



 CC-BY 4.0
CC-BY 4.0
1. Introduction
In Nature, enzymes are proteins that ensure the efficiency and selectivity of complex chemical transformations under physiological aqueous conditions. The key feature for catalysis is an appropriate macromolecular 3D folding to bring in close proximity catalytic partners (amino acid side-chains, co-factors …) while discriminating between potential substrates and assuring the final specificity outcome. Nucleic acids also display 3D complex structures with the unmatched advantages of a programable folding and dynamic renaturation processes (unlike proteins). Until the beginning of the 80s and the discovery of RNA acting as an enzyme with self-splicing properties (coining the term RNAzyme), the role of nucleic acids was thought to be only devoted to the storage and transfer of genetic information [1, 2]. DNA was not even primarily considered, assuming that an inert chemical nature would provide an evolutionary advantage by ensuring the absence of undesired alterations of genetic instructions. The major hurdles to overcome were four canonical nucleotides with limited functionalities (mostly involved into Watson and Crick base pairing) and no hydroxyl group at the 2′-position of the sugar. It took another decade to demonstrate that DNAzymes, single-stranded deoxyoligonucleotides (ODN) with no in vivo counterparts, were also capable of catalytic activities that could match those of enzymes [3, 4]. Both aptamers (oligonucleotides able to bind a selected target but with no catalytic properties) and DNAzymes could be selected in vitro by the iterative and powerful SELEX method [5, 6], relying on the use of unmodified nucleosides 5′-triphosphate (dNTP). These nucleotides are substrates of (mutant) DNA polymerases that can be integrated during the DNA amplification step of the polymerase chain reaction (PCR). It is wisely assumed that a library composed of 1015 sequences (obtained from a randomized region made of 25 nucleotides) is sufficient to identify a sequence of interest by exploring a sufficiently diverse chemical space [7]. To achieve such catalytic properties, DNAzymes were able to compensate for the absence of the 2′-hydroxyl group (in comparison with RNA counterparts) by a less restrictive sugar puckering, leading to complex architectures [8, 9, 10] or unusual secondary structures such as triplex structures [11]. DNA secondary structures adopt local conformations that could influence the pKa values of functional groups in the same magnitude as that observed in enzymes [12]. Taking these remarks into consideration, it has been experimentally confirmed that DNAzymes could be as efficient as their RNA counterparts in the Diels–Alder reaction [13]. Moreover, DNA displays intrinsic qualities in terms of fast and effective automated supported synthesis (SPS) combined with increased chemical stability compared with RNA. Nucleic-based biomimetic catalysis could definitively benefit from these former qualities combined with the DNA aptamer property and capacity to be selected owing to in vitro biomolecular methodologies.
However, it rapidly emerged that modifications of the native DNAzyme backbone were required when moving toward biological and/or clinical applications. Switching to a phosphorothioate linkage combined to a 3′-inverted nucleotide was proven to be efficient in avoiding nuclease degradation, relying on the solutions already developed for the antisens approach [14]. Most of the work was devoted to structural modifications of the sugar moiety, alongside the classical 2′-O-methyl modification. For example, DNAzymes based on XNAs (for Xenobiotic Nucleic Acid) [15, 16] and in particular on FANAs (2′-Fluoro-Arabino Nucleic Acid) were demonstrated to be less sensitive to nuclease digestion while maintaining catalytic RNA-ligating [17] or -cleaving activities [18, 19]. FANAzymes present the additional advantage of being obtained by in vitro evolution using non-engineered DNA polymerases. It is also of great interest to mention the ligase-catalyzed oligonucleotide polymerization (LOOPER) method developed by Hili’s laboratory [20]. Pentanucleotides anticodons encode modifications through functionalization of the 5′-end nucleotide with chemical scaffolds varying from alkyl chains and phenyl rings to organic functions while maintaining a 3′-endvariable dinucleotide sequence for degeneracy. The combination of SELEX and LOOPER processes allows the selection of functional aptamers against human alpha-thrombin bearing diverse chemical groups with high binding affinity and not relying on the usual G-quadruplex structuration. Although this approach was concentrated to improve aptamer affinities, it also emphasizes the added value of increasing the DNA chemical repertoire toward its functional role in terms of recognition and ultimately biomimetic catalysis. We decided to narrow the scope of the review to covalently modified monomers (either dNTP or phosphoramidites) that lead to functionalized (deoxy)oligonucleotides (FuON) with catalytic enhancement purposes to mimic biochemical reactions. We will then first describe FuON with enzyme-like properties that catalyze reactions on nucleic acids substrates, such as the extensively studied DNA catalyzed-RNA cleavage. The focus will then be shifted to a broader reactivity scope as described by the development of FuON, which displays peroxidase-mimicking activity and DNA-based proteases.
As the chemical strategies to introduce modifications have been reviewed in the literature by numerous groups [21, 22] and ourselves [23], we will only present the functionalized monomers of interest in charge of the catalytic activities.
2. Functionalized oligonucleotides as ribonuclease A mimics
Screening for new DNA catalysts candidates in the general context of degrading viral RNA in a sequence-specific fashion was inspired by the ribonuclease A (RNase A) responsible for cleaving the phosphodiester bond. The target, being a RNA substrate, presents the tremendous advantage of bypassing the recognition hurdle, as DNA is indeed complementary to RNA. RNase A catalyzes the cleavage of the P-5′-O-bond to give two RNA products bearing a 5′-OH termini and a 2′,3′-cyclic phosphodiester on the 3′-side of a pyrimidine nucleotide, which is further hydrolyzed to a 3′-monophosphate [24]. During the first step of the 2′-O-trans-phosphorylation, the imidazole ring of Histidine 12 (His12) acts as a general base to increase the nucleophilic character of the 2′-hydroxyl, allowing intramolecular in-line attack on the adjacent phosphorus atom. On the other hand, the leaving ability of the 5′-oxygen is increased because of the protonation provided by the imidazolium side chain of His19.
