, 1046, 1025, 863, 747. HRMS (GC EI, 70 eV) calculated for C13H13BrO2 [M+·] 280.00989, found 280.00933 (Diff.: −2 ppm).
5.2.6.3 3-((4S*,5R*)-5-(2-Bromophenyl)-2,2-dimethyl-1,3-dioxolan-4-yl)prop-2-yn-1-ol (25)
Propargyl alcohol 25 (colorless oil) was prepared following the general procedure C starting from terminal alkyne 24 (1.5 g, 5.49 mmol).
Rf = 0.17 (Heptane/EtOAc 7:3). 1H NMR (400 MHz, CDCl3) δ (ppm) = 7.68 (dd, 3J = 8 Hz, 4J = 1.6 Hz, 1H, H-6), 7.54 (dd, 3J = 8 Hz, 4J = 0.8 Hz, 1H, H-3), 7.36 (td, 3J = 7.6 Hz, 4J = 0.8 Hz, H-5), 7.19 (td, 3J = 8 Hz, 4J = 1.6 Hz, 1H, H-4), 5.51 (d, 3J = 6 Hz, 1H, H-7), 5.35 (dt, 3J = 6.4 Hz, 5J = 1.6 Hz, 1H, H-8), 4.02–3.91 (m, 2H, H-13), 1.70 (s, 3H, H-12 or 12′), 1.50 (s, 3H, H-12 or 12′), 0.96 (t, 3J = 6 Hz, 1H, OH). 13C NMR (100 MHz, CDCl3) δ (ppm) = 136.7 (C-1), 132.3 (C-3), 129.5 (C-4), 128.7 (C-6), 127.1 (C-5), 122.1 (C-2), 110.6 (C-11), 86.6 (C-10), 82.2 (C-9), 79.7 (C-7), 69.3 (C-8), 50.9 (C-13), 27.7 (C-12 or 12′), 26.1 (C-12 or 12′). IR (CDCl3) ν (cm−1) = 3423, 3061, 2886, 2935, 2861, 1569, 1381, 1232, 1067, 1025, 865, 755. HRMS (GC EI, 70 eV) calculated for C14H15BrO3 [M+·] 310.02046, found 310.01924 (Diff.: −3.93 ppm).
5.2.6.4 (4R*,5S*)-4-(2-Bromophenyl)-2,2-dimethyl-5-(3-(prop-2-yn-1-yloxy)prop-1-yn-1-yl)-1,3-dioxolane (26)
Compound 26 (colorless oil) was prepared following the general procedure D starting from propargyl alcohol 25 (449 mg, 1.44 mmol).
Rf = 0.55 (Heptane/Et2O 7:3). 1H NMR (400 MHz, CDCl3) δ (ppm) = 7.69 (dd, 3J = 8 Hz, 4J = 1.6 Hz, 1H, H-6), 7.54 (dd, 3J = 8 Hz, 4J = 1.2 Hz, 1H, H-3), 7.37 (td, 3J = 7.6 Hz, 4J = 1.2 Hz, H-5), 7.18 (td, 3J = 7.6 Hz, 4J = 1.6 Hz, 1H, H-4), 5.49 (d, 3J = 6 Hz, 1H, H-7), 5.38 (dt, 3J = 6 Hz, 5J = 2 Hz, 1H, H-8), 4.03 (d, 5J = 2 Hz, 2H, H-13), 3.74 (d, 4J = 2.4 Hz, 2H, H-14), 2.37 (t, 4J = 2.4 Hz, 1H, H-16), 1.70 (s, 3H, H-12 or 12′), 1.50 (s, 3H, H-12 or 12′). 13C NMR (100 MHz, CDCl3) δ (ppm) = 136.6 (C-1), 132.3 (C-3), 129.6 (C-4), 128.5 (C-6), 127.3 (C-5), 121.9 (C-2), 110.5 (C-11), 83.3 (C-9 or 10), 83.2 (C-9 or 10), 79.7 (C-7), 79.0 (C-15), 74.8 (C-16), 69.3 (C-8), 56.4 (C-13), 55.9 (C-14), 27.7 (C-12 or 12′), 26.1 (C-12 or 12′). IR (CDCl3) ν (cm−1) = 3293, 3074, 2987, 2936, 2892, 2855, 2196, 2119, 1569, 1471, 1441, 1381, 1338, 1233, 1166, 1123, 1070, 1048, 1027, 866, 756, 669. HRMS (ESI, 120 eV) calculated for C17H17BrO3 [M] 348.03611, found 348.03506 (Diff.: 2.8 ppm).
5.2.6.5 (3-((3-((4S*,5R*)-5-(2-Bromophenyl)-2,2-dimethyl-1,3-dioxolan-4-yl)prop-2-yn-1-yl)oxy)prop-1-yn-1-yl)trimethylsilane (1a)
Compound 1a (pale yellow oil) was prepared following the general procedure E starting from compound 26 (313 mg, 0.9 mmol).
Rf = 0.39 (Heptane/Et2O 95:5). 1H NMR (400 MHz, CDCl3) δ (ppm) = 7.68 (dd, 3J = 7.6 Hz, 4J = 0.8 Hz, 1H, H-6), 7.53 (dd, 3J = 8 Hz, 4J = 0.8 Hz, 1H, H-3), 7.36 (br t, 3J = 7.6 Hz, 1H, H-5), 7.18 (td, 3J = 8 Hz, 4J = 1.6 Hz, 1H, H-4), 5.49 (d, 3J = 6 Hz, 1H, H-7), 5.37 (dt, 3J = 6 Hz, 5J = 2 Hz, 1H, H-8), 4.01 (d, 5J = 2 Hz, 2H, H-13), 3.74 (s, 2H, H-14), 1.70 (s, 3H, H-12 or 12′), 1.50 (s, 3H, H-12 or 12′), 0.18 (s, 9H, TMS). 13C NMR (100 MHz, CDCl3) δ (ppm) = 136.6 (C-1), 132.3 (C-3), 129.6 (C-4), 128.6 (C-6), 127.3 (C-5), 121.9 (C-2), 110.5 (C-11), 100.8 (C-15), 91.9 (C-16), 83.5 (C-10), 83.1 (C-9), 79.7 (C-7), 69.3 (C-8), 56.8 (C-14), 56.4 (C-13), 27.7 (C-12 or 12′), 26.2 (C-12 or 12′), −0.04 (TMS). IR (CDCl3) ν (cm−1) = 3066, 2985, 2958, 2892, 2848, 2174, 1570, 1440, 1338, 1233, 1070, 920, 844, 756, 700. HRMS (ESI, 120 eV) calculated for C20H25BrO3Si [M] 420.07563, found 420.07475 (Diff.: 2.1 ppm).
5.2.6.6 (3-((3-((4S*,5R*)-5-(2-Bromophenyl)-2,2-dimethyl-1,3-dioxolan-4-yl)prop-2-yn-1-yl)oxy)prop-1-yn-1-yl)triethylsilane (1b)
Compound 1b (pale yellow oil) was prepared following the general procedure E starting from compound 26 (313 mg, 0.9 mmol).
Rf = 0.34 (Heptane/Et2O 95:5). 1H NMR (400 MHz, CDCl3) δ (ppm) = 7.68 (dd, 3J = 7.6 Hz, 4J = 1.6 Hz, 1H, H-6), 7.53 (dd, 3J = 8 Hz, 4J = 0.8 Hz, 1H, H-3), 7.36 (br t, 3J = 7.6 Hz, 1H, H-5), 7.17 (td, 3J = 7.6 Hz, 4J = 1.6 Hz, 1H, H-4), 5.49 (d, 3J = 6 Hz, 1H, H-7), 5.38 (dt, 3J = 6 Hz, 5J = 1.6 Hz, 1H, H-8), 4.03 (d, 5J = 1.6 Hz, 2H, H-13), 3.75 (s, 2H, H-14), 1.70 (s, 3H, H-12 or 12′), 1.50 (s, 3H, H-12 or 12′), 0.99 (t, 3J = 8 Hz, 9H, CH3-TES), 0.60 (q, 3J = 8 Hz, 6H, CH2-TES). 13C NMR (100 MHz, CDCl3) δ (ppm) = 136.6 (C-1), 132.3 (C-3), 129.5 (C-4), 128.6 (C-6), 127.3 (C-5), 121.9 (C-2), 110.5 (C-11), 101.8 (C-15), 89.3 (C-16), 83.6 (C-10), 83.0 (C-9), 79.7 (C-7), 69.4 (C-8), 56.7 (C-13), 56.2 (C-14), 27.7 (C-12 or 12′), 26.2 (C-12 or 12′), 7.6 (CH3-TES), 4.4 (CH2-TES). IR (CDCl3) ν (cm−1) = 2985, 2954, 2874, 2848, 1458, 1440, 1372, 1337, 1232, 1165, 1123, 1069, 999, 865, 725. HRMS (GC FI, 10,000 V) calculated for C23H31BrO3Si [M+·] 462.12258, found 462.12162 (Diff.: −2.09 ppm).
5.2.6.7 4-((3-((4S*,5R*)-5-(2-Bromophenyl)-2,2-dimethyl-1,3-dioxolan-4-yl)prop-2-yn-1-yl)oxy)but-2-yn-1-ol (1c)
A solution of compound 26 (150 mg, 0.43 mmol, 1 equiv) and EtMgBr (1 M in THF, 1 mL, 1 mmol, 2.5 equiv) in anhydrous THF (5 mL) was heated at 55 °C and stirred for 1 h. Paraformaldehyde (32 mg, 1.1 mmol, 2.5 mmol, 2.5 equiv) was added and the suspension was stirred at 55 °C for 2 h. TLC showed that the reaction was not completed. Paraformaldehyde (10 mg, 0.33 mmol) was added again and the reaction mixture was stirred at room temperature overnight. The mixture was quenched by addition of a saturated solution of NH4Cl (10 mL) and extracted with Et2O (3 × 10 mL). Combined organic layers were washed with water, brine, dried over Na2SO4, filtered, and concentrated under reduced pressure. Crude product was purified by flash column chromatography over silica gel (heptane/EtOAc 7:3) to afford the corresponding propargyl alcohol 1c as a yellow oil.
Rf = 0.26 (Heptane/EtOAc 7:3). 1H NMR (400 MHz, CDCl3) δ (ppm) = 7.68 (dd, 3J = 8 Hz, 4J = 1.6 Hz, 1H, H-6), 7.53 (dd, 3J = 8 Hz, 4J = 1.2 Hz, 1H, H-3), 7.37 (td, 3J = 7.6 Hz, 4J = 0.8 Hz, 1H, H-5), 7.19 (td, 3J = 8 Hz, 4J = 2 Hz, 1H, H-4), 5.49 (d, 3J = 6 Hz, 1H, H-7), 5.38 (dt, 3J = 6 Hz, 5J = 2 Hz, 1H, H-8), 4.29 (t, 5J = 1.6 Hz, 2H, H-14), 4.01 (d, 5J = 1.6 Hz, 2H, H-13), 3.85–3.74 (m, 2H, H-17), 1.70 (s, 3H, H-12 or 12′), 1.56 (br s, 1H, OH), 1.50 (s, 3H, H-12 or 12′). 13C NMR (100 MHz, CDCl3) δ (ppm) = 136.6 (C-1), 132.3 (C-3), 129.5 (C-4), 128.6 (C-6), 127.3 (C-5), 121.9 (C-2), 110.5 (C-11), 85.0 (C-15 or 16), 83.4 (C-10), 83.2 (C-9), 81.3 (C-15 or 16), 79.7 (C-7), 69.4 (C-8), 56.6 (C-13), 56.3 (C-17), 51.3 (C-14), 27.7 (C-12 or 12′), 26.1 (C-12 or 12′). IR (CDCl3) ν (cm−1) = 3422, 3066, 2987, 2933, 2854, 1439, 1373, 1231, 1121, 1067, 1018, 865, 755. HRMS (ESI, 120 eV) calculated for C18H19BrO4 [M] 378.04667, found 378.04712 (Diff.: −1.18 ppm).
5.2.6.8 (4R*,5S*)-4-(2-Bromophenyl)-2,2-dimethyl-5-(3-(pent-2-yn-1-yloxy)prop-1-yn-1-yl)-1,3-dioxolane (1d)
To a solution of propargyl alcohol 25 (257 mg, 0.82 mmol, 1 equiv) in CH2Cl2 (4.6 mL) were added 1-bromopent-2-yne (0.3 mL, 2.9 mmol, 3.6 equiv), n-Bu4NHSO4 (28 mg, 0.08 mmol, 0.1 equiv), and NaOH (1.5 mL, 50% aq). The reaction mixture was stirred at room temperature for 2 h and extracted with CH2Cl2 (3 × 10 mL). Combined organic layers were washed with water, brine, dried over Na2SO4, filtered, and concentrated under reduced pressure. Crude product was purified by flash column chromatography over silica gel (heptane/Et2O 96:4) to afford compound 1d (87%) as a pale yellow oil.
