

1 Introduction
Layered double hydroxides (LDHs) form a class of synthetic-layered metal-hydroxide minerals. Their synthesis in laboratory is nowadays well mastered and is relatively easy. Moreover, it is possible to easily monitor the synthetic pathways to yield a material with desired properties. One single layer of an LDH contains divalent cations octahedrally coordinated to hydroxide anions [1]. Some isomorphic substitutions of these divalent cations by trivalent cations result in permanent positive charge of the layer [1,2]. During the stacking along the c-axis, the layers' positive charge is balanced by hydrated anions [2–7]. These anions are in general easily exchangeable with more or less important variations in the interlayer distance depending on the size and the spatial arrangement of the guest anion. This therefore offers many possibilities in terms of LDH modification by intercalation of simple or complex anions. Literature reports the intercalation within LDHs of dyes, complexes, enzymes, and anionic surfactants [5,8–11]. These modifications facilitate and extend the application of LDHs in various fields including polymer fillers, flame retardants, and electrode materials [12–15]. One of the most important applications of LDHs is their use as adsorbent in pollution control for organic compounds such as pesticides [10,16–18]. Increased pollution by pesticides requires improved development of remediation and quantification methods. This last aspect useful in environmental science requires both sensitive and accurate tools for fast and quantitative analyses of pesticides. Following these lines, electrochemical methods are increasingly promising because they offer a wide variety of possibilities to build efficient sensors specific to a target compound or pollutant [15,19–21].
Methyl parathion (MP) is an organophosphate insecticide widely used in agriculture. It is considered as an extremely hazardous compound with harmful effects after inappropriate exposure of humans and for the environment [21–23]. Although there are precise limitations of the concentration of MP in the environment, this compound is frequently found in wastewater and crops [19,21]. Therefore the development of reliable, fast, and inexpensive quantification methods for MP remains a daily and important preoccupation for researchers in the fields of environmental science and analytical chemistry. In addition, MP decomposes rapidly and can generate highly toxic compounds such as 4-nitrophenol (4-NP), which is one of its most abundant hazardous metabolites [24]. Thus, the presence of MP in the environment is almost an indication of that of 4-NP [25,26]. Unfortunately, because these two compounds have the same electroactive site (the nitro group (-NO2)), this renders their differentiation difficult by means of electrochemistry. The development of electrochemical analytical tools capable of differentiating while separately quantifying these two species in a medium is of critical importance [25,26]. In electrochemistry, a strategy used to achieve such differentiation is to modify the working electrode with a material or a substance capable of promoting a preferential adsorption of one of these two compounds. By this mean, the signal of the species having the best affinity is greatly improved as compared with the less adsorbed [25–27].
In this work, a nickel-aluminum–based LDH (NiAl LDH) was synthesized and modified with a highly organophilic compound, bis(ethylhexyl)hydrogen phosphate (BEHP; Fig. 1a). The composite material obtained was expected to take advantage of its hydrophobicity to preferentially accumulate MP solubilized in aqueous solution. The synthesized materials were first characterized by X-ray diffraction (XRD), thermogravimetric analysis (TGA), and Fourier transform infrared (FTIR) and subsequently used as modifiers of a glassy carbon electrode (GCE). The modified electrodes were thus applied to the electrochemical quantification of MP in aqueous solution. An important part of this analytical application also includes the detection of MP in the presence of 4-NP at this modified electrode.
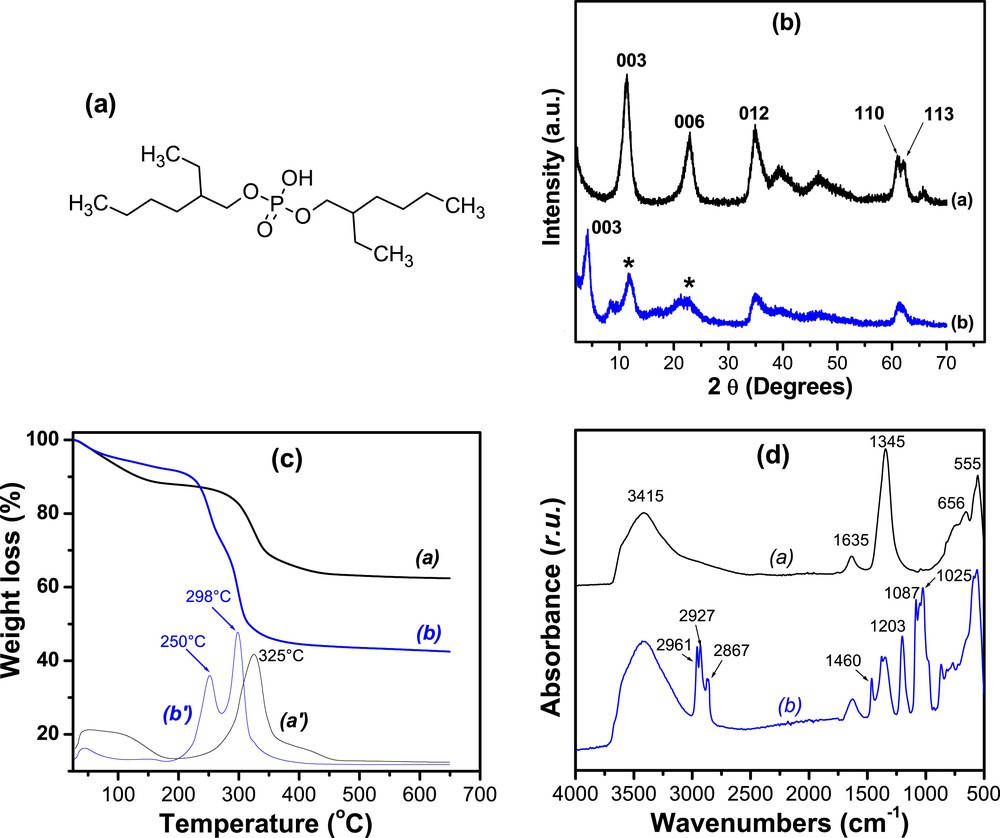
(a) Chemical structure of BEHP; (b) powdered XRD patterns of (curve a) NiAl and (curve b) NiAl–BEHP. (The sign ∗ indicates the peak of residual NiAl present in NiAl–BEHP); (c) TGA and DTG traces of (curves a and a′) NiAl and (curves b and b′) NiAl–BEHP; and (d) FTIR spectra of NiAl (curve a) and NiAl–BEHP (curve b).