Considering the key role of two histidine side chains in the general acid–base catalytic mechanism, the first modified DNAzyme selected to cleave RNA was named “16.2-11” and decorated with imidazole groups [25]. On a suitably protected 5-(3-aminopropenyl)-2′-deoxyuridine, the 4-imidazoleacrylic acid moiety was linked through an amide coupling to the primary amine before converting the resulting nucleoside either to its corresponding 5′-triphosphate 1 (for the SELEX process) or phosphoramidite 2, for the solid-phase synthesis of the most active DNAzymes (Figure 1).
C5-Functionalized nucleotides bearing an imidazole moiety (CE = cyanoethyl).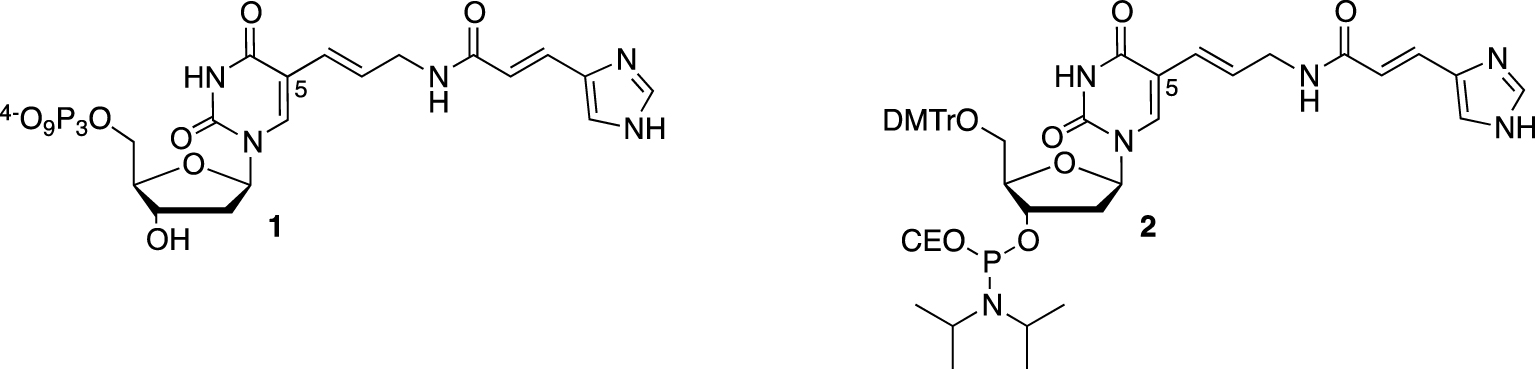
Introduction of modifications at the C5 position on pyrimidines (or at the N7 or C8 positions on the purine base) does not impair recognition by DNA polymerases during the enzymatic polymerization of dNTP inherent to the SELEX process. Moreover, Watson and Crick base pairing is also maintained during duplex formation, which is a key feature for efficient RNA recognition. The overall structure of such functionalized DNAzymes was described by a catalytic core containing four imidazole residues located close to the cleavage site and two binding arms complementary to the RNA substrate. Despite displaying high efficiency (kobs < 1.5 min−1), sequence specificity, and turnover (the DNA binding arms being short enough to denature the duplex structure once the RNA strand was cleaved), the presence of divalent metal ions (in particular Zn2+) was required. This metal ion dependency could be explained by Zn2+ ions either playing a structural role in folding the DNAzyme in an active conformation or being part of the mechanism, activating the 2′-hydroxyl or a molecule of water (or both). Although encouraging, this result indicated that imidazole modifications were not sufficient to bypass the need for metal cations catalysis.
Lysine 31 (Lys31) was also proposed to be involved in transition state stabilization, and the lack of a cationic function needed for the general acid–base mechanism might be the main reason for M2+-dependent catalysis. This rationale developed by Perrin’s laboratory was confirmed by the selection of the first independent DNAzyme 925-11 bearing both imidazole and ammonium functionalities with kobs < 0.2 min−1 [26, 27]. This approach relied on two dNTP 3 and 4 to introduce the different modifications on a purine or pyrimidine base, respectively (Figure 2).
Functionalized nucleotides used for the selection of RNase A mimics.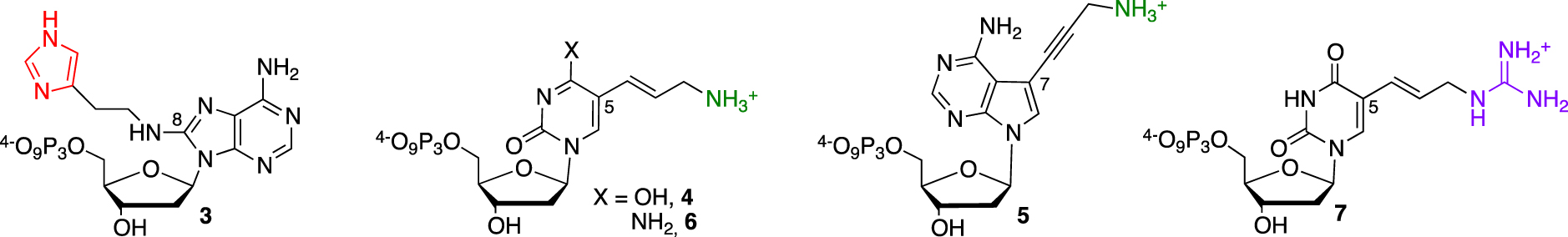
First, the substitution of the bromine atom by histamine onto 8-bromo-2-deoxyriboadenosine-5′-monophosphate and subsequent conversion led to the corresponding (8-(2-(4-imidazolyl)ethylamino)-2′-deoxyriboadenosine-5′-triphosphate (dAImTP) 3. Along with commercial 5-(3-aminoallyl)-2′-deoxyribouridine-5′-triphosphate (dUaaTP) 4, these compounds were used for in vitro selection. 925-11 was only able to cleave a single ribonucleotide bond embedded into a DNA substrate (Figure 3, left). To evaluate its trans-catalytic properties, M2+-independent 925-11 presenting a minimal yet active structure (represented in italic, Figure 3, left) was synthesized de novo by SPS from the corresponding phosphoramidites of 3 and 4. Kinetic studies indicated a kcat of ∼1.5 × 10−2 min−1, a 3-fold decrease compared with the previously observed cis cleavage. However, a high substrate specificity against the target d(GCGTGCC)rCd(GTCTGTT) was observed as a drastic reduced cleavage for degenerate sequences was measured.