Rf = 0.63 (Heptane/EtOAc 8:2). 1H NMR (400 MHz, CDCl3) δ (ppm) = 7.68 (dd, 3J = 8 Hz, 4J = 1.6 Hz, 1H, H-6), 7.53 (dd, 3J = 8 Hz, 4J = 1.2 Hz, 1H, H-3), 7.36 (td, 3J = 7.6 Hz, 4J = 1.2 Hz, 1H, H-5), 7.18 (td, 3J = 7.6 Hz, 4J = 1.6 Hz, 1H, H-4), 5.49 (d, 3J = 6 Hz, 1H, H-7), 5.37 (dt, 3J = 6 Hz, 5J = 1.6 Hz, 1H, H-8), 4.00 (d, 5J = 2 Hz, 2H, H-13), 3.74 (t, 3J = 2.4 Hz, 2H, H-14), 2.21 (qt, 3J = 7.6 Hz, 5J = 2 Hz, 2H, H-17), 1.70 (s, 3H, H-12 or 12′), 1.50 (s, 3H, H-12 or 12′), 1.14 (t, 3J = 7.2 Hz, 3H, H-18). 13C NMR (100 MHz, CDCl3) δ (ppm) = 136.6 (C-1), 132.3 (C-3), 129.5 (C-4), 128.6 (C-6), 127.3 (C-5), 121.9 (C-2), 110.5 (C-11), 88.8 (C-16), 83.8 (C-10), 82.8 (C-9), 79.7 (C-7), 74.6 (C-15), 69.4 (C-8), 56.6 (C-14), 56.2 (C-13), 27.7 (C-12 or 12′), 26.2 (C-12 or 12′), 13.9 (C-18), 12.6 (C-17). IR (CDCl3) ν (cm−1) = 2983, 2937, 2853, 2364, 2181, 1971, 1440, 1381, 1233, 1123, 1071, 1027, 866, 754. HRMS (ESI, 120 eV) calculated for C19H21BrO3 [M] 376.06741, found 376.06679 (Diff.: 1.64 ppm).
5.2.6.9 (((4S*,5R*)-5-(2-Bromo-5-methoxyphenyl)-2,2-dimethyl-1,3-dioxolan-4-yl)ethynyl)trimethylsilane (28)
Acetonide 28 (pale yellow oil) was prepared following the general procedure A starting from diol 27 [13] (1.967 g, 5.73 mmol).
Rf = 0.42 (Heptane/EtOAc 95:5). 1H NMR (400 MHz, CDCl3) δ (ppm) = 7.39 (d, 3J = 8.4 Hz, 1H, H-3), 7.23 (d, 4J = 3.2 Hz, 1H, H-6), 6.73 (dd, 3J = 8.4 Hz, 4J = 3.2 Hz, 1H, H-4), 5.44 (d, 3J = 6 Hz, 1H, H-7), 5.26 (d, 3J = 6 Hz, 1H, H-8), 3.81 (s, 3H, H-13), 1.70 (s, 3H, H-12 or 12′), 1.49 (s, 3H, H-12 or 12′), −0.07 (s, 9H, TMS). 13C NMR (100 MHz, CDCl3) δ (ppm) = 158.9 (C-5), 137.8 (C-1), 132.7 (C-3), 115.1 (C-4), 114.5 (C-6), 112.3 (C-2), 110.7 (C-11), 101.4 (C-9), 93.5 (C-10), 79.8 (C-7), 69.7 (C-8), 55.5 (C-13), 27.6 (C-12 or 12′), 26.3 (C-12 or 12′), −0.49 (TMS). IR (CDCl3) ν (cm−1) = 2988, 2958, 2900, 2179, 1472, 1231, 1066, 844. HRMS (GC EI, 70 eV) calculated for C17H23BrO3Si [M+·] 382.05998, found 382.05961 (Diff.: −0.99 ppm).
5.2.6.10 (4R*,5S*)-4-(2-Bromo-5-methoxyphenyl)-5-ethynyl-2,2-dimethyl-1,3-dioxolane (29)
Terminal alkyne 29 (white solid) was prepared following the general procedure B starting from silylated alkyne 28 (1.068 g, 2.79 mmol).
Rf = 0.31 (Heptane/EtOAc 95:5). 1H NMR (400 MHz, CDCl3) δ (ppm) = 7.40 (d, 3J = 8.8 Hz, 1H, H-3), 7.25 (d, 4J = 2.8 Hz, 1H, H-6), 6.74 (dd, 3J = 8.4 Hz, 4J = 3.2 Hz, 1H, H-4), 5.42 (d, 3J = 6 Hz, 1H, H-7), 5.31 (dd, 3J = 6 Hz, 4J = 2 Hz, 1H, H-8), 3.82 (s, 3H, H-13), 2.23 (d, 4J = 2.4 Hz, 1H, H-10), 1.70 (s, 3H, H-12 or 12′), 1.49 (s, 3H, H-12 or 12′). 13C NMR (100 MHz, CDCl3) δ (ppm) = 159 (C-5), 137.2 (C-1), 132.8 (C-3), 115.3 (C-4), 114.4 (C-6), 112.1 (C-2), 110.6 (C-11), 79.8 (C-9), 79.6 (C-7), 76.3 (C-10), 69.0 (C-8), 55.6 (C-13), 27.6 (C-12 or 12′), 26.3 (C-12 or 12′). IR (CDCl3) ν (cm−1) = 3292, 2987, 2937, 2837, 1575, 1472, 1228, 1161, 1068, 863. HRMS (GC EI, 70 eV) calculated for C14H15BrO3 [M+·] 310.02046, found 310.01832 (Diff.: −6.89 ppm). Mp = 58 °C.
5.2.6.11 3-((4S*,5R*)-5-(2-Bromo-5-methoxyphenyl)-2,2-dimethyl-1,3-dioxolan-4-yl)prop-2-yn-1-ol (30)
Propargyl alcohol 30 (pale yellow oil) was prepared following the general procedure C starting from terminal alkyne 29 (760 mg, 2.44 mmol).
Rf = 0.09 (Heptane/EtOAc 7:3). 1H NMR (400 MHz, CDCl3) δ (ppm) = 7.41 (d, 3J = 8.8 Hz, 1H, H-3), 7.25 (d, 4J = 3.2 Hz, 1H, H-6), 6.75 (dd, 3J = 8.8 Hz, 4J = 3.2 Hz, 1H, H-4), 5.46 (d, 3J = 6 Hz, 1H, H-7), 5.32 (dt, 3J = 6 Hz, 5J = 1.6 Hz, 1H, H-8), 4.00 (dd, 3J = 6 Hz, 5J = 1.6 Hz, 2H, H-14), 3.82 (s, 3H, H-13), 1.70 (s, 3H, H-12 or 12′), 1.49 (s, 3H, H-12 or 12′), 1.08 (t, 3J = 6 Hz, 1H, OH). 13C NMR (100 MHz, CDCl3) δ (ppm) = 158.9 (C-5), 137.7 (C-1), 132.8 (C-3), 115.2 (C-4), 114.5 (C-6), 112.4 (C-2), 110.6 (C-11), 86.6 (C-10), 79.7 (C-9), 79.6 (C-7), 69.3 (C-8), 55.7 (C-13), 51.0 (C-14), 27.7 (C-12 or 12′), 26.1 (C-12 or 12′). IR (CDCl3) ν (cm−1) = 3422, 2986, 2937, 2837, 2361, 2337, 1575, 1473, 1297, 1230, 1165, 1068, 864. HRMS (GC EI, 70 eV) calculated for C15H17BrO4 [M+·] 340.03102, found 340.03032 (Diff.: −2.05 ppm).
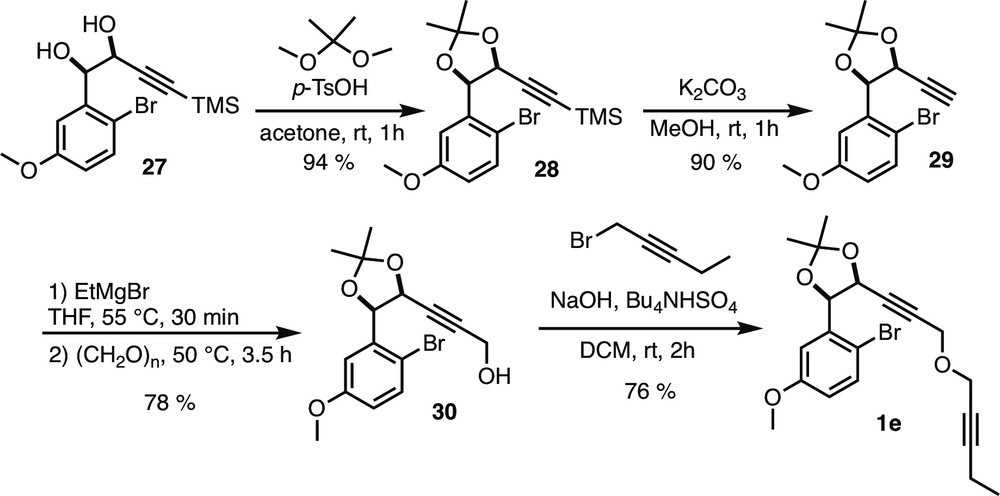
5.2.6.12 (4R*,5S*)-4-(2-Bromo-5-methoxyphenyl)-2,2-dimethyl-5-(3-(pent-2-yn-1-yloxy)prop-1-yn-1-yl)-1,3-dioxolane (1e)
To a solution of propargyl alcohol 30 (100 mg, 0.29 mmol, 1 equiv) in CH2Cl2 (1.6 mL) were added 1-bromopent-2-yne (0.1 mL, 0.98 mmol, 3.3 equiv), n-Bu4NHSO4 (10 mg, 0.03 mmol, 0.1 equiv), and NaOH (0.5 mL, 50% aq). The reaction mixture was stirred at room temperature for 4 h and extracted with CH2Cl2 (3 × 10 mL). Combined organic layers were washed with water, brine, dried over Na2SO4, filtered, and concentrated under reduced pressure. Crude product was purified by flash column chromatography over silica gel (heptane/Et2O 96:4) to afford compound 1e (76%) as a pale yellow oil.
Rf = 0.65 (Heptane/EtOAc 8:2). 1H NMR (400 MHz, CDCl3) δ (ppm) = 7.40 (d, 3J = 8.8 Hz, 1H, H-3), 7.24 (d, 4J = 2.8 Hz, 1H, H-6), 6.74 (dd, 3J = 8.8 Hz, 4J = 3.2 Hz, 1H, H-4), 5.43 (d, 3J = 6 Hz, 1H, H-7), 5.36 (dt, 3J = 6 Hz, 5J = 1.6 Hz, 1H, H-8), 4.03 (d, 5J = 1.6 Hz, 2H, H-14), 3.82 (s, 3H, H-13), 3.79–3.76 (m, 2H, H-15), 2.20 (qt, 3J = 7.2 Hz, 5J = 2.4 Hz, 2H, H-18), 1.70 (s, 3H, H-12 or 12′), 1.50 (s, 3H, H-12 or 12′), 1.13 (t, 3J = 7.6 Hz, 3H, H-19). 13C NMR (100 MHz, CDCl3) δ (ppm) = 159.1 (C-5), 137.6 (C-1), 132.9 (C-3), 115.4 (C-4), 114.1 (C-6), 112.2 (C-2), 110.5 (C-11), 88.8 (C-17), 83.7 (C-10), 82.8 (C-9), 79.6 (C-7), 74.6 (C-16), 69.4 (C-8), 56.6 (C-15), 56.3 (C-14), 55.6 (C-13), 27.7 (C-12 or 12′), 26.2 (C-12 or 12′), 13.9 (C-19), 12.6 (C-18). IR (CDCl3) ν (cm−1) = 2987, 2937, 2851, 1575, 1473, 1373, 1297, 1230, 1165, 1124, 1070, 1026, 864. HRMS (ESI, 120 eV) calculated for C20H23BrO4 [M] 406.07797, found 406.07766 (Diff.: 0.77 ppm).
5.2.6.13 (4R*,5S*)-4-(2-Bromophenyl)-2,2-dimethyl-5-(3-((3-phenylprop-2-yn-1-yl)oxy)prop-1-yn-1-yl)-1,3-dioxolane (1f)
To a solution of propargyl alcohol 25 (157 mg, 0.50 mmol, 1 equiv) in CH2Cl2 (2.8 mL) were added (3-chloroprop-1-yn-1-yl)benzene (0.24 mL, 1.7 mmol, 3.5 equiv), n-Bu4NHSO4 (18 mg, 0.05 mmol, 0.1 equiv), and NaOH (0.9 mL, 50% aq). The reaction mixture was stirred at room temperature for 2 h and extracted with CH2Cl2 (3 × 10 mL). Combined organic layers were washed with water, brine, dried over Na2SO4, filtered, and concentrated under reduced pressure. Crude product was purified by flash column chromatography over silica gel (heptane/Et2O 96:4) to afford compound 1f (87%) as a yellow oil.