2 Experimental section
2.1 Chemicals and reagents
All chemicals and reagents used in the electrochemical section were of analytical grade, and used as received. MP 98.7% and 4-NP 99.7% were purchased from Sigma–Aldrich. A stock solution of MP (10−2 mol L−1) was prepared in ethanol (95%). K3Fe(CN)6 (>99%) and BEHP (99%) both from Prolabo, used, respectively, as redox probe and NiAl modifier were reagent grade. A phosphate buffer solution (PBS) was used as a supporting electrolyte and was prepared by mixing molar solutions of potassium monohydrogen phosphate and potassium dihydrogen phosphate (Riedel-de-Haën). Al2(SO4)3, CaCl2, MgSO4, Ni(NO3)2, Al(NO3)3, and NaOH were of analytical grade. All of the aqueous solutions were prepared using deionized water.
2.2 Preparation of LDH and modified LDH
The NiAl LDH was prepared by coprecipitation method using boiling water, under inert atmosphere to avoid CO32− in the interlayer space [28]. In practice, solutions of nickel nitrate and aluminum nitrate in the molar ratio 3/1 were prepared by dissolving, respectively, 0.045 and 0.015 mol of the corresponding compounds in 50 mL of deionized water previously boiled, under nitrogen atmosphere. The mixture was titrated with 25 mL of a sodium hydroxide solution (2 M) under inert atmosphere. During the synthesis, the temperature was maintained at 25 °C and the pH was kept constant (10.1 ± 0.5). The resulting suspension was then stirred for 16 h, filtered, and the solid obtained was collected, washed, and dried in an oven at 70 °C for 24 h.
NiAl–BEHP was prepared by mixing NiAl with 10% (w/w) BEHP in alcoholic media following the method of Coronado et al. [8]. Briefly, 0.025 g of BEHP was added to 0.25 g of NiAl in 10 mL ethanol, the dispersion was stirred at 175 rpm for 48 h at room temperature. The solution was filtered and the collected solid was washed extensively with ethanol and dried in an oven at 70 °C for 24 h.
2.3 Material characterization
Powder XRD patterns were recorded on a Bruker D5005 diffractometer operating with Cu Kα radiation (λ = 1.54056 Å), using a generator with a voltage of 45 kV and a current of 40 mA.
TGA was performed using a TA instrument Q5000 under nitrogen flow (25 mL min−1) at a heating rate of 10 °C min−1. Approximately 20 mg of the clay material (NiAl or NiAl–BEHP) was placed on the thermobalance, which was purged with helium gas. The measurements were recorded between 50 and 700 °C with a temperature ramp of 10 °C min−1, and data analysis was performed using universal analysis 2000 software package. On each plot, the results were presented by the thermal gravimetric(TG) weight loss curve, and the first derivate of derivative of the thermogravimetric curve (DTG).
FTIR spectra were obtained in attenuated total reflectance (ATR) mode at room temperature in the spectral range 4000–500 cm−1, with a resolution of 4 cm−1 using an Alpha spectrometer from Bruker Optics.
2.4 Working electrode preparation, electrochemical equipment, and procedures
Before modification, the GCE (3 mm in diameter) was polished to obtain a mirror-like surface using alumina slurry on microcloth pads, rinsed thoroughly with deionized water, then sonicated in deionized water for 4 min in an ultrasonic bath, and dried in air. For the preparation of modified electrodes, a dispersion (3 g/L) of LDH or modified LDH was first prepared with deionized water. A volume of 5 μL of the dispersion was carefully deposited on the GCE surface using a micropipette and air-dried for 45 min. The pristine LDH and organo-LDH–modified electrodes referred to as GCE/NiAl and GCE/NiAl–BEHP, respectively, were used as working electrodes for electrochemical investigations.
Cyclic voltammetry and differential pulse voltammetry (DPV) measurements were conducted with a μ-autolab potentiostat equipped with the general purpose electrochemical system (GPES) electrochemical analysis system (Eco Chemie, Holland), and connected to a computer. The voltammograms were recorded under quiescent conditions, immediately after the immersion of the working electrode in a conventional single compartment cell containing the electrolyte and the analyte. The curves were recorded at room temperature, at a scan rate of 50 mV/s, unless otherwise stated. The electrochemical procedure for MP analysis by stripping voltammetry involved two successive steps: accumulation of MP at open circuit condition followed by a voltammetric detection in the same medium by DPV. Precisely, 25 mL of the supporting electrolyte was introduced in the voltammetric cell and the required volume of MP solution was added by a micropipette to obtain a precise concentration of MP. Then the solution was deaerated with nitrogen for 10 min. Preconcentration was achieved by dipping the working electrode in a cell containing the supporting electrolyte solution and MP. During this step, the solution was kept under mild constant stirring. After accumulation for a predetermined time, the DPV curves were recorded.
3 Results and discussion
3.1 Characterization
3.1.1 Structural characterization by XRD
The powder XRD pattern of the pristine material (Fig. 1b (curve a)) presents LDH characteristic peaks. The peaks at 11.4° and 22.8° (2θ) are assigned to the 003 and 006 reflections, respectively [29]. The first peak corresponds to a d003 of 7.75 Å. The presence of well-defined reflections 012, 110, and 113 was also observed, which are frequently used to confirm the good crystallinity of LDH [8,30]. After the reaction of NiAl with BEHP, the 003 and 006 reflections shifted to lower angles (Fig. 1b (curve b)). This usually reflects the increase in the interlayer distance, following the intercalation of bulky compound in the interlayer space of the material [8,30]. In this case, the calculated d003 is 21.02 Å, which corresponds to an increase on the order of 13.27 Å. This result confirms the effective intercalation of BEHP between the NiAl layers. As expected, the reflections 012, 110, and 113 remain unchanged, which is a proof that the reaction of BEHP with NiAl only promotes the swelling of the material [30]. On NiAl–BEHP XRD pattern, one also notices the presence of the 003 and 006 reflections of the unmodified NiAl (at 11.4° and 22.8°, 2θ respectively), which implies that the intercalation of BEHP was incomplete [8,30]. The intercalation performed for more than 16 h led instantly to a deintercalation of BEHP as a result of a driving force for the uptake process of the organic molecule from the interlayer space of the NiAl material. The poor solubility of BEHP in water could explain this result. Such a behavior could probably be prevented by operating in an organic solvent in which BEHP compound is more soluble (e.g., ethanol). However, a pronounced organophilic character of the obtained material could be a drawback of its use as electrode modifier for electrochemical analyses performed in aqueous solution. Also, the low resolution of the NiAl–BEHP XRD pattern also shows that the intercalation process reduces the crystallinity of LDH, probably by partial delamination [31].