Predicted secondary structures of (left) DNAzyme 925-11 [26] (in italic the mimimal catalytic core [27]) with a single ribophosphodiester substrate; (middle) DNAzyme clone 32 with an all-RNA target [28]; (right) DNAzyme 7-38-22t with an all-RNA target [29]. (Blue letters: ribonucleotides; black letters: deoxyribonucleotides; star colored letters: modified nucleotides; black arrows indicate the cleavage site.)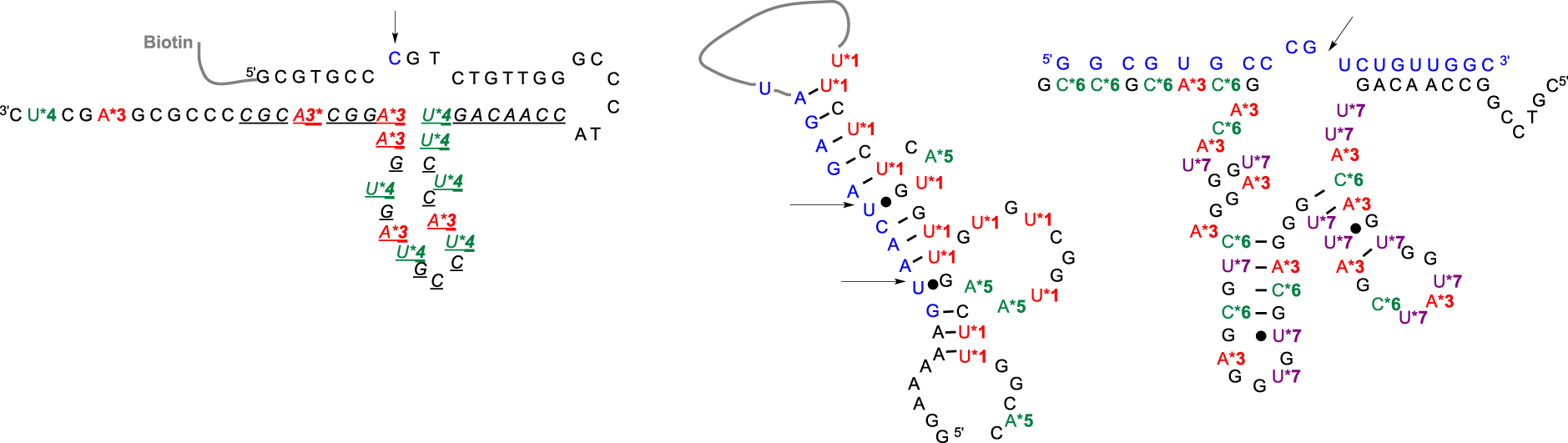
In contrast, subsequent work by Williams et al. led to the cleavage of substrates made only of RNA [28] (Figure 3, middle). They relied on previously described nucleotide 1 to introduce imidazole moieties and designed 7-aminopropynyl modified 7-deaza-dATP 5 (Figure 2). They observed that the introduction of modifications at this position on the adenine base was less destabilizing within a duplex, facilitating the PCR step, combined with a better tolerance of DNA polymerases toward a more rigid linker such as the propenyl group [30]. After a palladium-catalyzed coupling reaction between 7-deaza-7-iodo desoxyadenosine and N-propynyltrifluoracetamide followed by a deprotection step, the resulting nucleoside was converted into its corresponding dATP 5. This DNAzyme clone 32 did present a M2+-independent cis catalytic activity on a 12nt target RNA sequence with a weak kobs of 0.07 min−1, still a 105 rate improvement compared to the uncatalyzed RNA cleavage. The cleavage was site-specific, with the scissile bond located opposite the loop region presenting both imidazole and amino residues. The authors of those studies concluded on the unknown precise role of those appended functionalities, as they could either participate directly in the catalytic mechanism or be involved in the folding DNAzyme process leading to an active structuration, thus advocating for further investigations. Building on those first discoveries, other functionalized RNA-cleaving DNAzymes were developed as thoroughly reviewed by Hollenstein [31] and others [32, 33]. The long-term research work from Perrin’s laboratory was rewarded by the design of an efficient DNAzyme able to mimic RNase A catalysis in the absence of divalent metal cations. During selection, the authors combined the already developed 8-histaminyl-deoxyadenosine (dAimTP) 3, the 5-aminoallyl-deoxycytidine (dCaaTP) 6, an analog to dUaaTP, with 5-guanidinoallyl-deoxyuridine (dUgaTP) 7, presenting a cationic group known to increase thermostability by electrostatic complementarity with the phosphodiester negative charge (Figure 2).