Rf = 0.56 (Heptane/EtOAc 8:2). 1H NMR (400 MHz, CDCl3) δ (ppm) = 7.71 (dd, 3J = 7.6 Hz, 4J = 1.6 Hz, 1H, H-6), 7.54 (dd, 3J = 8 Hz, 4J = 1.2 Hz, 1H, H-3), 7.45–7.40 (m, 2H, H-18 & 18), 7.37 (td, 3J = 7.2 Hz, 4J = 0.8 Hz, 1H, H-5), 7.34–7.30 (m, 3H, H-19 & 19′ & 20), 7.17 (td, 3J = 8 Hz, 4J = 2 Hz, 1H, H-4), 5.51 (d, 3J = 6.4 Hz, 1H, H-7), 5.40 (dt, 3J = 6.4 Hz, 5J = 1.6 Hz, 1H, H-8), 4.09 (d, 5J = 1.6 Hz, 2H, H-13), 3.97 (s, 2H, H-14), 1.71 (s, 3H, H-12 or 12′), 1.51 (s, 3H, H-12 or 12′). 13C NMR (100 MHz, CDCl3) δ (ppm) = 136.7 (C-1), 132.3 (C-3), 131.9 (C-18 & 18′), 129.6 (C-4), 128.7 (C-6), 128.6 (C-20), 128.5 (C-19 & 19′), 127.4 (C-5), 122.7 (C-17), 121.9 (C-2), 110.5 (C-11), 86.7 (C-16), 84.4 (C-15), 83.5 (C-10), 83.2 (C-9), 79.7 (C-7), 69.4 (C-8), 56.7 (C-14), 56.4 (C-13), 27.7 (C-12 or 12′), 26.2 (C-12 or 12′). IR (CDCl3) ν (cm−1) = 2988, 2930, 2851, 1490, 1441, 1380, 1338, 1232, 1165, 1123, 1070, 1027, 866, 755, 691. HRMS (ESI, 120 eV) calculated for C23H21BrO3 [M] 424.06741, found 424.06761 (Diff.: −0.48 ppm).
5.2.6.14 ((3aR*,10cS*)-2,2-Dimethyl-3a,8,10,10c-tetrahydrofuro[3′,4′:3,4]acenaphtho[1,2-d][1,3]dioxol-7-yl)trimethylsilane (2a) and (3aR*,10cS*)-2,2-dimethyl-3a,8,10,10c-tetrahydrofuro[3′,4′:3,4]acenaphtho[1,2-d][1,3]dioxole (5)
Compounds 2a and 5 were obtained in a separable mixture (91%, ratio 2a/5 21:79) and were prepared following the general procedure F starting from substrate 1a (51 mg, 0.11 mmol).
Compound 2a (white solid): Rf = 0.17 (heptane/Et2O 9:1). 1H NMR (400 MHz, CDCl3) δ (ppm) = 7.98 (dd, 3J = 7.6 Hz, 4J = 1.2 Hz, 1H, H-4), 7.48 (m, 2H, H-5 & 6), 5.87 (d, 3J = 6 Hz, 1H, H-7), 5.79 (d, 3J = 6 Hz, 1H, H-8), 5.35–5.24 (m, 4H, H-13 & 14), 1.47 (s, 3H, H-12 or 12′), 1.09 (s, 3H, H-12 or 12′), 0.42 (s, 9H, TMS). 13C NMR (100 MHz, CDCl3) δ (ppm) = 148.0 (C-15), 141.3 (C-1), 136.6 (C-2), 135.4 (C-3), 134.1 (C-9), 133.3 (C-10), 128.2 (C-16), 127.7 (C-5 or 6), 126.6 (C-4), 121.3 (C-5 or 6), 112.9 (C-11), 82.5 (C-7), 81.1 (C-8), 74.4 (C-13 or 14), 70.7 (C-13 or 14), 27.4 (C-12 or 12′), 25.9 (C-12 or 12′), 1.9 (TMS). IR (CDCl3) ν (cm−1) = 3065, 2976, 2937, 2851, 1459, 1371, 1251, 1204, 1079, 1020, 923, 839, 770. HRMS (GC EI, 70 eV) calculated for C20H24O3Si [M+·] 340.14947, found 340.14955 (Diff.: 0.24 ppm). Mp = 163 °C. CCDC 1509122 contains the supplementary crystallographic data. These data can be obtained free of charge via http://www.ccdc.cam.ac.uk/.
Compound 5 (white solid): Rf = 0.11 (heptane/Et2O 9:1). 1H NMR (400 MHz, CDCl3) δ (ppm) = 7.76 (dd, 3J = 7.6 Hz, 4J = 1.2 Hz, 1H, H-4), 7.62 (br s, 1H, H-16), 7.58–7.52 (m, 2H, H-5 & 6), 5.97 (d, 3J = 6 Hz, 1H, H-7), 5.89 (d, 3J = 6 Hz, 1H, H-8), 5.33 (AB system, JAB = 13.2 Hz, Δ√ = 25.6 Hz, 2H, H-13), 5.25 (s, 2H, H-14), 1.54 (s, 3H, H-12 or 12′), 1.15 (s, 3H, H-12 or 12′). 13C NMR (100 MHz, CDCl3) δ (ppm) = 141.6 (C-15), 140.6 (C-1), 137.3 (C-2), 134.1 (C-10), 132.8 (C-9), 131.2 (C-3), 128.3 (C-5 or 6), 125.6 (C-4), 121.5 (C-5 or 6), 117.0 (C-16), 112.9 (C-11), 82.8 (C-7), 81.4 (C-8), 72.9 (C-14), 71.3 (C-13), 27.4 (C-12 or 12′), 25.9 (C-12 or 12′). IR (CDCl3) ν (cm−1) = 3057, 2897, 2857, 1469, 1377, 1253, 1202, 1063, 1010, 863, 767. HRMS (GC EI, 70 eV) calculated for C17H16BrO3 [M+·] 268.10994, found 268.10897 (Diff.: −3.65 ppm). Mp = 152 °C. CCDC 1509123 contains the supplementary crystallographic data. These data can be obtained free of charge via http://www.ccdc.cam.ac.uk/.
5.2.6.15 ((3aR*,10cS*)-2,2-dimethyl-3a,8,10,10c-tetrahydrofuro[3′,4′:3,4]acenaphtho[1,2-d][1,3]dioxol-7-yl)triethylsilane (2b)
Compounds 2b and 5 were obtained in a separable mixture (66%, ratio 2a/5 18:82) and were prepared following the general procedure F starting from substrate 1b (51 mg, 0.11 mmol).
Compound 2b (colorless oil): Rf = 0.14 (heptane/Et2O 9:1). 1H NMR (400 MHz, CDCl3) δ (ppm) = 8.08 (d, 3J = 8.1 Hz, 1H, H-4), 7.57–7.51 (m, 2H, H-6 & 5), 5.94 (d, 3J = 5.9 Hz, 1H, H-7), 5.87 (d, 3J = 5.7 Hz, 1H, H-8), 5.35–5.25 (m, 4H, H-13 & 14), 1.54 (s, 3H, H-12 or 12′), 1.17 (s, 3H, H-12 or 12′), 1.06–1.00 (m, 6H, CH2-TES), 0.97–0.92 (m, 9H, CH3-TES). 13C NMR (100 MHz, CDCl3) δ (ppm) = 149.4 (C-15), 141.3 (C-1), 136.6 (C-2), 136.0 (C-3), 134.0 (C-9), 133.3 (C-10), 127.7 (C-5), 126.5 (C-4), 126.2 (C-16), 121.3 (C-6), 112.9 (C-11), 82.5 (C-7), 81.1 (C-8), 74.6 (C-14), 70.7 (C-13), 27.4 (C-12 or 12′), 25.9 (C-12 or 12′), 7.8 (TES-CH3), 5.3 (TES-CH2). IR (CDCl3) ν (cm−1) = 3070, 2954, 2935, 2909, 2874, 1459, 1380, 1371, 1250, 1205, 1078, 1020, 1001, 924, 766, 729, 697. HRMS (GC FI, 10,000 V) calculated for C23H30O3Si [M+·] 382.19642, found 382.19662 (Diff.: 0.52 ppm).
5.2.6.16 ((3aR*,10cS*)-2,2-Dimethyl-3a,8,10,10c-tetrahydrofuro[3′,4′:3,4]acenaphtho[1,2-d][1,3]dioxol-7-yl)methanol (2c)
Compound 2c (white solid) was prepared following the general procedure F starting from substrate 1c (44 mg, 0.11 mmol).
Rf = 0.39 (Heptane/EtOAc 5:5). 1H NMR (400 MHz, CDCl3) δ (ppm) = 8.04–8.00 (m, 1H, H-4), 7.61–7.58 (m, 2H, H-5 & 6), 5.96 (d, 3J = 6 Hz, 1H, H-7), 5.85 (d, 3J = 5.6 Hz, 1H, H-8), 5.39–5.26 (m, 4H, H-13 & 14), 5.06 (s, 1H, H-17), 1.53 (s, 3H, H-12 or 12′), 1.13 (s, 3H, H-12 or 12′). 13C NMR (100 MHz, CDCl3) δ (ppm) = 141.1 (C-1), 140.5 (C-15), 137.8 (C-2), 134.2 (C-10), 133.0 (C-9), 129.6 (C-3), 128.6 (C-6), 127.7 (C-16), 122.4 (C-4), 121.7 (C-5), 113.0 (C-11), 82.8 (C-7), 81.0 (C-8), 72.4 (C-14), 71.5 (C-13), 59.9 (C-17), 27.4 (C-12 or 12′), 26.0 (C-12 or 12′). IR (CDCl3) ν (cm−1) = 3421, 2985, 2934, 2852, 1726, 1468, 1373, 1254, 1204, 1038, 914, 732. HRMS (GC FI, 10,000 V) calculated for C18H18O4 [M+·] 298.12051, found 298.12069 (Diff.: 0.18 ppm).
5.2.6.17 (3aR*,10cS*)-7-Ethyl-2,2-dimethyl-3a,8,10,10c-tetrahydrofuro[3′,4′:3,4]acenaphtho[1,2-d][1,3]dioxole (2d)
Compound 2d (white solid) was prepared following the general procedure F starting from substrate 1d (50 mg, 0.13 mmol).
Rf = 0.21 (Heptane/Et2O 8:2). 1H NMR (400 MHz, CDCl3) δ (ppm) = 7.92–7.87 (m, 1H, H-4), 7.59–7.54 (m, 2H, H-5 & 6), 5.96 (d, 3J = 6 Hz, 1H, H-7), 5.86 (d, 3J = 6 Hz, 1H, H-8), 5.35 (AB system, JAB = 13.2 Hz, Δ√ = 26.9 Hz, 2H, H-13), 5.25 (s, 2H, H-14), 3.02–2.88 (m, 2H, H-17), 1.54 (s, 3H, H-12 or 12′), 1.28 (t, 3J = 7.6 Hz, 3H, H-18), 1.16 (s, 3H, H-12 or 12′). 13C NMR (100 MHz, CDCl3) δ (ppm) = 141.1 (C-1), 138.6 (C-15), 137.9 (C-2), 133.8 (C-10), 132.2 (C-16), 130.5 (C-9), 129.7 (C-3), 127.8 (C-6), 122.2 (C-4), 121.3 (C-5), 112.7 (C-11), 82.8 (C-7), 81.1 (C-8), 72.4 (C-14), 72.0 (C-13), 27.4 (C-12 or 12′), 25.9 (C-12 or 12′), 22.8 (C-17), 14.6 (C-18). IR (CDCl3) ν (cm−1) = 3061, 2967, 2933, 2873, 2848, 1463, 1372, 1252, 1204, 1059, 766. HRMS (ESI, 120 eV) calculated for C19H20O3 [M] 296.14124, found 296.14135 (Diff.: −0.35 ppm).
5.2.6.18 (3aR*,10cS*)-7-Ethyl-5-methoxy-2,2-dimethyl-3a,8,10,10c-tetrahydrofuro[3′,4′:3,4]acenaphtho[1,2-d][1,3]dioxole (2e)
Compound 2e (orange solid) was prepared following the general procedure F starting from substrate 1e (45 mg, 0.11 mmol).