3.1.2 Structural characterization by TGA
The thermal analysis of NiAl before and after modification is presented in Fig. 1c. NiAl shows a progressive mass loss of 11.3% in the temperature range between 25 and 170 °C, attributed to the loss of water molecules adsorbed on the external surface of the material or in the interlayer surfaces. From 250 to 450 °C, there is a significant mass loss (25%) in two stages, corresponding to the dehydroxylation and the loss of nitrate ions [32,33]. The first event is fast and characterized by a well-defined DTG with a peak at 325 °C, whereas the second is much slower with a broad and poorly defined peak centered at 400 °C. After BEHP intercalation, important changes in the thermal behavior of the material are observed. The loss of water molecules at temperatures less than 170 °C represents only 6% of the material. The mass loss of 10% centered at 252 °C is attributed to the decomposition of BEHP [30,34,35]. The dehydroxylation step occurs at a much lower temperature (298 °C instead of 325 °C). The significant decrease in the amount of water in NiAl–BEHP is probably because of the partial removal of hydrated nitrate anions from the interlayer space during the intercalation of BEHP. The amount of residual water is mainly attributed to hydrated nitrate anions cointercalated with BEHP and the fraction of nonintercalated LDH still present in the composite as shown in XRD results. The decrease in the dehydroxylation temperature after intercalation is very common in the case of hydroxylated lamellar materials, including aluminosilicates [36,37]. Indeed, the intercalated compound facilitates heat diffusion between the layers, which can lead to a remarkable lowering of the dehydroxylation temperature [38].
3.1.3 Characterization by FTIR
In Fig. 1d, the FTIR spectrum of NiAl shows a wide band centered at 3415 cm−1 characteristic of the stretching vibration of hydrogen-bonded physisorbed and intercalated water molecules [3,37]. Similarly, the band at 1635 cm−1 is assigned to the bending deformations of these water molecules. The very intense band at 1345 cm−1 is due to nitrate ions present in the interlayer space [29,30,39]. The band characteristic of metal–oxygen bond stretching appears at 655 cm−1 and the sharp band at 555 cm−1 is caused by various lattice vibrations associated with metal hydroxide sheets [30,39].
Upon intercalation of BEHP, new bands associated with the presence of the organic modifier are observed. It is the case of the intense aliphatic C–H stretching vibration bands between 2850 and 2970 cm−1. The band at 1460 cm−1 corresponds to the bending vibration of these aliphatic C–H bonds [30,40]. The less intense band at 1203 cm−1 is assigned to the PO bond. The bands between 1080 and 1030 cm−1 are due to the P–O–C bond [30,41]. The significant decrease in intensity of the bands corresponding to nitrate and that of the bending vibration of water molecules is also observed. This clearly shows that BEHP is intercalated in the anionic form by replacing the hydrated nitrate. This observation confirms the results of the thermal analyses, which indicated a significant decrease in the amount of water in the material after modification with BEHP.
3.1.4 Structural characterization by electrochemistry
The reactivity of a material at a given modified electrode strongly depends on the properties of that material. Thus, to get precise information about the influence of the functional groups of the organo-LDH, this material was characterized by ion-exchange voltammetry, by the means of film-modified electrodes. Yet, permselectivity studies based on ion exchange properties constitute a proper mean to characterize the ability of an electrode material, especially when this later can enhance the local concentration of an electrochemical probe because of favorable interactions [42]. Cyclic voltammetry is a convenient method for the monitoring of ion exchange properties of electrode materials. Fig. 2a presents the signal recorded on the bare GCE between −0.3 and 0.6 V, formed by a stable, fast (ΔE = 100 mV), and reversible (iPc/iPa = 1.08) electrochemical signal, which was attributed to the electronic transformation involving [Fe(CN)6]3−/4− redox system [19]. When the electrode was modified by NiAl (Fig. 2b), during the first scan, a poorly intense signal was observed, because of the barrier effect caused by the nonconductive NiAl film. Because this material is an excellent anionic exchanger, a gradual increase in the intensities of the signals with the number of scans was noticed. The saturation of the film results in the superimposition of the signals after about 50 scans. This result is a proof that the LDH film accumulates [Fe(CN)6]3− ions by anion exchange mechanism [43]. At equilibrium, the signal obtained is approximately 2-fold more intense than that recorded on the bare GCE (Fig. 2d). The NiAl film locally increases the concentration of [Fe(CN)6]3− near the surface of the GCE by accumulation through anion exchange.
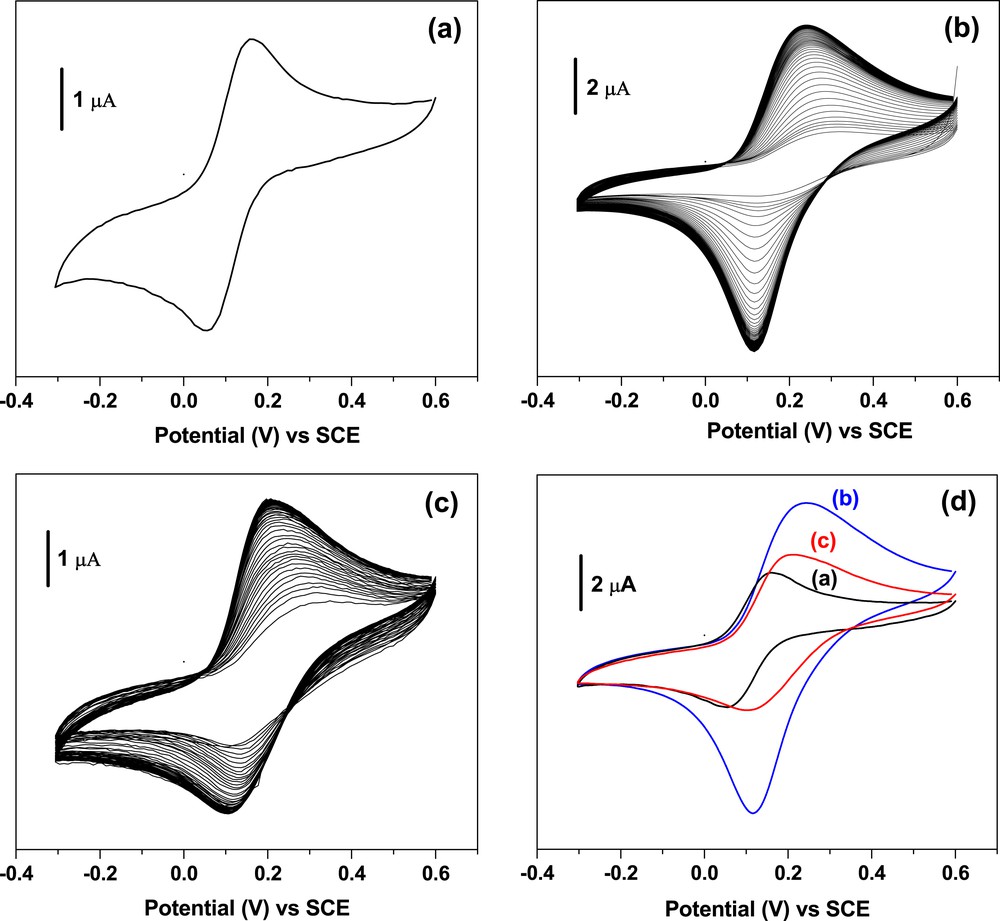
Multicyclic voltammograms of 10−4 M [Fe(CN)6]3− recorded in 0.1 M NaCl on (a) bare GCE, (b) GCE/NiAl, and (c) GCE/NiAl–BEHP. (d) Superimposition of voltammograms at equilibrium recorded on (curve a) bare GCE, (curve b) GCE/NiAl, and (curve c) GCE/NiAl–BEHP. Potential scan rate: 100 mV/s. SCE, saturated calomel electrode.