DNAzyme 7-38-22 was found to cleave a 19nt long all-RNA substrate in the absence of M2+ ions and with multiple turnovers, displaying a rate constant of 4.9 min−1 for self-cleavage [29]. Designed to easily allow the evaluation of trans-catalysis, the DNazyme was able to cleave (kcat = 0.27 min−1 with multiple turn-overs) with a 20-fold selectivity the UG bond of a 19nt all-RNA substrate embedded into a four bases bulge that was located (as usually observed) in front of a fully modified catalytic core (Figure 3, right). They then demonstrated that the presence of an imidazole ring, which is thought to be required as a key player for acid–base catalysis, could be bypassed by solely introducing both cationic amine and guanidine functions to select self-cleaving DNAzymes with higher activities than some imidazole-decorated DNAzymes [34].
Site-selective modification of the catalytic core of 10-23 DNAzyme was explored through SPS by introducing functionalized 2′-deoxy(deaza)adenosine phosphoramidites with imidazole, guanidinium, or amino group either at the C-6, C-7 or C-8 onto the purine base. The “chemical evolution” of the A9 or A12 position confirmed the necessity of maintaining the Watson and Crick base pairing (as expected), whereas other modifications did not improve cleaving activity [35]. Other phosphoramidites bearing different linkers with a terminal amino group at the 2′-position were synthesized to evaluate the impact of the modifications of the known conserved positions of the 8-17 DNAzyme. The catalytic effects of a Ca2+-mediated reaction were found to be limited [36].
Apart from the progress that could arise from the optimization of SELEX conditions, there is also room for chemical input in designing new functionalized nucleotides with different linkers or even functionalities, considering that no clear rationale has emerged to pinpoint the beneficial positions to modify a given DNAzyme.
As indicated, RNA cleavage mediated by DNA usually requires the presence of bivalent cations. Consequently, DNAzymes have been selected for metal detection and biosensing purposes; however, when unmodified, metal selectivity remains low [37]. The introduction of additional coordination sites via an organic ligand onto the ODN could improve such selectivity. This approach was recently described using a glycil-histidine functionalized tertiary amine introduced onto a RNA-cleaving DNAzyme, which enhanced selectivity toward the desired divalent metallic ions [38]. Oxidation also requires a metallic center in charge of the catalytic function. A recent example from Martell’s group demonstrated the use of a DNA double helix as a scaffold to spatially pre-organize two co-catalysts (4-carboxy-TEMPO and 4,4′-dicarboxy-2,2′-bipyridine) involved in synergistic Cu-TEMPO alcohol oxidation [39]. Conjugated through an amide link at the 5′-end or 3′-end, respectively, of a matched DNA duplex, both are positioned at an optimal distance, as confirmed by the 70-fold activity enhancement in catalyst turnover number compared with the unscaffolded oxidation reaction; however, no specificity for the various tested alcohol substrates was observed. We decided to focus on the field of peroxidase-like mimics because studies have exemplified the intrinsic DNA properties in terms of structuration, recognition, and catalysis, which are combined in functionalized oligonucleotides purposefully designed to recognize their substrates and perform the subsequent catalytic reaction.
3. Functionalized oligonucleotides as horseradish peroxidase and phosphatase mimics
Hemin/G-quadruplex structures were well-known to act as horseradish peroxidase (HRP)-mimicking catalysts [40]. These unmodified short single-stranded oligonucleotides present sub-micromolar affinities toward the hemin metallofactor they were selected against. The DNA backbone is organized in a highly ordered G-quadruplex structure, acting as an apoprotein mimic by interacting in a supramolecular fashion through π–π stacking interactions with the hemin in charge of the enhanced (NADH) peroxidase activities [41]. Most are used for biosensing applications as the oxidation of substrates, either ABTS (2,2′-azinobis(3-ethylbenzothiazoline-6-sulfonic acid) by H2O2 to ABTS∙+ or luminol induces a color change or generates chemoluminescence, respectively [42].
Willner’s team developed an elegant concept based initially originated from combining a DNAzyme (in charge of the catalytic activity) with an aptamer with a short TATA sequence between the two nucleic parts. In this case, the aptamer is recognized by hydrogen bonding its substrate to oxidize, acting as a binding pocket. They anticipated that close proximity between partners would be beneficial, inspired by RNA-cleaving DNAzymes in which the catalyst and RNA target substrate are kept in close proximity through Watson and Crick base pairing. Those “nucleoaptazymes” (as called) mimic the main features in the active site of native enzymes, namely catalysis and recognition. To the hemin/G-quadruplex horseradish peroxidase-mimicking DNAzyme was tethered one aptamer selected against L-DOPA to perform H2O2-mediated oxidation, leading to the chromophoric aminochrome with a 20-fold enhanced activity for the modified DNAzyme [43]. One of the key features is the facilitated release of the product after oxidation due to its decreased affinity toward the aptamer pocket, allowing a possible multiple turnover, as observed in native enzymes. A modest enantioselectivity was also observed for the oxidation of L-DOPA against its D-enantiomer, which was correlated with differences in affinities. However, the development of efficient unmodified nucleoaptazymes, as defined by Willner’s team, faced two issues [44]. First, even close proximity between partners is not sufficient to ensure an optimum (catalytic) conformation. Second, despite a relatively furnished catalog of DNAzymes and aptamer sequences, finding relevant DNAzyme–aptamer couples that can perform a designed reaction is not trivial.
To overcome this former limitation, the same team proposed a new family of functionalized nucleoaptazymes by combining the dopamine binding aptamer (DBA) sequence while substituting the DNAzyme part by a metallic ligand such as Fe3+ or Cu2+-terpyridine derivatives to perform the oxidation of L-dopamine, mimicking the catechol oxidase activity (Scheme 1) [45]. These oligonucleotides were functionalized by introducing a single metal complex ligand to chelate cupric or ferric ions in the right oxidation state because the native enzyme presents an oxidized CuII–CuII met state center with histidine side chains as proximal ligands in both metal binding sites [46]. Those FuON were synthetized through a convertible approach by reacting the 5′- or 3′-amino DBA sequence with the corresponding 4′-carboxylic acid of 2,2′:6′,2′′-terpyridine, resulting in the formation of a stable amide bond between both partners.