Rf = 0.20 (Heptane/Et2O 8:2). 1H NMR (400 MHz, CDCl3) δ (ppm) = 7.25 (d, 4J = 2 Hz, 1H, H-arom), 7.17 (d, 3J = 2 Hz, 1H, H-arom), 5.87 (d, 3J = 6 Hz, 1H, H-7), 5.84 (d, 3J = 6 Hz, 1H, H-8), 5.32 (AB system, JAB = 13.2 Hz, Δ√ = 26.5 Hz, 2H, H-13), 5.23 (s, 2H, H-14), 3.95 (s, 3H, H-19), 2.97–2.83 (m, 2H, H-17), 1.53 (s, 3H, H-12 or 12′), 1.28 (t, 3J = 7.6 Hz, 3H, H-18), 1.15 (s, 3H, H-12 or 12′). 13C NMR (100 MHz, CDCl3) δ (ppm) = 160.5 (C-5), 142.7 (C-9), 139.4 (C-15), 133.5 (C-2), 131.4 (C-10), 130.9 (C-16), 130.4 (C-3 & 1), 113.5 (C-6), 112.9 (C-11), 102.1 (C-4), 82.5 (C-7), 81.4 (C-8), 72.4 (C-14), 72.0 (C-13), 55.9 (C-19), 27.5 (C-12 or 12′), 26.0 (C-12 or 12′), 22.9 (C-17), 14.2 (C-18). IR (CDCl3) ν (cm−1) = 2965, 2934, 2872, 1760, 1625, 1603, 1458, 1412, 1236, 1202, 1157, 1059, 1040. HRMS (ESI, 120 eV) calculated for C20H22O4 [M] 326.15181, found 326.15304 (Diff.: −3.78 ppm).
5.2.6.19 (3aR*,10cS*)-2,2-Dimethyl-7-phenyl-3a,8,10,10c-tetrahydrofuro[3′,4′:3,4]acenaphtho[1,2-d][1,3]dioxole (2f)
Compound 2f was prepared following the general procedure F starting from substrate 1f (52.3 mg, 0.12 mmol).
Rf = 0.22 (Heptane/Et2O 8:2). 1H NMR (400 MHz, CDCl3) δ (ppm) = 7.74–7.67 (m, 1 ou 2H, H-arom), 7.64 (d, 3J = 8 Hz, 1H, H-arom), 7.59 (d, 3J = 8 Hz, 1H, H-arom), 7.55–7.31 (m, 4 ou 5H, H-arom), 6.01 (d, 3J = 6 Hz, 1H, H-7), 5.93 (d, 3J = 6 Hz, 1H, H-8), 5.40 (AB system, JAB = 13.2 Hz, Δ√ = 26.0 Hz, 2H, H-13), 5.09 (m, 2H, H-14), 1.57 (s, 3H, H-12 or 12′), 1.22 (s, 3H, H-12 or 12′).
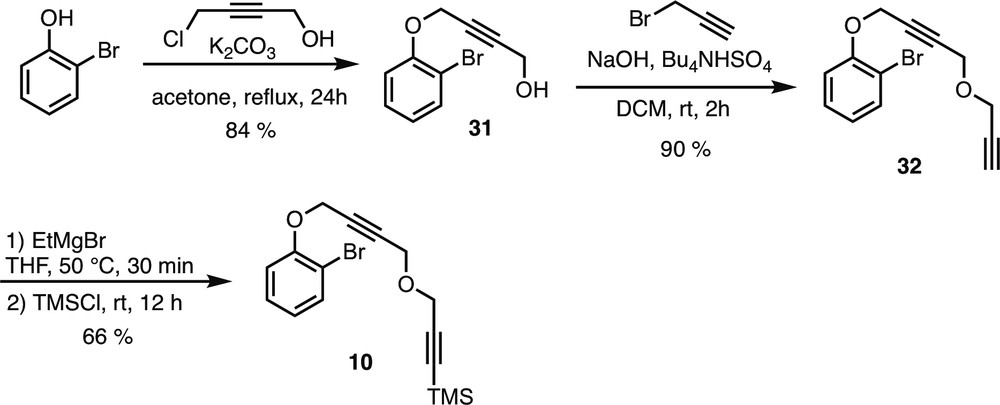
5.2.6.20 4-(2-Bromophenoxy)but-2-yn-1-ol (31)
To a suspension of K2CO3 (2 g, 14.5 mmol, 5 equiv) in acetone (18 mL) was added 2-bromophenol (524 mg, 3.0 mmol, 1.04 equiv) and 4-chlorobut-2-yn-1-ol (0.25 mL, 2.9 mmol, 1 equiv) under argon atmosphere at room temperature. The reaction mixture was refluxed and stirred for 24 h and then quenched with water (10 mL) and extracted with EtOAc (3 × 10 mL). The organic layers were washed with a saturated aqueous solution of NaHCO3 (2 × 10 mL) and brine (10 mL). The organic layers were then dried over Na2SO4, filtered, and concentrated in vacuo. The crude product was purified by flash column chromatography on silica gel (pentane/EtOAc 80:20) to afford compound 31 as an orange oil.
Rf = 0.22 (Pentane/EtOAc 8:2). 1H NMR (400 MHz, CDCl3) δ (ppm) = 7.56 (dd, 3J = 7.6 Hz, 4J = 1.6 Hz, 1H, H-3), 7.30–7.25 (m, 2H, H-5 & 4), 7.04 (dd, 3J = 8.4 Hz, 4J = 1.6 Hz, 1H, H-6), 4.82 (t, 5J = 2 Hz, 2H, H-7), 4.31 (dt, 3J = 6.4 Hz, 5J = 1.6 Hz, 2H, H-10), 1.55 (t, 3J = 6 Hz, 1H, OH). Spectral data were in accordance with literature data [14].
5.2.6.21 1-Bromo-2-((4-(prop-2-yn-1-yloxy)but-2-yn-1-yl)oxy)benzene (32)
Compound 32 (yellow oil) was prepared following the general procedure D starting from propargyl alcohol 31 (180 mg, 0.75 mmol).
Rf = 0.21 (Heptane/EtOAc 96:4). 1H NMR (400 MHz, CDCl3) δ (ppm) = 7.55 (dd, 3J = 8 Hz, 4J = 1.6 Hz, 1H, H-3), 7.30–7.25 (m, 1H, H-5), 7.05 (dd, 3J = 8 Hz, 4J = 1.2 Hz, 1H, H-6), 6.89 (td, 3J = 7.6 Hz, 4J = 1.2 Hz, 1H, H-4), 4.83 (t, 5J = 1.6 Hz, 2H, H-7), 4.30 (t, 5J = 1.6 Hz, 2H, H-10), 4.21 (d, 4J = 2.4 Hz, 2H, H-11), 2.44 (t, 4J = 2.4 Hz, 1H, H-13). 13C NMR (100 MHz, CDCl3) δ (ppm) = 154.2 (C-1), 133.7 (C-3), 128.4 (C-5), 123.0 (C-4), 114.5 (C-6), 112.7 (C-2), 83.3 (C-9), 81.5 (C-8), 78.9 (C-12), 75.3 (C-13), 57.3 (C-7), 56.9 (C-10), 56.7 (C-11). IR (CDCl3) ν (cm−1) = 2959, 2852, 2367, 1478, 1250, 844. HRMS (ESI, 120 eV) calculated for C13H11BrO2 [M] 277.99424, found 277.99404 (Diff.: 0.74 ppm).
5.2.6.22 (3-((4-(2-Bromophenoxy)but-2-yn-1-yl)oxy)prop-1-yn-1-yl)trimethylsilane (10)
Compound 10 (yellow oil) was prepared following the general procedure E starting from propargyl ether 32 (167.5 mg, 0.6 mmol).
Rf = 0.47 (Heptane/EtOAc 9:1). 1H NMR (400 MHz, CDCl3) δ (ppm) = 7.55 (dd, 3J = 8 Hz, 4J = 1.6 Hz, 1H, H-3), 7.30–7.25 (m, 1H, H-5), 7.05 (dd, 3J = 8 Hz, 4J = 1.2 Hz, 1H, H-6), 6.89 (td, 3J = 7.6 Hz, 4J = 1.2 Hz, 1H, H-4), 4.83 (t, 5J = 1.6 Hz, 2H, H-7), 4.28 (t, 5J = 1.6 Hz, 2H, H-10), 4.20 (s, 2H, H-11), 0.18 (s, 9H, TMS). 13C NMR (100 MHz, CDCl3) δ (ppm) = 154.2 (C-1), 133.7 (C-3), 128.5 (C-5), 123.0 (C-4), 114.5 (C-6), 112.7 (C-2), 100.5 (C-12), 92.4 (C-13), 83.5 (C-9), 81.3 (C-8), 57.6 (C-7), 57.3 (C-10), 56.8 (C-11), −0.06 (TMS). IR (CDCl3) ν (cm−1) = 2959, 2852, 2367, 1478, 1250, 844. HRMS (ESI, 120 eV) calculated for C16H19BrO2Si [M] 350.03377, found 350.03295 (Diff.: 2.33 ppm).
5.2.6.23 7,9-Dihydro-1H-naphtho[1,8-bc:6,7-c′]difuran (11)
A solution of compound 10 (43 mg, 0.12 mmol, 1 equiv) and Cs2CO3 (197 mg, 0.60 mmol, 5 equiv) in 1,4-dioxane (1 mL) was degassed with argon. A solution of Pd(OAc)2 (5 mg, 22 μmol, 0.18 equiv) and P(OPh)3 (9 μL, 36 μmol, 0.30 equiv) in dioxane (1 mL) was then added. The reaction mixture was degassed again and heated for 30 min at 100 °C under microwave irradiation. The mixture was passed through a short Celite pad, concentrated in vacuo, and purified by flash column chromatography over silica gel (pentane/EtOAc 96:4) to afford compound 11 (65%), as a white solid.
Naphthalene 53: Rf = 0.41 (heptane/EtOAc 9:1). 1H NMR (400 MHz, CDCl3) δ (ppm) = 7.46 (br s, 1H, H-13), 7.36 (t, 3J = 8 Hz, 1H, H-5), 7.25–7.19 (m, 1H, H-4), 6.70 (d, 3J = 7.2 Hz, 1H, H-6), 5.69 (s, 2H, H-7), 5.22 (s, 2H, H-11), 5.1 (s, 2H, H-10). 13C NMR (100 MHz, CDCl3) δ (ppm) = 161.8 (Cquat), 141.9 (Cquat), 131.8 (Cquat), 130.7 (Cquat), 129.8 (Cquat), 129.6 (C-5), 128.9 (Cquat), 127.8 (Cquat), 115.6 (C-4), 115.4 (Cquat), 114.7 (C-13), 100.7 (C-6), 76.0 (C-7), 73.0 (C-11), 71.3 (C-10). HRMS (GC EI, 70 eV) calculated for C13H10O2 [M+·] 198.06808, found 198.06980 (Diff.: 1.73 mmu).
5.2.6.24 4-((1-Bromonaphthalen-2-yl)oxy)but-2-yn-1-ol (33)
To a suspension of K2CO3 (15 g, 108 mmol, 4.8 equiv) in acetone (144 mL) was added 1-bromo-2-naphthol (5 g, 22 mmol, 1 equiv) and 4-chlorobut-2-yn-1-ol (2.1 mL, 24 mmol, 1.09 equiv) under argon atmosphere at room temperature. The reaction mixture was refluxed and stirred under argon atmosphere for 24 h. The reaction was quenched with water (150 mL) and extracted with EtOAc (3 × 100 mL). The combined organic layer was washed with a saturated solution of NaHCO3 and brine. Then it was dried over Na2SO4, filtered, and concentrated in vacuo. The crude product was purified by flash column chromatography on silica gel (heptane/EtOAc 8:2) to afford compound 33 as a white solid.
Rf = 0.21 (Pentane/EtOAc 8:2). 1H NMR (400 MHz, CDCl3) δ (ppm) = 8.24 (dd, 3J = 8.4 Hz, 4J = 0.8 Hz, 1H, H-4), 7.83–7.79 (m, 2H, H-9 & 7), 7.60–7.56 (m, 1H, H-5), 7.45–7.41 (m, 1H, H-6), 7.37 (d, 3J = 9.2 Hz, 1H, H-10), 4.94 (t, 5J = 1.6 Hz, 2H, H-11), 4.30 (dt, 3J = 6 Hz, 5J = 1.6 Hz, 2H, H-14), 1.63 (t, 3J = 6 Hz, 1H, OH). 13C NMR (100 MHz, CDCl3) δ (ppm) = 152.3 (C-1), 133.3 (C-3), 130.7 (C-8), 128.9 (C-9), 128.2 (C-7), 127.9 (C-5), 126.5 (C-4), 125.0 (C-6), 115.9 (C-10), 110.5 (C-2), 86.4 (C-12), 80.6 (C-13), 58.2 (C-11), 51.2 (C-14). IR (CDCl3) ν (cm−1) = 3332, 3063, 2924, 2865, 1624, 1594, 1503, 1464, 1351, 1333, 1267, 1224, 1132, 1049, 1019, 801, 764, 747. HRMS (GC FI, 10,000 V) calculated for C14H11BrO2 [M+·] 289.99424, found 289.99327 (Diff.: −3.36 ppm). Mp = 76 °C.