By covering the GCE by a film of NiAl–BEHP (Fig. 2c), a behavior similar to that obtained on NiAl film was observed. However, lower current intensities and faster film saturation were obtained. The lower currents can be explained by the decrease in the anion exchange capacity of the modified material. This shows that BEHP intercalates in the anionic form and when compared with nitrate ions (in NiAl) displays poor exchange ability. The observed accumulation is probably because of the unmodified LDH fraction present in the composite as shown by XRD and FTIR results. Overlaying the signals at equilibrium (Fig. 2d) showed that the electrode modified with NiAl–BEHP has a signal of intensity comparable to that of the bare electrode and thus approximately 2-fold less than that recorded on the NiAl film electrode.
3.2 Application of NiAl–BEHP as electrode material for the determination of MP
3.2.1 MP behavior on bare and modified GCEs
Multisweep cyclic voltammograms of a solution of MP were recorded on GCE and on both modified electrodes (Fig. 3). The three electrodes exhibit the typical reported electrochemical signal of MP [19,44]. During the first reduction scan, only the irreversible reduction of the nitro group to the hydroxylamine group (-NHOH) according to reaction (1) was observed. During the reverse oxidation scan, the oxidation of this hydroxylamine group to yield the nitroso group (-NO) according to reaction (2) was noticed. During the second scan, the nitroso group was converted to the parent hydroxylamine group. There was also a fast decrease in the signal intensities assigned to the reduction of the nitro group. This reduction has been widely reported in the literature and is associated with the reduction of the active surface of the electrode, caused by the adsorption of MP reduction products [19,22,44,45].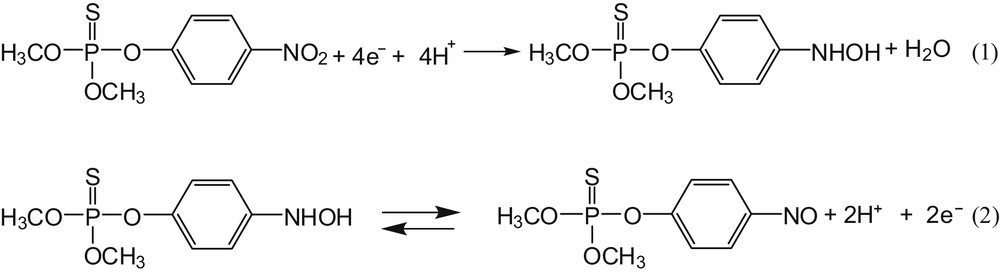
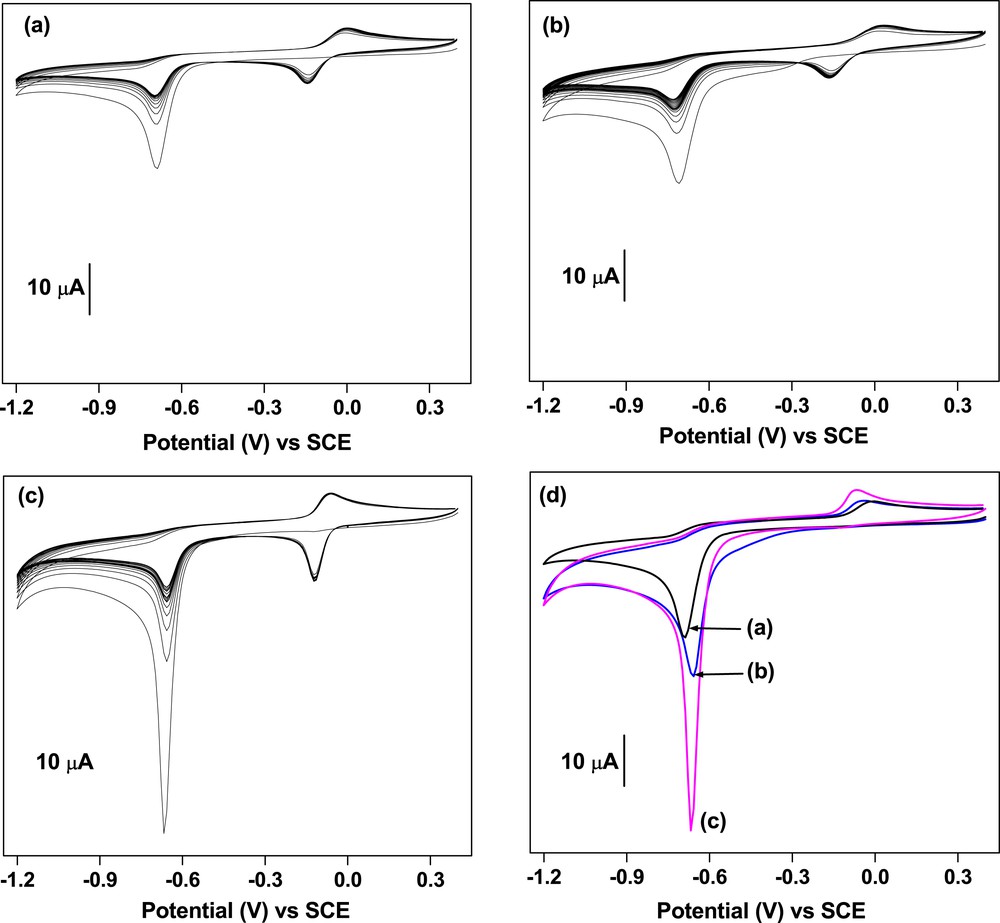
Multisweep cyclic voltammograms of 5 × 10−5 M MP recorded in 0.1 M PBS (pH 7) on (a) bare GCE, (b) GCE/NiAl, and (c) GCE/NiAl–BEHP. (d) Superimposition of the first scans recorded on (curve a) GCE, (curve b) GCE/NiAl, and (curve c) GCE/NiAl–BEHP. Potential scan rate: 100 mV/s.