Schematic representation of a nucleoaptazyme constituted by a Dopamine Binding Aptamer linked to a Cu2+-terpyridine moiety at its 5′-end by a 4T sequence.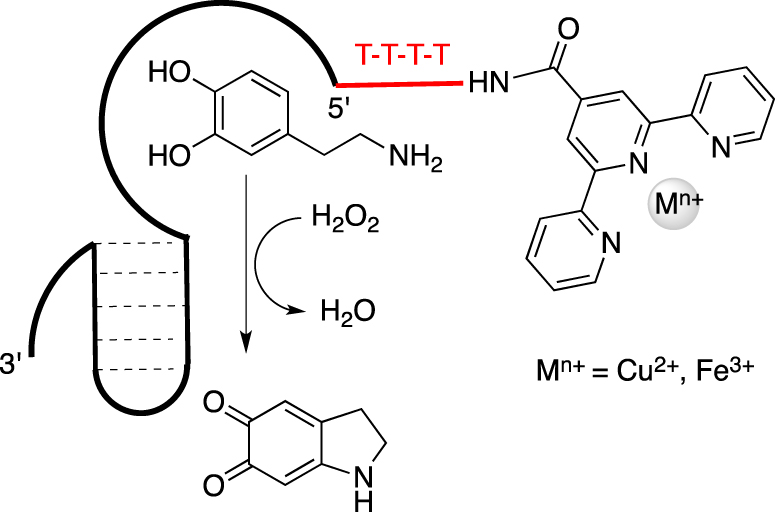
They demonstrated the beneficial presence of a covalent flexible linker made of four thymidines (T4) between the metallic part and the aptamer binding site because oxidation does not occur efficiently when both sites are separated.
The best nucleoaptazymes present the T4 linker at the 5′-end with a kcat of 40 × 10−4 s−1 and 267 × 10−4 s−1 for the Cu2+- and Fe3+-terpyridine complexes, respectively. This result highlights the impact on catalysis of the nature of the metal ion within the complex. As expected, the spatial organization adopted by the tethered partners also played a key role in the catalytic enhancement, as confirmed by the molecular dynamic simulations. An application of this concept was described for the bioconjugation of tyrosine derivatives using N-methyl luminol. The impact of this aptamer-assisted catalysis could be demonstrated by the 12-fold enhancement observed when the aptamer sequence binds to the tyrosine substrate [47].
The presence of metal ions is also crucial in the phosphatase mimics field because the native mechanism relies on the presence of a Zn2+ dinuclear site. The Zn2+ ions coordinate and activate the hydroxyl side chain of the serine that attacks the phosphorus atom of the monophosphoester bond while maintaining the right geometry for substrate binding through a hydrogen bonding network [48, 49]. Several teams have investigated the design of these challenging DNAzymes as phosphatase mimics. Unfunctionalized DNAzymes were previously demonstrated by Silverman’s team to be either in charge of the phosphorylation of the tyrosine side chain of a peptide substrate or able to catalyze the reverse process, i.e., the dephosphorylation step with multiple turnovers and a kobs of 0.19 min−1 [50, 51]. Building on the nucleoaptazyme concept, Willner’s team also designed phosphatase mimics with a bis-Zn2+-pyridyl-salen-type complex tethered to an ATP aptamer [52]. Relying again on amine bond formation, an ATP aptamer (pdb 1AW4) bearing an amino function at the 5′- or 3′-end was covalently linked to a bis-Zn2+-pyridyl-salen-type complex. In this case, an optimal distance composed of a dithymidine linker at the 3′-end between the two partners is sufficient to ensure both flexibility and cooperative proximity. As the promising measured kcat (7 min−1) shows a 103 times lower efficiency than the native enzyme, it appeared that the future FuON would still have room for improvement.
4. Functionalized oligonucleotides as serine protease mimics
As illustrated with the last described biomimetic approach and for asymmetric chemical reactions, most of the literature on DNA-based catalysis relies on oligonucleotides using metallic co-factors [53]. Alongside some recent developments for Ribonuclease A mimics, other examples based only on organic co-factors in a protein-like fashion were developed for the DNA-based proteases field. We now describe the synthetic efforts in this area led by the functionalization of oligonucleotides with amino acid-like functions to hydrolyze the amide bond.
α-Chymotrypsin is one of the most representative members of the serine proteases family that uses three amino acids constitutive of its catalytic triad, serine 195 (Ser195), histidine 57 (His57), and aspartate 102 (Asp102), in a well-known cooperative mechanism (Scheme 2) [54].