5.2.6.25 1-Bromo-2-((4-(prop-2-yn-1-yloxy)but-2-yn-1-yl)oxy)naphthalene (34)
Compound 34 (yellow solid) was prepared following the general procedure D starting from propargyl alcohol 33 (543 mg, 1.9 mmol).
Rf = 0.29 (Heptane/EtOAc 9:1). 1H NMR (400 MHz, CDCl3) δ (ppm) = 8.24 (br dd, 3J = 8.8 Hz, 4J = 1.2 Hz, 1H, H-4), 7.83–7.79 (m, 2H, H-9 & 7), 7.60–7.56 (m, 1H, H-5), 7.45–7.41 (m, 1H, H-6), 7.39 (d, 3J = 8.8 Hz, 1H, H-10), 4.95 (t, 5J = 2 Hz, 2H, H-11), 4.30 (t, 5J = 2 Hz, 2H, H-14), 4.19 (d, 4J = 2.4 Hz, 2H, H-15), 2.42 (t, 4J = 2.4 Hz, 1H, H-17). 13C NMR (100 MHz, CDCl3) δ (ppm) = 152.3 (C-1), 133.3 (C-3), 130.6 (C-8), 128.9 (C-9), 128.2 (C-7), 127.9 (C-5), 126.6 (C-4), 125.0 (C-6), 116.1 (C-10), 110.7 (C-2), 83.4 (C-12 or 13), 81.8 (C-12 or 13), 78.8 (C-16), 75.2 (C-17), 58.3 (C-11), 56.9 (C-14), 56.7 (C-15). IR (CDCl3) ν (cm−1) = 3290, 3064, 2949, 2918, 2898, 2853, 1624, 1594, 1502, 1464, 1348, 1332, 1266, 1223, 1149, 1127, 1079, 1048, 1020, 801, 765, 747, 642, 520, 413. HRMS (ESI, 120 eV) calculated for C17H13BrO2 [M] 328.00989, found 328.01071 (Diff.: −2.49 ppm). Mp = 57 °C.
5.2.6.26 (3-((4-((1-Bromonaphthalen-2-yl)oxy)but-2-yn-1-yl)oxy)prop-1-yn-1-yl)trimethylsilane (3a)
Compound 3a (colorless oil) was prepared following the general procedure E starting from compound 34 (200 mg, 0.61 mmol).
Rf = 0.46 (Heptane/EtOAc 9:1). 1H NMR (400 MHz, CDCl3) δ (ppm) = 8.24 (br d, 3J = 8.8 Hz, 1H, H-4), 7.81 (br t, 3J = 8.8 Hz, 2H, H-9 & 7), 7.60–7.56 (m, 1H, H-5), 7.45–7.41 (m, 1H, H-6), 7.39 (d, 3J = 8.8 Hz, 1H, H-10), 4.95 (t, 5J = 2 Hz, 2H, H-11), 4.28 (t, 5J = 2 Hz, 2H, H-14), 4.19 (s, 2H, H-15), 0.17 (s, 9H, TMS). 13C NMR (100 MHz, CDCl3) δ (ppm) = 152.3 (C-1), 133.3 (C-3), 130.6 (C-8), 128.9 (C-9), 128.2 (C-7), 127.9 (C-5), 126.6 (C-4), 124.9 (C-6), 116.1 (C-10), 110.6 (C-2), 100.5 (C-16), 92.4 (C-17), 83.4 (C-12 or 13), 81.8 (C-12 or 13), 58.3 (C-11), 57.6 (C-15), 56.9 (C-14), −0.06 (TMS). IR (CDCl3) ν (cm−1) = 3066, 2958, 2896, 2851, 2225, 2173, 1624, 1595, 1502, 1464, 1349, 1250, 1224, 1126, 1080, 1048, 1020, 1000, 842, 80, 762, 644, 528. HRMS (ESI, 120 eV) calculated for C20H21BrO2Si [M] 400.04942, found 400.05059 (Diff.: −2.94 ppm).
5.2.6.27 3,6-Dihydro-5H-1,4-dioxacyclopenta[h]naphtho[2,1,8-cde]azulen-6-yl(trimethyl)silane (12)
A solution of substrate 3a (49.4 mg, 0.12 mmol, 1 equiv) in 1,4-dioxane (1.5 mL) was degassed. Then Pd(OAc)2 (4.4 mg, 20 μmol, 0.16 equiv), P(OPh)3 (10 μL, 38 μmol, 0.31 equiv), and Cs2CO3 (200 mg, 0.61 mmol, 5 equiv) were added. The reaction mixture was degassed again and heated in a sealed tube at 100 °C for 30 min. The mixture was passed through a short Celite pad, concentrated in vacuo, and purified by preparative TLC (UV254) on silica gel (toluene/CH2Cl2 9:1) to afford compound 12 as an orange/pink oil.
Rf = 0.44 (Heptane/EtOAc 9:1). 1H NMR (400 MHz, CDCl3) δ (ppm) = 7.73 (dd, 3J = 6.8 Hz, 4J = 0.8 Hz, 1H, H-7), 7.64 (AB system, JAB = 7.2 Hz, Δ√ = 50.7 Hz, 2H, H-9 & 10), 7.44 (s, 1H, H-11), 7.37 (dd, 3J = 6.8 Hz, 3J = 6 Hz, 1H, H-6), 5.60 (br d, 1H, H-5), 5.14–5.10 (m, 1H, H-14 or 14′), 4.88–4.84 (m, 1H, H-14 or 14′), 4.78–4.76 (m, 2H, H-15), 3.40 (s, 1H, H-17), −0.21 (s, 9H, TMS). 13C NMR (100 MHz, CDCl3) δ (ppm) = 152.9 (C-1), 138.3 (C-11), 134.6 (C-16), 133.1 (C-4), 131.6 (C-8), 127.5 (C-3), 126.8 (C-7 or 9), 126.7 (C-7 or 9), 125.1 (C-6), 120.8 (C-13 or 2), 120.7 (C-13 or 2), 117.7 (C-12), 112.4 (C-10), 79.8 (C-15), 76.6 (C-14), 39.9 (C-17), −1.77 (TMS). MS (GC EI, 70 eV) 320.04 ([M+·], 27%), 72.94 (100%).
5.2.6.28 3,6-Dihydro-5H-1,4-dioxacyclopenta[h]naphtho[2,1,8-cde]azulene (13)
A solution of substrate 3a (39 mg, 0.1 mmol, 1 equiv) in toluene (1.25 mL) was degassed. Then Pd(OAc)2 (3.5 mg, 16 μmol, 0.16 equiv), P(OPh)3 (8 μL, 29 μmol, 0.3 equiv), and Cs2CO3 (157 mg, 0.48 mmol, 5 equiv) were added. The reaction mixture was degassed again and heated in a sealed tube at 110 °C for 30 min. The mixture was passed through a short Celite pad, concentrated in vacuo, and purified by prep. The crude product was directly purified by flash column chromatography on silica gel (heptane/EtOAc 95:5) to afford compound 13 as an orange oil.
Rf = 0.31 (Heptane/EtOAc 9:1). 1H NMR (400 MHz, CDCl3) δ (ppm) = 7.85 (d, 3J = 8 Hz, 1H, H-7), 7.71 (AB system, JAB = 8.8 Hz, Δ√ = 50.4 Hz, 2H, H-9 & 10), 7.52 (s, 1H, H-11), 7.44 (br t, 3J = 7.6 Hz, 1H, H-6), 7.35 (br dd, 3J = 7.2 Hz, 4J = 0.8 Hz, 1H, H-5), 5.05–5.02 (m, 2H, H-14), 4.84–4.82 (m, 2H, H-15), 4.10 (s, 2H, H-17). 13C NMR (100 MHz, CDCl3) δ (ppm) = 153.2 (C-1), 138.8 (C-11), 132.2 (C-13 & 16), 131.7 (C-8), 131.1 (C-3), 128.1 (C-5), 127.9 (C-7), 126.8 (C-9), 125.2 (C-6), 123.5 (C-4), 119.8 (C-2), 116.8 (C-12), 112.5 (C-10), 79.6 (C-15), 76.6 (C-14), 33.7 (C-17). IR (CDCl3) ν (cm−1) = 2953, 2925, 2849, 1248, 1106, 841. HRMS (GC EI, 70 eV) calculated for C17H12O2 [M+·] 248.08373, found 248.08617 (Diff.: 9.82 ppm).
5.2.6.29 (2-Bromo-3-methylphenyl)methanol (35)
A solution of 2-bromo-3-methylbenzoic acid (3.7 g, 17.2 mmol, 1 equiv), 2,2-dimethoxypropane (9 g, 86.3 mmol, 5 equiv), and hydrochloric acid (12 M, 2.9 mL, 34.4 mmol, 2 equiv) in methanol (44 mL) was refluxed overnight. After cooling, the solvent was removed in vacuo. The residue was dissolved in Et2O and washed with water. Organic extract was dried over Na2SO4, filtered, and concentrated in vacuo. Crude product was purified by flash column chromatography over silica gel (heptane/Et2O 97:3) to give the methyl 2-bromo-3-methylbenzoate (3.3 g, 96%) as a colorless oil.
Rf = 0.37 (Pentane/Et2O 95:5). 1H NMR (400 MHz, CDCl3) δ (ppm) = 7.47–7.45 (m, 1H, H-6), 7.34 (ddd, 3J = 7.6 Hz, 4J = 2 Hz, 5J = 0.8 Hz, 1H, H-4), 7.24 (br t, 3J = 7.6 Hz, 1H, H-5), 3.93 (s, 3H, H-9), 2.46 (s, 3H, H-7). 13C NMR (100 MHz, CDCl3) δ (ppm) = 167.9 (C-8), 139.9 (C-3), 134.2 (C-1), 133.2 (C-4), 128.0 (C-6), 127.0 (C-5), 123.2 (C-2), 52.6 (C-9), 24.0 (C-7). IR (CDCl3) ν (cm−1) = 2951, 1729, 1433, 1293, 1193, 1142, 1029, 973, 874, 789, 752, 701, 573. MS (GC EI, 70 eV) 227.79 ([M+·], 38%), 196.79 (100%), 168.81 (23%), 88.92 (43%).
To a suspension of LiBH4 (10 mg, 0.48 mmol, 1.1 equiv) in THF (0.4 mL) was added the methyl 2-bromo-3-methylbenzoate (100 mg, 0.44 mmol, equiv) in Et2O (1.3 mL) at 0 °C. The reaction mixture was then stirred at room temperature overnight, quenched with 0.5 M HCl to pH 6–7, and extracted with Et2O. Combined organic layers were washed with brine, dried over Na2SO4, filtered, and concentrated under reduced pressure. Crude product was purified by flash column chromatography over silica gel (pentane/Et2O 8:2) to give the (2-bromo-3-methylphenyl)methanol 35 (75 mg, 85%) as a white powder.
Rf = 0.16 (Pentane/Et2O 9:1). 1H NMR (400 MHz, CDCl3) δ (ppm) = 7.31–7.29 (m, 1H, H-6), 7.23 (br t, 3J = 7.6 Hz, 1H, H-5), 7.19 (dd, 3J = 7.6 Hz, 4J = 1.6 Hz, 1H, H-4), 4.76 (d, 3J = 6 Hz, 1H, H-8), 2.43 (s, 3H, H-7), 2.09 (t, 3J = 6.4 Hz, 1H, OH). 13C NMR (100 MHz, CDCl3) δ (ppm) = 140.2 (C-1), 138.6 (C-3), 130.2 (C-4), 127.3 (C-5), 126.5 (C-6), 125.3 (C-2), 65.9 (C-8), 23.5 (C-7). IR (CDCl3) ν (cm−1) = 3311, 2973, 2918, 2857, 1576, 1452, 1408, 1372, 1349, 1240, 1103, 1056, 1025, 989, 769, 618. HRMS (GC FI, 10,000 V) calculated for C8H9BrO [M+·] 199.98368, found 199.98480 (Diff.: −5.63 ppm). Mp = 75 °C.
5.2.6.30 2-Bromo-3-methylbenzaldehyde (36)
A solution of the (2-bromo-3-methylphenyl)methanol 35 (100 mg, 0.49 mmol, 1 equiv), PCC (161 mg, 0.75 mmol, 1.5 equiv), and Celite (0.48 mg) in anhydrous CH2Cl2 (4.5 mL) was stirred at room temperature overnight. The mixture was quenched by addition of brine and extracted with Et2O. Combined organic layers were washed with water, brine, dried over Na2SO4, filtered, and concentrated under reduced pressure. Crude product was passed through a silica/Celite plug and concentrated to give 36 (94 mg, 95%) as a white solid.