However, there are some remarkable differences in the electrochemical behavior of MP at different electrodes. As compared with GCE/NiAl–BEHP, the signals recorded on bare GCE and GCE/NiAl are less intense. Indeed, the current associated with the reduction of the nitro group on GCE/NiAl–BEHP was 2-fold more intense. Further observations of Fig. 3d show that the peak potential has shifted to less cathodic values (−690 mV for GCE and −666 mV for GCE/NiAl–BEHP). This potential displacement combined with the important gain in current intensities suggests a beneficial effect of the presence of the film on the signal of MP. This advantageous effect is explained by the presence of NiAl–BEHP, which improves the organophilic nature of the working electrode. Because MP is a highly organophilic compound (its solubility in water is only 24 mg/L), there will be a strong attraction (through van der Waals interactions) between this compound and the NiAl–BEHP film [19], inducing an accumulation of the pesticide at the vicinity of the active surface of the electrode. It was also noticed that the reversible system is better defined and more intense on GCE/NiAl–BEHP. Moreover, on this electrode, this system is very stable (only small variation in current intensities was observed after several cycles). Such stability could be judiciously exploited for the quantification of MP in aqueous solution.
On GCE/NiAl–BEHP, by varying the scan rate in the range of 10–200 mV/s, excellent linearity was obtained when plotting the peak currents associated with nitro group reduction and those associated with oxidation of the hydroxylamine group as a function of the square root of the scanning rate (Fig. SI 1). According to Randles Sevcik's equation, this is a proof that both processes are controlled by mass transfer through the modified film.
As concluding remarks, one can notice that NiAl–BEHP can be used as electrode modifier to build an electrochemical sensor for MP detection. DPV is one of the most advanced and efficient techniques usually used for the detection at low concentrations of inorganic and organic compounds [46]. This technique was hereafter exploited for the sensing of MP.
3.2.2 Quantitative analysis of MP
The improvement in the electrochemical signal of MP when the NiAl–BEHP film was coated at the surface of GCE motivated its use for the quantitative electroanalysis of this pesticide. We were first interested in both the reversible signal (-NO/-NHOH system) and that corresponding to the reduction of the nitro group, although the latter is very unstable. However, good reproducibility of this signal was obtained when the electrochemical cell was slightly agitated after the recording of a signal. Only 2.8% variation of the signal was obtained for a series of 10 consecutive measurements. This is a proof that the chemical species that reduces the activity of the electrode are weakly adsorbed. Because the reduction signal of the reversible system was more intense than the oxidation one (as shown by cyclic voltammetry results), it was therefore used for the quantitative analyses experiments. However, this signal was not observed when performing a direct potential scan in the suitable potential range (between 0.25 and −0.40 V), because the nitroso group that produces this signal was not initially present in the electrolytic solution. To solve this problem, a procedure consisting of an electrolysis step at −0.8 V immediately followed by scanning step in the potential range 0.25 to −0.4 V was used. During the electrolysis step, there is the reduction of the nitro group to yield the hydroxylamine group at the vicinity of the electrode surface according to reaction (1) (see Section 3.2.1.). During the scanning step, hydroxylamine group is first oxidized to nitroso group followed by its reduction to hydroxylamine (reaction (2)). Preliminary results on the reproducibility of this signal were performed to justify its potential use as a quantitative tool for MP detection. A series of 10 consecutive signals were recorded in a 0.1 M PBS (pH 6.67) containing 5 μM MP (Fig. SI 2). The current intensities show a variation of less than 0.7%. This confirms the exceptional stability of the signal of nitroso reduction on GCE/NiAl–BEHP.
Some experimental parameters have been investigated to optimize MP detection. They include the pH of the electrolytic solution, the electrolysis potential, and the electrolysis time.
3.2.2.1 Effect of the pH on MP signal
The pH range investigated was between 4 and 11. The results obtained are shown in Fig. 4: a variation in the pH has shown a notable effect on the reduction and peak currents of both the nitro and the nitroso groups. In contrast to the nitroso group, the peaks are intense in acidic media in the case of the reduction of the nitro group (Fig. 4a and b). The observed effect is a displacement of the peak potentials toward more cathodic values (Fig. 4a and b). This effect on the potential was expected with respect to the electrochemical reactions that involve the consumption of protons to achieve the reduction reactions. Under these conditions, proton-rich media is expected to favor electron transfer at the electrode surface (which results in less cathodic reduction potentials in acidic media). In the particular case of the reduction of the nitroso group, the plot of the peak potential as a function of the pH of the electrolytic solution is a straight line whose slope is −0.048 V/pH unit. This value is close to the standard value of −0.059 V/pH, indicating that equal numbers of electrons and protons are involved in the electrochemical reaction. In the case of the nitro group, however, a poor linearity (R2 = 0.94) is observed in the pH range 4–11, with a slope of 0.015 V/pH. This result is not in agreement with the proposed electrochemical reaction (2) [47,48], which predicts an equivalent number of electrons and protons exchanged during the process. This can be due to some variation in the reaction mechanism depending on the pH of the electrochemical solution. Also, the poor variation in the peak potential at pH values greater than 8 (as compared with lower pH values) could explain such a result.
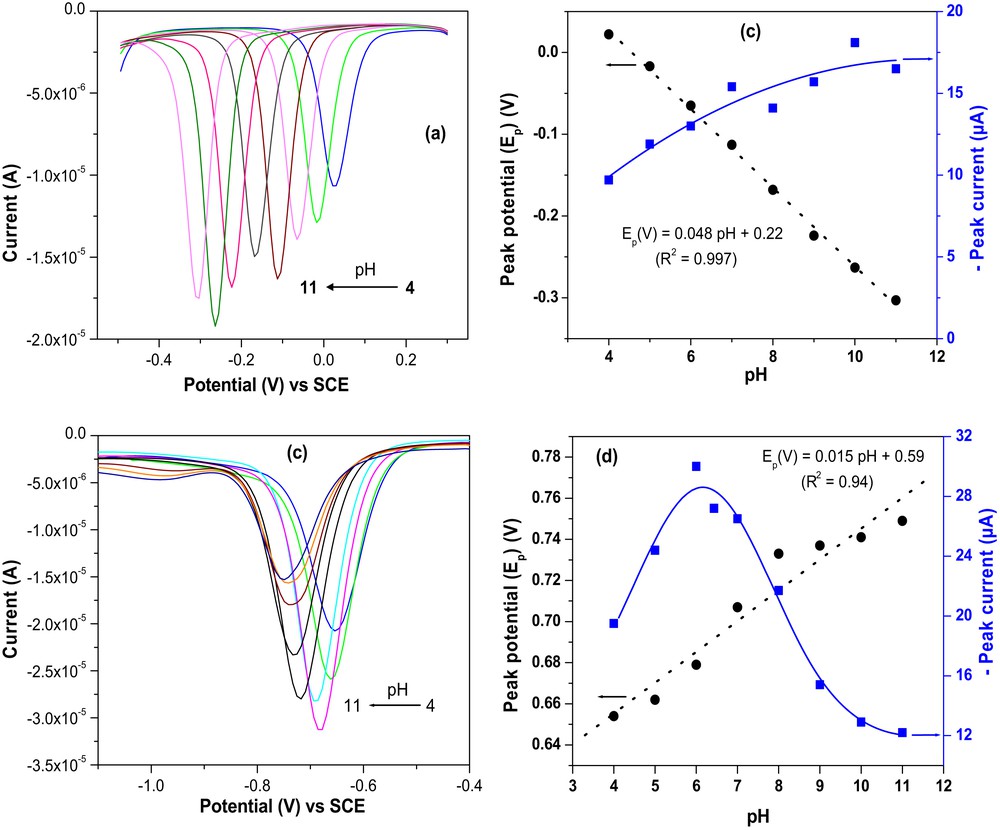
Effect of pH of the detection media on the reduction of (a) 5 × 10−5 M of nitro and (b) 5 × 10−5 M of nitroso groups on GCE/NiAl–BEHP. Influence of the pH on the peak potential and peak intensity (c,d).