Accepted mechanism for chymotrypsin-like serine proteases.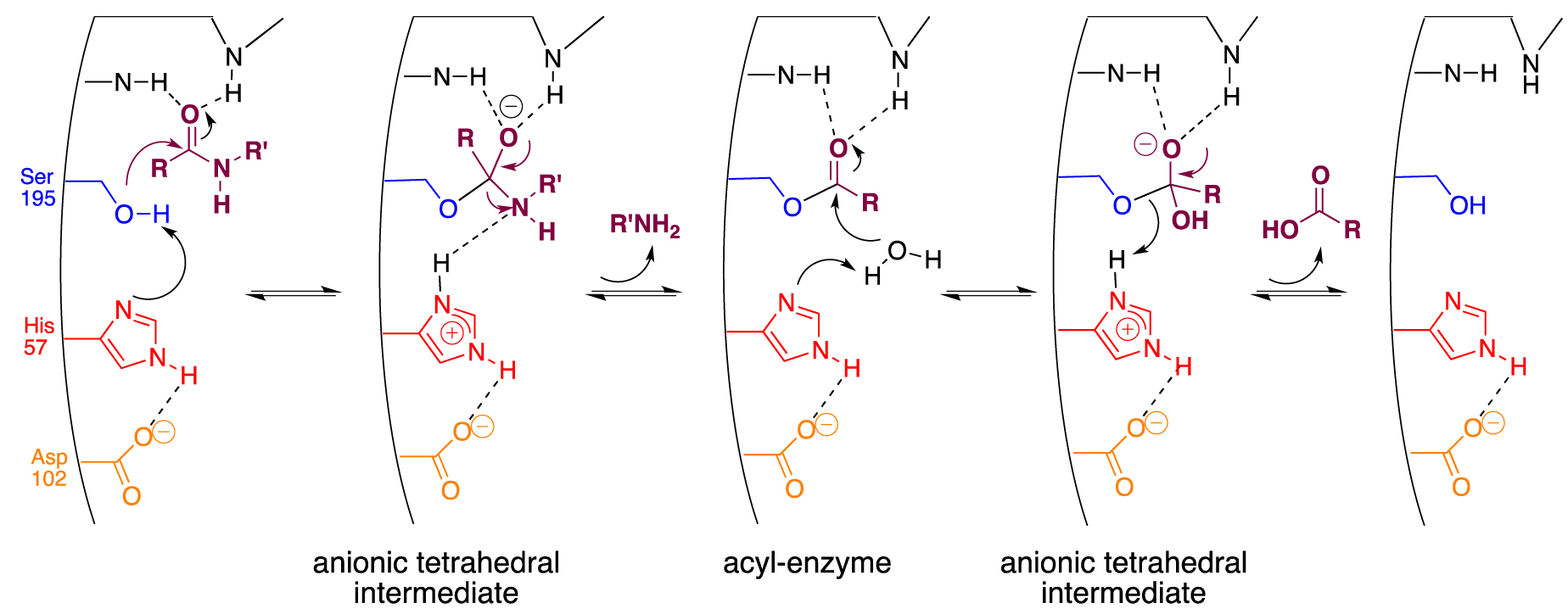
Acting as a general base catalyst, the proximal imidazole ring from His57 enhances the nucleophilic character of the hydroxyl of Ser195, which is consequently sufficiently reactive to attack the carbonyl center of the peptide bond. The resulting anionic tetrahedral intermediate is stabilized in the so-called oxyanion hole by hydrogen bonds originating from the NH of the peptide backbone. It then collapses into a covalent acyl-enzyme complex, liberating the N-part of the peptide. The resulting (ester) acyl-enzyme is now more easily hydrolyzed than the previous amide bond. Thanks to the general acid catalysis of His57, under its protonated form stabilized by Asp 102, the active serine is finally regenerated following the liberation of the C-part of the peptide. Although the protease mimics field has been active for decades, no synthetic scaffolds have been able to match the enzyme efficiency [55]. Enzymatic catalysis and chemical hydrolysis are strictly two different processes. The first process introduces a two-step sequence to lower energy barriers through covalent acyl enzyme intermediates. The former is impaired by chemical factors such as amide bond stabilization by resonance, the combined poor nucleophily of water, and the poor ability of the amine to function as a good leaving group. Diverging from small organic catalysts or micellar systems, DNA was indeed expected to be a suitable scaffold because the nucleic part of the ribosome performs the reverse reaction, i.e., peptide bond formation [56]. Moreover, with the difference of peptide-based mimics that folding could neither be accurately predicted nor undergo any renaturation process, DNA has the unmatched ability to (re)fold into any designed secondary structures that could mimic the active site of enzymes. Finally, as stated earlier, phosphoramidite-based ON synthesis allows the precise introduction of amino acid residues alongside the nucleic backbone to reproduce the putative spatial organization of the catalytic triad.
Most of the efforts were concentrated on the SELEX approach, exploring de facto an expanded chemical space to screen for a functional nucleic catalyst. The RNAzyme field, which relies only on unmodified NTP, remains underexplored [57]. Dr. Hollenstein’s work was aimed at the functionalization of dNTP, such as modified 7-deaza-2′-deoxyadenosine bearing at the 7-position a propargylamido-histamine moiety (dAHsTP) 8 to act as a histidine surrogate (Figure 4). Two 5-modified-2′-deoxyribopyrimidine-5′-triphosphates, either bearing a valeric acid moiety in the cytosine series (dCValTP) 9 or a pentynol arm in the uracil series (dUPOHTP) 10, completed the set of functionalized dNTP, being mimics of aspartate and serine side chains, respectively [58].