Rf = 0.57 (Pentane/Et2O 95:5). 1H NMR (400 MHz, CDCl3) δ (ppm) = 10.45 (d, 4J = 0.8 Hz, 1H, H-8), 7.75–7.73 (m, 1H, H-6), 7.48 (ddd, 3J = 7.6 Hz, 4J = 1.6 Hz, 5J = 0.8 Hz, 1H, H-4), 7.32 (br t, 3J = 7.6 Hz, 1H, H-5), 2.48 (s, 3H, H-7). 13C NMR (125 MHz, CDCl3) δ (ppm) = 192.8 (C-8), 139.7 (C-3), 136.4 (C-4), 134.1 (C-1), 129.7 (C-2), 127.52 (C-5 or 6), 127.45 (C-5 or 6), 23.0 (C-7). IR (CDCl3) ν (cm−1) = 3339, 2982, 2920, 2866, 1889, 1695, 1674, 1573, 1448, 1373, 1237, 1031, 912, 779, 690, 536. HRMS (GC FI, 10,000 V) calculated for C8H7BrO [M+·] 197.96803, found 197.96767 (Diff.: −1.80 ppm). Mp = 50 °C.
5.2.6.31 (((4S*,5R*)-5-(2-Bromo-3-methylphenyl)-2,2-dimethyl-1,3-dioxolan-4-yl)ethynyl)triethylsilane (38)
A solution of n-butyllithium (1.75 M in hexanes, 1.2 equiv) was added dropwise to a dried round-bottom flask containing the triethyl(3-((2-methoxypropan-2-yl)oxy)prop-1-yn-1-yl)silane (1.2 equiv) in anhydrous THF (1.40 M) at −78 °C. The reaction was stirred for 30 min at −78 °C. A solution of 2-bromo-3-methylbenzaldehyde 36 (2.2 g, 11.05 mmol, 1 equiv.) in anhydrous THF (0.61 M) was then added at −78 °C. The mixture was stirred for 2 h at −78 °C and then quenched by addition of saturated aqueous NaHCO3 solution and water. After extraction with Et2O, combined organic layers were washed with water, brine, dried over MgSO4, filtered, and concentrated under reduced pressure.
Resulting oil was dissolved in methanol (0.35 M) containing PPTS (0.1 equiv). The solution was stirred for 1 h at room temperature and quenched by the addition of brine. The mixture was extracted with Et2O and combined extract was washed with water, brine, dried over MgSO4, filtered, and concentrated under reduced pressure. The anti and syn diols 37 compounds were not characterized because of inseparable impurities. Acetonide 38 (pale yellow oil) was prepared following the general procedure A starting from diol 37 (anti diol) (51 mg, 0.14 mmol).
Rf = 0.84 (Heptane/EtOAc 8:2). 1H NMR (400 MHz, CDCl3) δ (ppm) = 7.50 (dd, 3J = 7.8 Hz, 3J = 1.7 Hz, 1H, H-6), 7.22 (br t, 3J = 7.6 Hz, 1H, H-5), 7.17 (dd, 3J = 7.6 Hz, 3J = 2 Hz, 1H, H-4), 5.52 (d, 3J = 6.2 Hz, 1H, H-8), 5.38 (d, 3J = 6.1 Hz, 1H, H-9), 2.40 (s, 3H, H-7), 1.71 (s, 3H, H-13 or 13′), 1.50 (s, 3H, H-13 or 13′), 0.76 (br t, 3J = 8 Hz, 9H, H-15), 0.37–0.31 (m, 6H, H-14). 13C NMR (100 MHz, CDCl3) δ (ppm) = 137.8 (C-3), 137.3 (C-1), 130.2 (C-4), 126.9 (C-5), 126.2 (C-6), 124.4 (C-2), 110.4 (C-12), 102.6 (C-10), 90.6 (C-11), 80.3 (C-8), 69.8 (C-9), 27.6 (C-13 or 13′), 26.4 (C-13 or 13′), 23.5 (C-7), 7.3 (C-15), 4.1 (C-14). IR (CDCl3) ν (cm−1) = 2986, 2954, 2911, 2875, 1456, 1380, 1233, 1162, 1077, 1023, 864, 768, 726. HRMS (GC FI, 10,000 V) calculated for C20H29BrO2Si [M+·] 408.11202, found 408.10995 (Diff.: −5.08 ppm).
5.2.6.32 (4R*,5S*)-4-(2-Bromo-3-methylphenyl)-5-ethynyl-2,2-dimethyl-1,3-dioxolane (39)
TBAF (1 M in THF, 5.6 mL, 5.6 mmol, 2.2 equiv) was added dropwise to a cooled solution of compound 38 (1.05 g, 2.57 mmol, 1 equiv) in anhydrous THF (25 mL) at 0 °C. The reaction mixture was stirred 10 min at 0 °C. The mixture was quenched by addition of a saturated aqueous solution of NH4Cl (30 mL). After extraction with EtOAc (3 × 30 mL), the combined organic layers were washed with water, brine, dried over Na2SO4, filtered, and concentrated in vacuo. The crude product was purified by flash column chromatography on silica gel (pentane/Et2O 99:1) to give compound 39 (88%) as a colorless oil.
Rf = 0.35 (Heptane/EtOAc 97:3). 1H NMR (400 MHz, CDCl3) δ (ppm) = 7.54 (dd, 3J = 7.6 Hz, 4J = 2 Hz, 1H, H-6), 7.26 (br t, 3J = 7.2 Hz, 1H, H-5), 7.21 (dd, 3J = 7.6 Hz, 4J = 1.6 Hz, 1H, H-4), 5.52 (d, 3J = 5.9 Hz, 1H, H-8), 5.36 (dd, 3J = 6 Hz, 4J = 2.4 Hz, 1H, H-9), 2.43 (s, 3H, H-7), 2.20 (d, 4J = 2.4 Hz, 1H, H-11), 1.71 (s, 3H, H-13 or 13′), 1.50 (s, 3H, H-13 or 13′). 13C NMR (100 MHz, CDCl3) δ (ppm) = 137.9 (C-3), 136.5 (C-1), 130.5 (C-4), 126.9 (C-5), 126.2 (C-6), 124.4 (C-2), 110.5 (C-12), 80.0 (C-8), 76.2 (C-11), 69.0 (C-9), 27.7 (C-13 or 13′), 26.3 (C-13 or 13′), 23.6 (C-7). IR (CDCl3) ν (cm−1) = 3294, 2986, 2935, 1577, 1454, 1372, 1231, 1160, 1078, 1025, 863, 753, 657. HRMS (GC FI, 10,000 V) calculated for C14H15BrO2 [M+·] 294.02554, found 294.02699 (Diff.: 4.92 ppm).
5.2.6.33 3-((4S*,5R*)-5-(2-Bromo-3-methylphenyl)-2,2-dimethyl-1,3-dioxolan-4-yl)prop-2-yn-1-ol (40)
Propargyl alcohol 40 (colorless oil) was prepared following the general procedure C starting from terminal alkyne 39 (660 mg, 2.24 mmol).
Rf = 0.38 (Pentane/Et2O 7:3). 1H NMR (400 MHz, CDCl3) δ (ppm) = 7.51 (dd, 3J = 7.2 Hz, 4J = 1.6 Hz, 1H, H-6), 7.25 (br t, 3J = 7.6 Hz, 1H, H-5), 7.20 (dd, 3J = 7.2 Hz, 4J = 1.6 Hz, 1H, H-4), 5.56 (d, 3J = 6 Hz, 1H, H-8), 5.36 (td, 3J = 6 Hz, 5J = 2 Hz, 1H, H-9), 4.00–3.91 (m, 2H, H-14), 2.43 (s, 3H, H-7), 1.70 (s, 3H, H-13 or 13′), 1.50 (s, 3H, H-13 or 13′). 13C NMR (100 MHz, CDCl3) δ (ppm) = 138.0 (C-3), 137.0 (C-1), 130.5 (C-4), 126.7 (C-5), 126.1 (C-6), 124.6 (C-2), 110.4 (C-12), 86.5 (C-11), 82.4 (C-10), 80.2 (C-8), 69.2 (C-9), 50.9 (C-14), 29.8 (C-13 or 13′), 27.7 (C-13 or 13′), 23.6 (C-7). IR (CDCl3) ν (cm−1) = 3422, 2985, 2930, 2856, 1453, 1373, 1230, 1128, 1107, 1024, 865, 778. HRMS (GC FI, 10,000 V) calculated for C15H17BrO3 [M+·] 324.03611, found 324.03674 (Diff.: 1.96 ppm).
5.2.6.34 (4R*,5S*)-4-(2-Bromo-3-methylphenyl)-2,2-dimethyl-5-(3-(prop-2-yn-1-yloxy)prop-1-yn-1-yl)-1,3-dioxolane (41)
Compound 41 (colorless oil) was prepared following the general procedure D starting from propargyl alcohol 40 (630 mg, 1.94 mmol).
Rf = 0.24 (Pentane/Et2O 95:5). 1H NMR (400 MHz, CDCl3) δ (ppm) = 7.51 (dd, 3J = 7.6 Hz, 4J = 1.6 Hz, 1H, H-6), 7.26 (br t, 3J = 7.6 Hz, 1H, H-5), 7.20 (dd, 3J = 7.2 Hz, 4J = 1.2 Hz, 1H, H-4), 5.54 (d, 3J = 6 Hz, 1H, H-8), 5.41 (dt, 3J = 6 Hz, 5J = 1.6 Hz, 1H, H-9), 4.07–3.98 (m, 2H, H-14), 3.70 (doublet of AB system, JAB = 16 Hz, 5J = 2.4 Hz, Δ√ = 9 Hz, 2H, H-15), 2.43 (s, 3H, H-7), 2.36 (t, 4J = 2.4 Hz, 1H, H-17), 1.69 (s, 3H, H-13 or 13′), 1.50 (s, 3H, H-13 or 13′). 13C NMR (100 MHz, CDCl3) δ (ppm) = 138.1 (C-3), 137.0 (C-1), 130.5 (C-4), 127.0 (C-5), 126.0 (C-6), 124.4 (C-2), 110.4 (C-12), 83.4 (C-10), 83.0 (C-11), 80.1 (C-8), 79.1 (C-16), 74.7 (C-17), 69.3 (C-9), 56.4 (C-14), 55.8 (C-15), 27.7 (C-13 or 13′), 26.2 (C-13 or 13′), 23.6 (C-7). IR (CDCl3) ν (cm−1) = 3929, 2986, 2936, 2896, 2848, 1453, 1372, 1335, 1229, 1075, 1025, 861, 778, 653. HRMS (GC FI, 10,000 V) calculated for C18H19BrO3 [M+·] 362.05176, found 362.04955 (Diff.: −6.11 ppm).
5.2.6.35 (3-((3-((4S*,5R*)-5-(2-Bromo-3-methylphenyl)-2,2-dimethyl-1,3-dioxolan-4-yl)prop-2-yn-1-yl)oxy)prop-1-yn-1-yl)trimethylsilane (3b)
Compound 3b (colorless oil) was prepared following the general procedure E starting from compound 41 (590 mg, 1.62 mmol).
Rf = 0.13 (Pentane/Et2O 97:3). 1H NMR (400 MHz, CDCl3) δ (ppm) = 7.51 (br d, 3J = 7.6 Hz, 1H, H-6), 7.26 (t, 3J = 7.6 Hz, 1H, H-5), 7.20 (br d, 3J = 7.6 Hz, 1H, H-4), 5.54 (d, 3J = 6 Hz, 1H, H-8), 5.40 (dt, 3J = 6 Hz, 5J = 2 Hz, 1H, H-9), 4.05–3.96 (m, 2H, H-14), 3.69 (AB system, JAB = 15.6 Hz, Δ√ = 8.1 Hz, 2H, H-15), 2.43 (s, 3H, H-7), 1.69 (s, 3H, H-13 or 13′), 1.50 (s, 3H, H-13 or 13′), 0.18 (s, 9H, TMS). 13C NMR (100 MHz, CDCl3) δ (ppm) = 138.1 (C-3), 137.0 (C-1), 130.5 (C-4), 127.0 (C-5), 126.0 (C-6), 124.4 (C-2), 110.4 (C-12), 100.8 (C-16), 91.8 (C-17), 83.3 (C-10 or 11), 83.2 (C-10 or 11), 80.1 (C-8), 69.4 (C-9), 56.7 (C-14), 56.5 (C-15), 27.7 (C-13 or 13′), 26.2 (C-13 or 13′), 23.6 (C-7), −0.03 (TMS). IR (CDCl3) ν (cm−1) = 2958, 1453, 1372, 1336, 1249, 1231, 1166, 1125, 1077, 1026, 999, 840, 776, 759, 652. HRMS (ESI, 120 eV) calculated for C21H27BrO3Si [M] 434.09128, found 434.09222 (Diff.: −2.16 ppm).