There is also a pH effect on the measured current intensities. For the nitroso group, the general trend is a slight increase in the reduction current with an increase of pH. For the nitro group, current intensities are higher in acidic media and drop sharply when pH becomes basic. The best compromise for the pH of the electrolytic solution corresponds to the values close to 7. For this reason, for the further experiments, the PBS was used at pH value close to 7 (6.67).
3.2.2.2 Effect of the electrolysis potential and electrolysis time on the reduction signal of the nitroso group
Signal recording of the nitroso group reduction needs a crucial electrolysis step to quantitatively generate the hydroxylamine group. Thus, the effects of the electrolysis potential and the electrolysis time have been scrutinized. The results obtained are shown in Fig. 5: one can realize that a potential lower than −0.6 V is required to obtained a quantitative electrolysis of MP. However, for more cathodic potentials, a gradual decrease in signal intensity is observed, probably because of competitive reactions (such as water hydrolysis with hydrogen evolution) that favor the fast diffusion of the reduced MP from the electrode surface to the bulk solution (Fig. 5a). A potential of −0.8 V was selected as the optimal to investigate other parameters. This value is in agreement with the peak potential of the MP obtained with cyclic voltammetry studies (−0.67 V). It is also noted that the peak current increases rapidly with the electrolysis time in the first 5 s (Fig. 5b). Surprisingly, at the time greater than 20 s there is a slow and gradual decrease in the reduction current. Although one can expect that a long electrolysis time ensures the quantitative conversion of the MP, very high electrolysis time can promote the diffusion of the electrolyzed species from the surface of the electrode to the bulk solution. For this purpose, 10 s was selected as the optimal electrolysis time.

Effect of (a) electrolysis potential and (b) electrolysis time on the peak intensity reduction of nitroso groups. DPV was performed for 5 × 10−5 M MP in 0.1 M PBS at pH 7.
3.2.2.3 Calibration curves and interference study
The calibration curves plotted for nitro and nitroso group reduction in the concentration range of 0.5–12 μM are reported in Fig. 6. For low MP concentrations (0.5–3.5 μM), the method was more sensitive when the nitro group is used (Fig. SI 3). The sensitivity recorded was 0.79 μA μM−1, a value 5.6-fold higher than that obtained when the reduction of the nitroso group was used (0.14 μA μM−1). This sensitivity decreases at high MP concentration (3.5–7 μM) only when the nitro group was considered (Fig. SI 3). The resulting sensitivity drops down to a value of 0.48 μA μM−1. This value is still 3.4-fold greater than the sensitivity obtained for nitroso group reduction. Although the sensitivity obtained with the nitroso group is smaller, the range of linearity of the sensor is much greater (0.5–12 μM). This is an undeniable advantage because for applications in real environmental systems, such a signal will be less sensitive to interfering species and moreover, will be effective even for high MP concentrated solutions without the need for a prior dilution step. The reduction of this function was exclusively used for further experiments. The detection limit (DL) of the investigated analyte, defined as the MP concentration yielding an analytical peak equal to the minimum detectable one, can be calculated as DL = 3Sb/m [49] where Sb (μA) is the standard deviation of the blank and m (μA μM−1) is the slope of the linear regression equation. In this work, the estimated DL was 2.28 × 10−8 and 12.4 × 10−8 mol L−1 for nitro and nitroso groups, respectively. A comparison of the performance of GCE/NiAl–BEHP including the limit of detection and the linear range with those reported in the literature is shown in Table 1. These results indicate that the proposed sensor exhibited DLs lower than those reported by some previous works with the same aims.
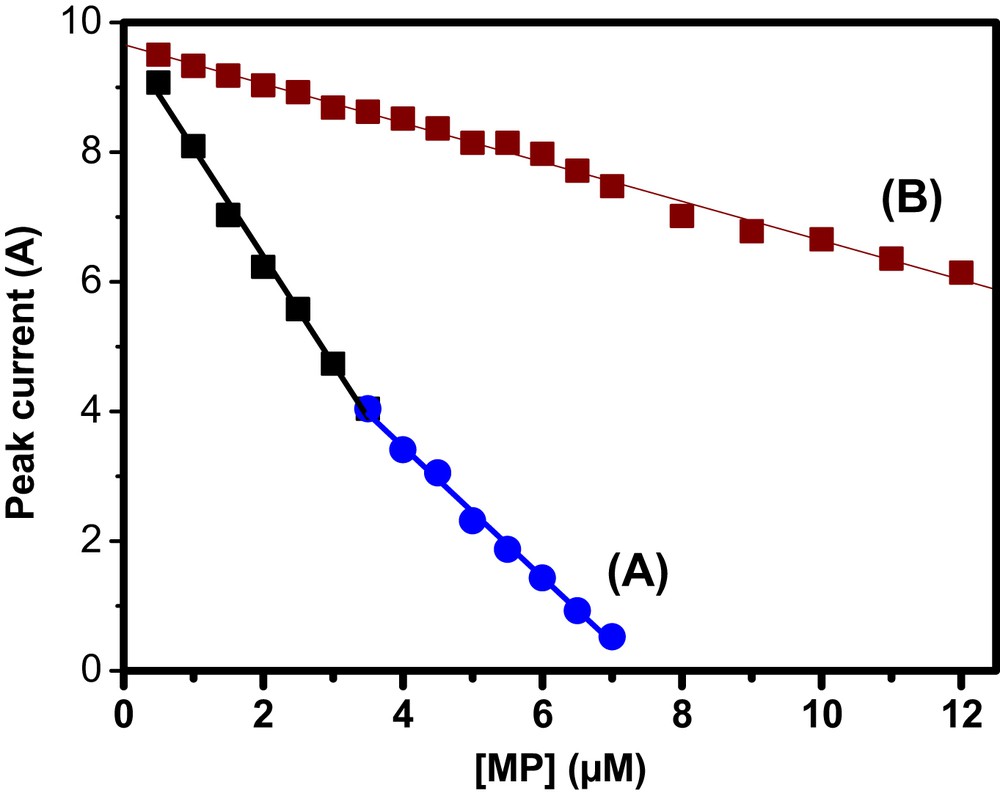
Calibration curves obtained under optimized conditions in 0.1 M PBS (pH 6.7) for (curve A) nitro groups [MP] = 0.5–7 μM) and (curve B) nitroso groups ([MP] = 0.5–12 μM).