Functionalized dNTP with imidazole, carboxylate, or hydroxyl functions.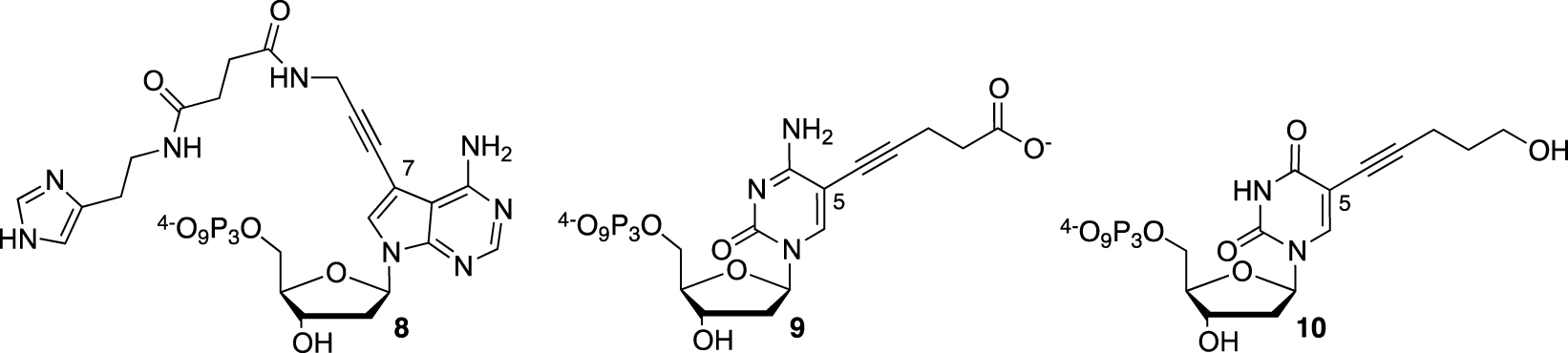
They were obtained using a Sonogashira cross-coupling step starting from their commercial 5-iodo-deoxynucleoside counterparts, followed by the necessary protection/deprotection steps to introduce the 5′-triphosphate group required for the SELEX process. Although the modified dNTPs were well tolerated by DNA polymerases, as demonstrated during primer extension experiments, they were less accepted during the PCR amplification step. Unfortunately, no SELEX attempt in the search for a DNAzyme with amidolytic properties was described.
The key player in the field was Silverman’s group [59], which has devoted important efforts to developing DNA-based protease mimics and overcoming some pitfalls associated with the SELEX process. First, they were urged to design a sophisticated selection and “product capture” process to avoid the selection of DNA-cleaving DNAzyme, as previously observed [60]. By embedding the amide bond into a nucleic substrate facing the 40-nucleotide random loop region, they reported in 2013 the selection of unmodified DNAzymes performing esters hydrolysis (kobs = 0.54 h−1) leading to a moderate 240-fold rate enhancement compared with basal hydrolysis in the required presence of divalent cations such as Mg2+ [61]. Eventually, although a kobs of 3.1 h−1 was measured for aromatic amide hydrolysis, no DNAzymes could be selected for cleaving the amide bond of aliphatic substrates. They then postulated that a better nucleophile than water might be required, such as an amino group. They confirmed their hypothesis in a subsequent publication starting from commercial 5-hydroxymethyl-, 5-amino-allyl- or (E)-5-(2-carboxyvinyl)-2′-deoxyuridine-5′-triphosphates 11, 12, and 13 (Scheme 3).
Capture process (left) and C5-functionalized deoxyuridine 5′-triphosphates (right).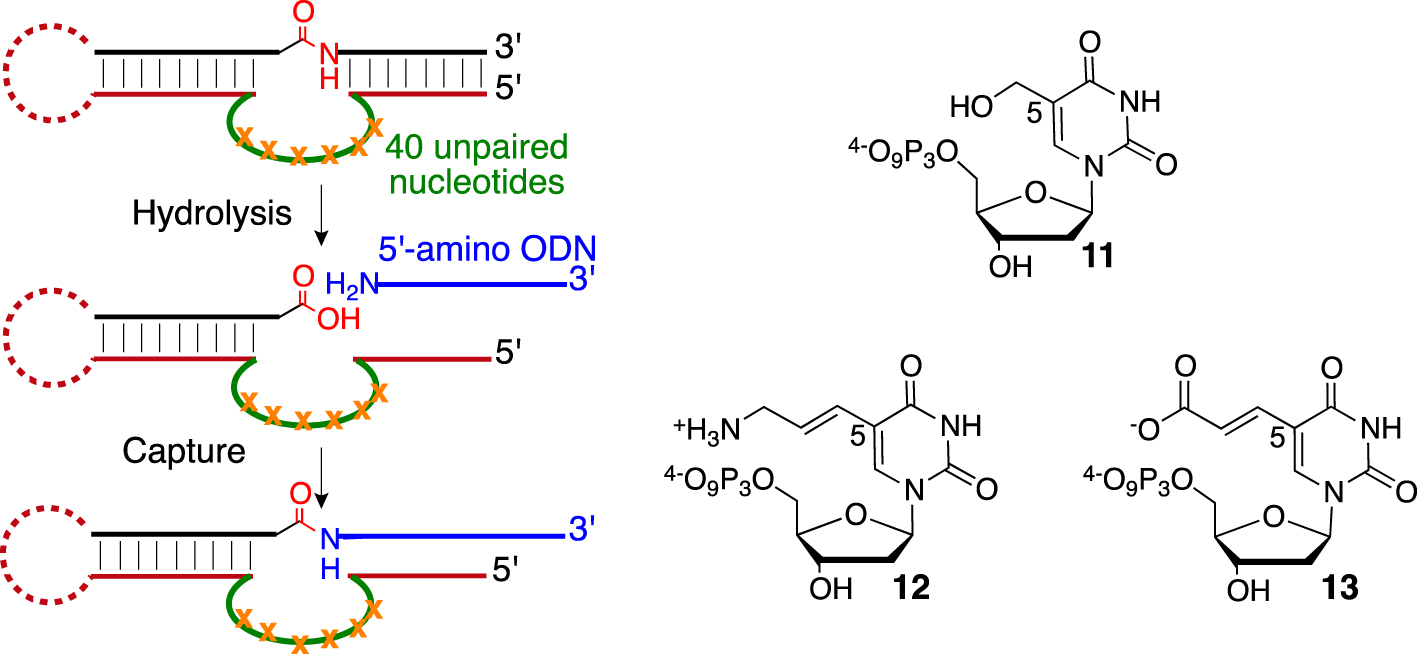
After rounds of SELEX optimization using one C5-substituted dUTP at the time, they were able to isolate the most potent DNAzymes decorated with hydroxyl groups with hydrolysis activity (kobs = 0.1–0.2 h−1) on an aliphatic amide embedded between two DNA fragments [62]. The two sequences leading to the most potent DNAzymes were re-synthetized with no modifications for control purposes. Surprisingly, they still presented substantial catalytic activity, although such unmodified sequences did not arise during the previous selections. This twist of events highlights the fact that SELEX is undoubtedly a powerful method, but it remains a tedious task prone to selection bias.