5.2.6.36 ((8bR*,11aS*)-10,10-Dimethyl-3,5,8b,11a-tetrahydro-1H-benzo[3,4]furo[3′,4′:7,8]azuleno[1,2-d][1,3]dioxol-4-yl)trimethylsilane (4b)
A solution of substrate 3b (50 mg, 0.12 mmol, 1 equiv) in diisopropylamine (1 mL) was degassed with argon. Pd(PPh3)4 (6.7 mg, 6 μmol, 0.05 equiv) was added. The reaction mixture was degassed again and heated at 100 °C for 22 h (no microwave irradiation). The mixture was filtered through Celite and concentrated in vacuo. Further purification by flash column chromatography over silica gel (pentane/Et2O 9:1) afforded a separable mixture of compounds 4b (12%, pale yellow oil) and 14 (19%, colorless oil).
Rf = 0.18 (Pentane/Et2O 9:1). 1H NMR (400 MHz, CDCl3) δ (ppm) = 7.43 (t, 3J = 7.2 Hz, 1H, H-5), 7.37 (d, 3J = 7.2 Hz, 1H, H-6), 7.11 (d, 3J = 7.6 Hz, 1H, H-4), 5.67 (d, 3J = 6 Hz, 1H, H-8), 5.34 (d, 3J = 5.6 Hz, 1H, H-9), 4.89 (AB system, JAB = 13.6 Hz, Δ√ = 70.3 Hz, 2H, H-14), 4.57–4.49 (m, 2H, H-15), 3.26 (AB system, JAB = 13.6 Hz, Δ√ = 56 Hz, 2H, H-7), 1.49 (s, 3H, H-13 or 13′), 1.26 (s, 3H, H-13 or 13′), 0.22 (s, 9H, TMS). 13C NMR (100 MHz, CDCl3) δ (ppm) = 146.3 (C-16), 139.6 (C-1), 138.7 (C-2), 136.8 (C-11), 135.2 (C-10), 131.7 (C-5), 130.0 (C-3), 128.7 (C-17), 127.6 (C-4), 123.2 (C-6), 112.8 (C-12), 82.4 (C-8), 81.4 (C-9), 72.6 (C-15), 70.6 (C-14), 36.0 (C-7), 27.6 (C-13 or 13′), 26.4 (C-13 or 13′), −0.4 (TMS). IR (CDCl3) ν (cm−1) = 2987, 2949, 2933, 2853, 2835, 1455, 1379, 1371, 1249, 1206, 1159, 1144, 1063, 1036, 837, 764, 689. HRMS (GC FI, 10,000 V) calculated for C21H26O3Si [M+·] 354.16512, found 354.16623 (Diff.: 3.12 ppm).
5.2.6.37 (8bR*,11aS*)-10,10-Dimethyl-3,5,8b,11a-tetrahydro-1H-benzo[3,4]furo[3′,4′:7,8]azuleno[1,2-d][1,3]dioxole (14)
Desilylated compound 14 was prepared following general procedure F starting from substrate 3b (50 mg, 0.11 mmol).
Rf = 0.12 (Pentane/Et2O 9:1). 1H NMR (400 MHz, CDCl3) δ (ppm) = 7.41–7.35 (m, 2H, H-5 & 6), 7.05 (br d, 3J = 7.2 Hz, 1H, H-4), 5.61 (d, 3J = 5.6 Hz, 1H, H-8), 5.53–5.50 (m, 1H, H-17), 5.28 (d, 3J = 5.6 Hz, 1H, H-9), 4.88 (AB system, JAB = 13.6 Hz, Δ√ = 60.5 Hz, 2H, H-14), 4.52–4.44 (m, 2H, H-15), 3.50–3.36 (m, 2H, H-7), 1.48 (s, 3H, H-13 or 13′), 1.27 (s, 3H, H-13 or 13′). 13C NMR (100 MHz, CDCl3) δ (ppm) = 140.5 (C-1), 139.0 (C-16), 138.5 (C-2), 135.9 (C-11), 134.3 (C-10), 131.6 (C-5), 130.2 (C-3), 128.9 (C-4), 123.9 (C-6), 115 (C-17), 112.7 (C-12), 82.1 (C-8), 80.9 (C-9), 72.6 (C-15), 71.5 (C-14), 32.9 (C-7), 27.6 (C-13 or 13′), 26.4 (C-13 or 13′). IR (CDCl3) ν (cm−1) = 2984, 2930, 2852, 1456, 1371, 1248, 1206, 1158, 1061, 920, 868, 836, 769. HRMS (GC FI, 10,000 V) calculated for C18H18O3 [M+·] 282.12559, found 282.12623 (Diff.: 2.25 ppm).
5.2.6.38 2-Bromo-3,6-dimethylphenol (42)
Compound 42 is already known and was synthesized following the literature method [15]. Compound 42 was not separated from 2,4-dibromo-3,6-dimethylphenol.
5.2.6.39 2-Bromo-1,4-dimethyl-3-(prop-2-yn-1-yloxy)benzene (43)
Potassium carbonate (241 mg, 1.74 mmol, 5 equiv) and propargyl bromide (80% in toluene, 45 μL, 1.2 equiv) were added to the mixture of 42 (68 mg, 0.34 mmol, 1 equiv) and 2,4-dibromo-3,6-dimethylphenol in acetone (1.75 mL). The resulting mixture was stirred and refluxed overnight. The reaction was quenched by filtration and the solvent was concentrated in vacuo. The crude product was purified by flash column chromatography on silica gel (heptane/EtOAc 99:1) to give a separable mixture of compounds 43 and 2,6-dibromo-1,4-dimethyl-3-(prop-2-yn-1-yloxy)benzene. Compound 43 was obtained in 79% yield as a white solid from 42.
Rf = 0.20 (Heptane 100%). 1H NMR (400 MHz, CDCl3) δ (ppm) = 6.98 (AB system, JAB = 8 Hz, Δ√ = 33 Hz, 2H, H-4 & 5), 4.65 (d, 4J = 2.4 Hz, 2H, H-9), 2.52 (t, 4J = 2.4 Hz, 1H, H-11), 2.38 (s, 3H, H-8), 2.36 (s, 3H, H-7). 13C NMR (100 MHz, CDCl3) δ (ppm) = 153.6 (C-1), 137.4 (C-3), 130.9 (C-6), 129.6 (C-5), 126.7 (C-4), 120.1 (C-2), 79.1 (C-10), 75.4 (C-11), 60.1 (C-9), 23.2 (C-8), 17.0 (C-7). IR (CDCl3) ν (cm−1) = 3296, 3061, 3021, 2950, 2924, 2863, 2123, 1478, 1455, 1359, 1271, 1169, 1134, 1042, 1009, 991, 807, 672, 635. HRMS (GC FI, 10,000 V) calculated for C11H11BrO [M+·] 237.99933, found 237.99897 (Diff.: −1.51 ppm). Mp = 53 °C.
5.2.6.40 4-(2-Bromo-3,6-dimethylphenoxy)but-2-yn-1-ol (44)
Propargyl alcohol 44 (white solid) was prepared following the general procedure C starting from terminal alkyne 43 (423 mg, 1.77 mmol).
Rf = 0.25 (Heptane/EtOAc 8:2). 1H NMR (400 MHz, CDCl3) δ (ppm) = 6.97 (AB system, JAB = 7.6 Hz, Δ√ = 34.0 Hz, 2H, H-4 & 5), 4.66 (t, 5J = 1.6 Hz, 2H, H-9), 4.33 (m, 2H, H-12), 2.37 (s, 3H, H-8), 2.35 (s, 3H, H-7). 13C NMR (100 MHz, CDCl3) δ (ppm) = 153.5 (C-1), 137.4 (C-3), 130.8 (C-6), 129.6 (C-5), 126.6 (C-4), 120.2 (C-2), 85.5 (C-10), 81.3 (C-11), 60.3 (C-9), 51.3 (C-12), 23.1 (C-8), 16.3 (C-7). IR (CDCl3) ν (cm−1) = 3335, 2922, 2862, 1478, 1453, 1358, 1261, 1169, 1133, 1038, 1006, 976, 805, 638, 520. HRMS (ESI, 120 eV) calculated for C12H13BrO2 [M] 268.00989, found 268.01035 (Diff.: −1.71 ppm). Mp = 51 °C.
5.2.6.41 2-Bromo-1,4-dimethyl-3-((4-(prop-2-yn-1-yloxy)but-2-yn-1-yl)oxy)benzene (45)
Terminal alkyne 45 (pale yellow oil) was prepared following the general procedure D starting from propargyl alcohol 44 (352 mg, 1.21 mmol).
Rf = 0.65 (Heptane/EtOAc 8:2). 1H NMR (400 MHz, CDCl3) δ (ppm) = 6.97 (AB system, JAB = 7.6 Hz, Δ√ = 34.0 Hz, 2H, H-4 & 5), 4.70 (t, 5J = 2 Hz, 2H, H-9), 4.31 (t, 5J = 1.6 Hz, 2H, H-12), 4.21 (d, 4J = 2.4 Hz, 2H, H-13), 2.45 (t, 4J = 2.4 Hz, 1H, H-15), 2.37 (s, 3H, H-8), 2.35 (s, 3H, H-7). 13C NMR (100 MHz, CDCl3) δ (ppm) = 153.5 (C-1), 137.4 (C-3), 130.9 (C-6), 129.6 (C-5), 126.6 (C-4), 120.2 (C-2), 82.5 (C-10 & 11), 79.0 (C-14), 75.1 (C-15), 60.3 (C-9), 56.9 (C-12), 56.6 (C-13), 23.2 (C-8), 17.0 (C-7). IR (CDCl3) ν (cm−1) = 3292, 2923, 2856, 1478, 1455, 1357, 1262, 1134, 1080, 1040, 981, 932, 808, 638. HRMS (ESI, 120 eV) calculated for C15H15BrO2 [M] 306.02554, found 306.02521 (Diff.: 1.1 ppm).
5.2.6.42 (3-((4-(2-Bromo-3,6-dimethylphenoxy)but-2-yn-1-yl)oxy)prop-1-yn-1-yl)trimethylsilane (3c)
A solution of terminal alkyne 45 (750 mg, 2.44 mmol, 1 equiv) and EtMgBr (1 M in THF, 6.5 mL, 6.5 mmol, 2.7 equiv) in anhydrous THF (27 mL) was heated at 51 °C and stirred for 1 h. Trimethylsilyl chloride (freshly distilled, 0.4 mL, 3.13 mmol, 1.3 equiv) was added and the suspension was stirred at 53 °C for 2.5 h. The mixture was quenched by addition of a saturated aqueous solution of NaHCO3 (30 mL) and extracted with Et2O (3 × 30 mL). Combined organic layers were washed with water, brine, dried over Na2SO4, filtered, and concentrated under reduced pressure. Crude product was purified by flash column chromatography over silica gel (heptane/EtOAc 98:2) to afford 3c as a pale yellow oil.
Rf = 0.22 (Heptane/EtOAc 98:2). 1H NMR (400 MHz, CDCl3) δ (ppm) = 6.97 (AB system, JAB = 8 Hz, Δ√ = 33.9 Hz, 2H, H-4 & 5), 4.70 (t, 5J = 2 Hz, 2H, H-9), 4.29 (t, 5J = 2 Hz, 2H, H-12), 4.19 (s, 2H, H-13), 2.37 (s, 3H, H-8), 2.35 (s, 3H, H-7), 0.19 (s, 9H, TMS). 13C NMR (100 MHz, CDCl3) δ (ppm) = 153.5 (C-1), 137.4 (C-3), 130.9 (C-6), 129.6 (C-5), 126.6 (C-4), 120.2 (C-2), 100.7 (C-14), 92.3 (C-15), 82.7 (C-10), 82.3 (C-11), 60.3 (C-9), 57.5 (C-13), 56.9 (C-12), 23.2 (C-8), 17.0 (C-7), −0.04 (TMS). IR (CDCl3) ν (cm−1) = 2958, 2924, 2896, 2851, 1478, 1455, 1346, 1250, 1134, 1082, 1040, 1001, 844, 761. HRMS (ESI, 120 eV) calculated for C18H23BrO2Si [M] 378.06507, found 378.06490 (Diff.: 0.46 ppm).
5.2.6.43 Trimethyl(3-methyl-6,7,8,10-tetrahydrofuro[3′,4′:6,7]cyclohepta[1,2,3-cd][1]benzofuran-7-yl)silane (19)
A solution of Pd(OAc)2 (4.4 mg, 20 μmol, 0.12 equiv) and P(OPh)3 (10 μL, 0.038 mmol, 0.24 equiv) in anhydrous 1,4-dioxane (0.2 mL) was degassed. Then substrate 3c (60 mg, 0.16 mmol, 1 equiv) in anhydrous 1,4-dioxane (1.7 mL) and Cs2CO3 (258 mg, 0.79 mmol, 5 equiv) were added. The reaction mixture was degassed again and heated at 100 °C for 1.5 h under microwave irradiation. The mixture was passed through a short Celite pad, concentrated in vacuo, and purified by flash column chromatography over silica gel (heptane/EtOAc 98:2) to afford compound 19 (40%) as a pale yellow lace.