Comparison of the efficiency of some modified electrodes in the electrochemical determination of MP.
| Electrode configuration | Linear range (μmol/L) | Detection limit (μmol/L) | Reference |
| Smectite-gemini surfactant/GCE | 0.1–8.5 | 0.07 | [19] |
| ZrO2 NPs/CPEa | 0.019–1.14 | 0.0076 | [22] |
| 1,2-Tetradecanediol/kaolinite/GCE | 2–14 | 0.09 | [44] |
| Hydroxyapatite nanopowder/GCE | 1–300 | 0.6 | [48] |
| Mercury dropping electrode | 0–114 | 15.3 | [50] |
| Unmodified CPE | 1–60 | 0.05 | [51] |
| C18-modified CPE | 0–0.046 | 0.030 | [52] |
| Heteropolyacid-montmorillonite clay–modified/GCE | 0.076–2.66 | 0.076 | [53] |
| GCE/GRGO/[Co(bpy)3]b | 0.05–1700 | 0.0029 | [54] |
| AChE-SF/MWNTsc/GCE | 3.5–2000 | 0.5 | [55] |
| Nafion-coated/GCE | 0–15 | 0.05 | [56] |
| LDHs-GNsd/GCE | 0.0114–0.19 | 0.0023 | [57] |
| Organobentonite modified/GCE | 0–200 | 1 | [58] |
| SiNPs/MWNTs/AuNPse/Au | 0.0038–19 | 0.0011 | [59] |
| ZrO2 nanoparticles modified/Au | 0.02–0.38 | 0.011 | [60] |
| GCE/NiAl–BEHP | 0.5–3.5 | 0.023 | This work |
a ZrO2 NPs/CPE: ZrO2 nanoparticles–modified carbon paste electrode.
b GRGO/[Co(bpy)3]: reduced graphene oxide/cobalt 2,2′-bipyridine.
c AChE-SF/MWNTs: acetylcholinesterase–silk fibroin/multiwall carbon nanotubes.
d GNs: graphene nanosheets.
e SiNPs/MWNTs/AuNPs: silica nanoparticles/multiwall carbon nanotubes/gold nanoparticles.
The performance of the proposed sensor for the quantification of MP in the presence of some potential interfering chemical compounds including Al2(SO4)3, MgSO4, and CaCl2 that could be found with MP in real samples has been studied. MP concentration was set at 50 μM, and the concentration of each interfering compound was progressively set at 50, 500, and 5000 μM (i.e., factors of the MP initial concentration of 1, 10, and 100). The experiments were carried out using only the reduction of the nitroso function. The results obtained show that these chemical species do not interfere when their concentration is equal or 10 times greater than the MP concentration. For much higher concentration (100 times), a decrease in the expected current was observed. Indeed, there was a current drop of about 12% for Al2(SO4)3, 8% for CaCl2, and 24% for MgSO4.
Using the best experimental conditions, the voltammetric method was applied to the determination of MP in a river water sample (collected downtown Yaounde, Cameroon) using the standard addition method. A volume of 25 mL of the river water sample was first analyzed on the basis of the optimized parameters established herein, and MP was not detected. By spiking the collected sample with 3 μM of MP and then submitting it to direct analysis by DPV, a signal related to MP was clearly detected. The exploitation of the calibration graph allowed us to recover 98% of the spiked value, which is 2.94 μM. The relative standard deviation on five assayed samples was 1.18. This indicated that the proposed method could serve for the analytical determination of MP in the sample.
3.2.2.4 Detection of MP in the presence of 4-NP
4-NP is the most important degradation compound of MP. It results from MP hydrolysis in basic media as shown in reaction (3). Therefore, MP solution after a long storage time is likely to contain significant amounts of 4-NP as degradation byproduct. Given the chemical similarity between these two compounds, an analytical tool capable of distinguishing MP from 4-NP is of key importance.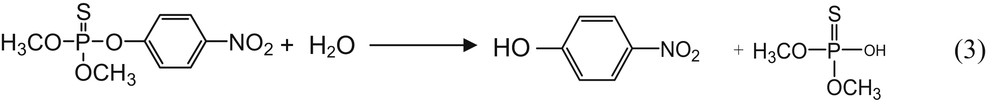
During preliminary experiments, we found that in a solution containing an equimolar mixture of the two compounds, the signal of the nitro group gives two signals with closed peak potentials (ΔE = 120 mV). On the other hand, by applying the strategy using the nitroso group reduction (electrolysis step followed by the recording of the nitroso group signal), two distinct signals well separated with ΔE = 174 mV were obtained (Fig. 7). The less intense signal of 4-NP was attributed to its hydrophilic character that limits its accumulation on the surface of the electrode modified by a hydrophobic material.