Rf = 0.44 (Heptane/EtOAc 9:1). 1H NMR (400 MHz, CDCl3) δ (ppm) = 7.38 (s, 1H, H-9), 6.95 (AB system, JAB = 7.6 Hz, Δ√ = 45.6 Hz, 2H, H-4 & 5), 5.08–5.03 (m, 1H, H-12 or 12′), 4.96–4.92 (m, 1H, H-12 or 12′), 4.76–4.67 (m, 2H, H-13), 3.35 (br d, 2J = 15.2 Hz, 1H, H-8 or 8′), 3.05 (dd, 2J = 15.2 Hz, 4J = 4 Hz, 1H, H-8 or 8′), 2.49 (s, 3H, H-7), 2.16 (br t, 3J = 3.6 Hz, 1H, H-15), −0.24 (s, 9H, TMS). 13C NMR (100 MHz, CDCl3) δ (ppm) = 154.1 (C-1), 138.9 (C-14), 138.3 (C-9), 133.2 (C-3), 125.7 (C-5), 125.2 (C-2), 122.9 (C-4), 120.9 (C-10 or 11), 119.6 (C-6), 116.5 (C-10 or 11), 80.2 (C-13), 76.5 (C-12), 33.3 (C-8), 29.9 (C-15), 14.9 (C-7), −1.58 (TMS). IR (CDCl3) ν (cm−1) = 2951, 2925, 2842, 1751, 1248, 1105, 1069, 838, 776. HRMS (GC EI, 70 eV) calculated for C18H22O2Si [M+·] 298.13891, found 298.14060 (Diff.: 5.68 ppm). Mp = 103 °C.
5.2.6.44 2-Bromo-1-methyl-3-((prop-2-yn-1-yloxy)methyl)benzene (46)
Compound 46 (pale yellow oil) was prepared following general procedure D starting from benzyl alcohol 35 (2.286 g, 11.37 mmol).
Rf = 0.58 (Heptane/EtOAc 9:1). 1H NMR (500 MHz, CDCl3) δ (ppm) = 7.31 (br d, 3J = 7.5 Hz, 1H, H-6), 7.22 (br t, 3J = 7 Hz, 1H, H-5), 7.18 (dd, 3J = 7.5 Hz, 4J = 1.5 Hz, 1H, H-4), 4.70 (s, 2H, H-8), 4.27 (d, 4J = 2.5 Hz, 2H, H-9), 2.49 (br t, 4J = 2.5 Hz, 1H, H-11), 2.43 (s, 3H, H-7). 13C NMR (125 MHz, CDCl3) δ (ppm) = 138.6 (C-3), 137.3 (C-1), 130.2 (C-4), 127.1 (C-5), 126.8 (C-6), 125.6 (C-2), 79.7 (C-10), 74.9 (C-11), 71.8 (C-8), 58.0 (C-9), 23.6 (C-7). IR (CDCl3) ν (cm−1) = 3295, 3054, 2977, 2949, 2924, 2852, 1578, 1453, 1348, 1252, 1108, 1081, 1027, 773, 669, 633. HRMS (GC EI, 70 eV) calculated for C11H11BrO [M+·] 237.99933, found 237.99883 (Diff.: −2.09 ppm).
5.2.6.45 4-((2-Bromo-3-methylbenzyl)oxy)but-2-yn-1-ol (47)
Propargyl alcohol 47 (pale yellow oil) was prepared following the general procedure C starting from benzyl propargyl ether 46 (2.583 g, 10.8 mmol).
Rf = 0.22 (Heptane/EtOAc 8:2). 1H NMR (500 MHz, CDCl3) δ (ppm) = 7.30 (dd, 3J = 7.5 Hz, 4J = 1.5 Hz, 1H, H-6), 7.21 (t, 3J = 7.5 Hz, 1H, H-5), 7.18 (dd, 3J = 7.5 Hz, 4J = 2 Hz, 1H, H-4), 4.68 (s, 2H, H-8), 4.34 (dt, 3J = 6 Hz, 5J = 2 Hz, 2H, H-12), 4.31 (t, 5J = 1.5 Hz, 2H, H-9), 2.42 (s, 3H, H-7), 1.60 (t, 3J = 6 Hz, 1H, OH). 13C NMR (125 MHz, CDCl3) δ (ppm) = 138.6 (C-3), 137.3 (C-1), 130.2 (C-4), 127.1 (C-5), 126.8 (C-6), 125.6 (C-2), 85.0 (C-11), 81.9 (C-10), 72.0 (C-8), 58.2 (C-9), 51.3 (C-12), 23.6 (C-7). IR (CDCl3) ν (cm−1) = 3364, 3047, 2920, 2856, 1578, 1452, 1380, 1347, 1252, 1124, 1100, 1077, 1024, 774. HRMS (GC EI, 70 eV) calculated for C12H13BrO2 [M+·] 268.00989, found 268.00918 (Diff.: −2.66 ppm).
5.2.6.46 2-Bromo-1-methyl-3-(((4-(prop-2-yn-1-yloxy)but-2-yn-1-yl)oxy)methyl)benzene (48)
Propargyl ether 48 (pale yellow oil) was prepared following the general procedure D starting from propargyl alcohol 47 (2.16 g, 8.03 mmol).
Rf = 0.44 (Heptane/EtOAc 90:10). 1H NMR (500 MHz, CDCl3) δ (ppm) = 7.30 (dd, 3J = 7.5 Hz, 4J = 1.5 Hz, 1H, H-6), 7.21 (t, 3J = 7.5 Hz, 1H, H-5), 7.18 (dd, 3J = 7.5 Hz, 4J = 1.5 Hz, 1H, H-4), 4.68 (s, 2H, H-8), 4.34 (t, 5J = 1.5 Hz, 2H, H-12), 4.32 (t, 5J = 1.5 Hz, 2H, H-9), 4.27 (d, 4J = 2 Hz, 2H, H-13), 2.45 (t, 4J = 2.5 Hz, 1H, H-15), 2.42 (s, 3H, H-7). 13C NMR (125 MHz, CDCl3) δ (ppm) = 138.6 (C-3), 137.3 (C-1), 130.2 (C-4), 127.1 (C-5), 126.8 (C-6), 125.6 (C-2), 83.0 (C-10 or 11), 81.9 (C-10 or 11), 79.0 (C-14), 75.2 (C-15), 72.0 (C-8), 58.2 (C-9), 57.0 (C-12), 56.7 (C-13), 23.6 (C-7). IR (CDCl3) ν (cm−1) = 3292, 3050, 2952, 2898, 2853, 1579, 1454, 1441, 1343, 1249, 1073, 1026, 932, 887, 774, 669, 635. HRMS (ESI, 120 eV) calculated for C15H15BrO2 [M] 306.02554, found 306.02541 (Diff.: 0.43 ppm).
5.2.6.47 (3-((4-((2-Bromo-3-methylbenzyl)oxy)but-2-yn-1-yl)oxy)prop-1-yn-1-yl)trimethylsilane (3d)
Silylated compound 3d (colorless oil) was prepared following the general procedure E starting from terminal alkyne 48 (2.16 g, 8.03 mmol).
Rf = 0.38 (Heptane/EtOAc 95:5). 1H NMR (400 MHz, CDCl3) δ (ppm) = 7.30 (dd, 3J = 7.2 Hz, 4J = 2 Hz, 1H, H-6), 7.23–7.17 (m, 2H, H-5 & 4), 4.68 (s, 2H, H-8), 4.31 (s, 4H, H-9 & 12), 4.26 (s, 2H, H-13), 2.42 (s, 3H, H-7), 0.18 (s, 9H, TMS). 13C NMR (100 MHz, CDCl3) δ (ppm) = 138.3 (C-3), 137.0 (C-1), 129.9 (C-4), 126.8 (C-5), 126.5 (C-6), 125.3 (C-2), 100.4 (C-14), 92.0 (C-15), 82.6 (C-10 or 11), 81.8 (C-10 or 11), 71.6 (C-8), 57.9 (C-9), 57.2 (C-13), 56.6 (C-12), 23.3 (C-7), −0.37 (TMS). IR (CDCl3) ν (cm−1) = 3048, 2957, 2850, 2173, 1579, 1343, 1249, 1074, 1026, 999, 840, 760. HRMS (GC EI, 70 eV) calculated for C18H23BrO2Si [M+·] 378.06507, found 378.06527 (Diff.: 0.53 ppm).
5.2.6.48 (Z)-Trimethyl(3-(2-(5-methylisochroman-4-ylidene)ethoxy)prop-1-yn-1-yl)silane (20) and 3,7,9,11-tetrahydro-1H-furo[3′,4′:6,7]cyclohepta[1,2,3-de]isochromene (21).
A solution of silylated substrate 3d (51 mg, 0.13 mmol, 1 equiv) in 1,4-dioxane (1.6 mL) was degassed thoroughly. Then Pd(PPh3)4 (15 mg, 13 μmol, 0.1 equiv) and Cs2CO3 (58 mg, 0.18 mmol, 1.3 equiv) were added. The reaction mixture was degassed again and the mixture was heated at 100 °C for 2 × 30 min under microwaves irradiation. The mixture was then filtered through Celite and the solvent was evaporated under reduced pressure. Further purification by flash column chromatography on silica gel (heptane/EtOAc 9:1) gave three compounds, which seemed to be compounds 20 and 21.
Compound 21 (colorless oil): Rf = 0.24 (heptane/EtOAc 90:10). 1H NMR (400 MHz, CDCl3) δ (ppm) = 7.32 (t, 3J = 7.2 Hz, 1H, H-5), 7.10 (d, 3J = 7.6 Hz, 1H, H-4), 7.02 (d, 3J = 7.6 Hz, 1H, H-6), 5.67 (t, 3J = 6.4 Hz, 1H, H-15), 4.69 (s, 2H, H-8), 4.68 (s, 2H, H-12), 4.59 (s, 2H, H-9), 4.46 (d, 4J = 1.6 Hz, 2H, H-13), 3.12 (d, 3J = 6.4 Hz, 2H, H-7). 13C NMR (100 MHz, CDCl3) δ (ppm) = 138.6 (C-14), 134.8 (C-1), 133.6 (C-3), 133.1 (C-11), 130.6 (C-2), 128.9 (C-5), 128.1 (C-10), 127.5 (C-4), 122.4 (C-6), 117.6 (C-15), 71.5 (C-13), 70.4 (C-12), 69.0 (C-8), 68.6 (C-9), 34.5 (C-7). MS (GC–MS) (CI+, NH3): [M + H]+ = 227, [M + NH4]+ = 244.
Compound 20 (yellow oil): Rf = 0.35 (heptane/EtOAc 9:1). 1H NMR (400 MHz, CDCl3) δ (ppm) = 7.19 (t, 3J = 7.6 Hz, 1H, H-5), 7.11 (d, 3J = 7.6 Hz, 1H, H-4), 7.04 (d, 3J = 7.6 Hz, 1H, H-6), 5.50 (t, 3J = 2.4 Hz, 1H, H-11), 4.64 (dd, 2J = 13.2 Hz, 3J = 2.0 Hz, 1H, H-12a or 12b), 4.56–4.51 (m, 4H, H-8a or 8b, 12a or 12b & 13), 4.44–4.36 (m, 2H, H-8a or 8b & 9a or 9b), 4.22 (d, 3J = 12.8 Hz, 1H, H-9a or 9b), 2.26 (s, 3H, H-7), 0.02 (TMS). 13C NMR (100 MHz, CDCl3) δ (ppm) = 150.6 (Cquat), 139.3 (Cquat), 134.8 (Cquat), 124.2 (Cquat), 129.5 (C-4), 127.4 (C-5), 122.7 (C-6), 122.5 (C-11), 81.9 (C-14), 80.7 (C-15), 73.1 (C-13), 71.1 (C-12), 68.1 (C-8), 67.7 (C-9), 20.2 (C-7), −0.55 (TMS). MS (GC–MS) (CI+, NH3): [M + H]+ = 301, [M + NH4]+ = 318.
Acknowledgments
The authors gratefully acknowledge the support of the University of Strasbourg Institute for Advanced Study (USIAS), the University of Strasbourg, and the ‘Centre national de la recherche scientifique’ (CNRS), and the Pierre Fabre Laboratories (J.J.).
Appendix A Supplementary data
The following are the supplementary data related to this article: Synthesis of starting material 1a. Synthesis of starting material 1e. Synthesis of starting material 10. Synthesis of starting material 3a. Synthesis of starting material 3b. Synthesis of starting material 3c. Synthesis of starting material 3d.