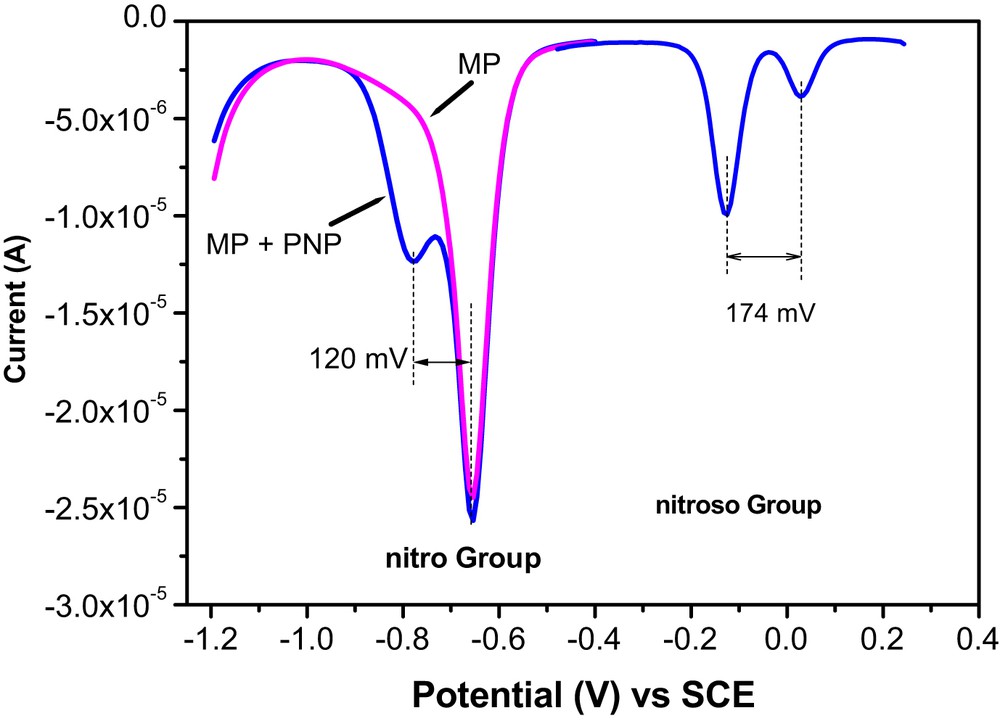
DPV curves recorded in optimized conditions for equimolar mixture of 50 μM MP + 4-NP nitro group and nitroso on GCE/NiAl–BEHP.
When the concentration of 4-NP was set at 1 μM, the MP current increases linearly with concentration in the range of 0.5–10 μM (Fig. 8a). The sensitivity and the DL obtained for MP were 0.13 μA μM−1 and 14 × 10−8 M, respectively. However, when MP concentration was set at 1 μM and 4-NP concentration varied in the range of 0.5–10 μM, a poor correlation (R2 = 0.95) was obtained for the plot of the intensity of current as a function of the concentration of 4-NP (result not shown). When the concentration of the two chemical species was varied simultaneously (Fig. 8b), MP showed an excellent linear increase in the current as a function of the concentration (R2 = 0.99) as compared with 4-NP (R2 = 0.94).
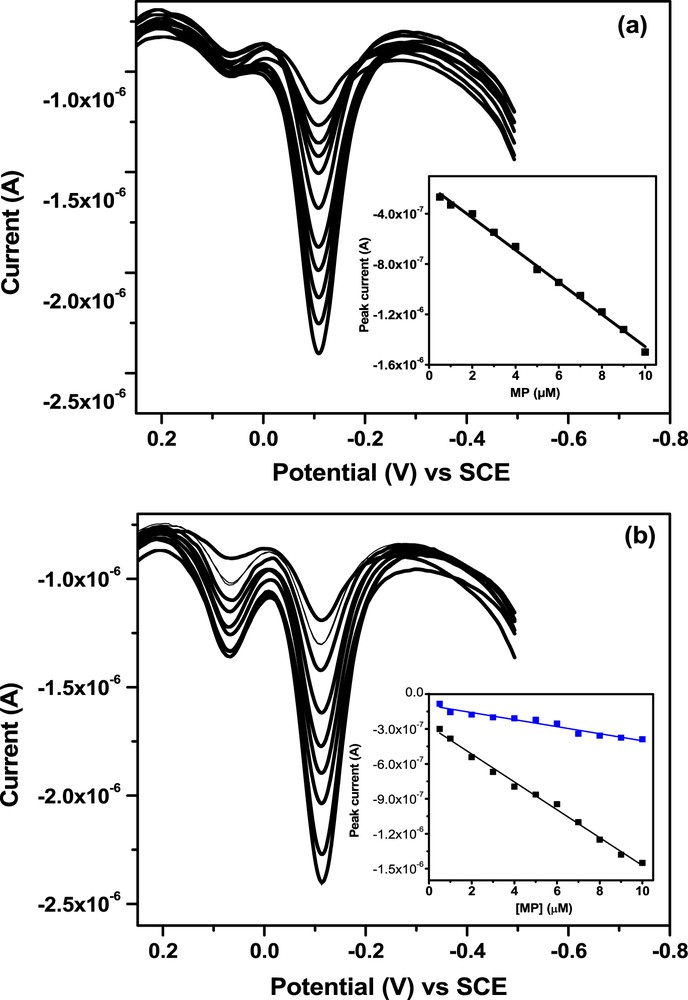
(a) DPV curves recorded under optimized conditions for the detection of MP in the presence of 4-NP at a constant concentration of 4-NP (1 μM) and (b) simultaneous variation in the two species concentration between 0.5 and 10 μM. The inset in (b) shows the calibration curve of 4-NP (blue) and MP (black).
The resulted sensitivities and DLs were −0.12 μA μM−1 and 15 × 10−8 mol L−1 for MP and −0.03 μA μM−1 and 59 × 10−8 mol L−1 for 4-NP. This result clearly shows that the hydrophobic nature of the electrode material favors the electrochemical detection of hydrophobic compounds as compared to the hydrophilic ones.
4 Conclusions
Harmful effects of pesticide upon exposure explain the development of reliable and efficient sensors for their identification and quantification in natural environments. In this context, the intercalation of BEHP in the interlayer space of a NiAl LDH yields an organic–inorganic hybrid material used to build an electrochemical sensor effective for the analysis of MP in water at a trace level. By exclusively using the reduction of the nitroso function, by far more stable as a quantification tool, it was found that
- • when the concentration of some interfering species likely to be found in the same natural environments as MP was equal or 50 times greater than MP concentration, the sensor was poorly affected;
- • it was possible to differentiate MP from 4-NP, one of its most abundant degradation products, which presents similar electroactive sites;
- • the presence of 4-NP caused only a minor decrease in the performance of the sensor for MP quantification (the sensitivity decreases from 0.14 to 0.13 μA μM−1); and
- • the quantification of 4-NP was not accurate (R2 = 0.94) due to the hydrophobicity of the electrode material, which prevented the accumulation of the highly hydrophilic 4-NP.
These results showed that a judicious choice of the intercalated compound in the NiAl material can allow a good separation of the electrochemical signals of chemically similar species. Such an approach could find an excellent application in the elaboration of solid stationary phases in liquid chromatography.
Acknowledgments
Financial support from The Academy of Sciences for the Developing World (TWAS) for the Advancement of Science in Developing Countries (grant no. RGA 16-515 RG/CHE/AF/AC_G1) is gratefully acknowledged. The authors thank the International Science Programme (ISP, Sweden) for its support to the African Network of Electroanalytical Chemists (ANEC).