The complementary bottom-up approach is the SPS of functionalized ON based on the phosphoramidite strategy. Even if the chemical space explored cannot be as extensive as the one offered by SELEX, DNA-based structures mimicking the active site of the serine protease could be designed through careful and deliberate choice of the positions to be modified.
Attempts were made to design esterase mimics by solely using an imidazole pendant as the nucleophilic catalyst. 5-Ethylnyl-imidazole-deoxyuridine-3′-phosphoramidites were incorporated during SPS and organized within duplexes, leading to structures bearing up to three imidazole-modified positions. However, none of the duplexes displayed catalytic activity above that of imidazole, which was used as a buffer control. Relying also solely on imidazole modification, Gorin’s team used an approach similar to the “nucleoaptazyme” strategy developed by Willner’s laboratory [63]. They designed DNA-conjugated small-molecule catalysts (DCats) by functionalizing with an imidazole group two cholic acid aptamers folded in a three-way junction (3WJ) secondary structure (Scheme 4). At different positions alongside each arm of the junction, commercially available 2′-deoxycytidine or thymidine 3′-phosphoramidites bearing a C6-amino-linker at the C5 position were introduced and then conjugated with disuccinimidyl glutarate to histamine.
Schematic representation of the “DCat” approach based on a cholic acid aptamer linked to an imidazole moiety.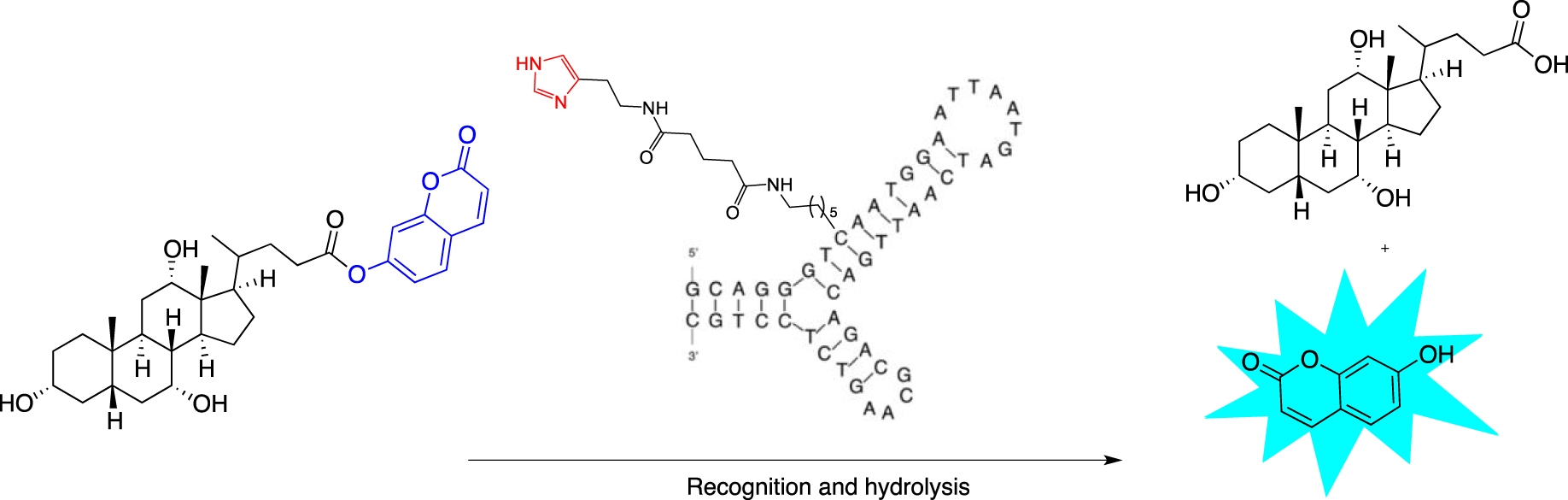
Up to ten DCats were obtained by modulating the position of the modification alongside the 3WJ to evaluate their ability to cleave after binding cholic acid-derived umbelliferone esters and the possible impact on the recognition process. This substrate was chosen because the cholic acid part is supposed to be recognized at the structure core, while the fluorogenic release of the umbelliferone allows the monitoring of the reaction evolution. The best DCat displayed a 100-fold enhancement esterase rate compared with the free equimolar imidazole but only when it was correctly folded, as corroborated by a turn on-off experiment with a toehold displacement.
However, only Madder’s team developed 2′-functionalized deoxyuridine phosphoramidites 14, 15, and 16 bearing amino acid side chain-like residues such as suitably protected carboxylic acid, imidazole, and alcohol functions, respectively, which were tethered to the oligonucleotide via an amide bond (Figure 5) [64]. After solid-phase synthesis, up to three modifications could be introduced by strand, and the rigid but programable duplex structure was used to organize the residues along the major groove of the helix to obtain up to six different sequences, which were then combined into their corresponding duplexes [65]. Analysis of the thermal denaturation studies indicated that each introduction has a negative impact of −5 °C on the thermal value, although one mismatched duplex displayed a 𝛥Tm of −5.3 °C (compared to an expected 𝛥Tm of ≈−15 °C), compensating and largely overcoming the destabilization effect. This result could be explained by the fact that the imidazole and carboxylate moieties, facing each other, were able to interact and stabilize the structures through electrostatic interactions. Despite this chemical effort, no proteolytic activity has been reported.
2′-functionalized nucleotides bearing amino acid side chain-like residues and further introduced in oligonucleotides organized within a duplex structure.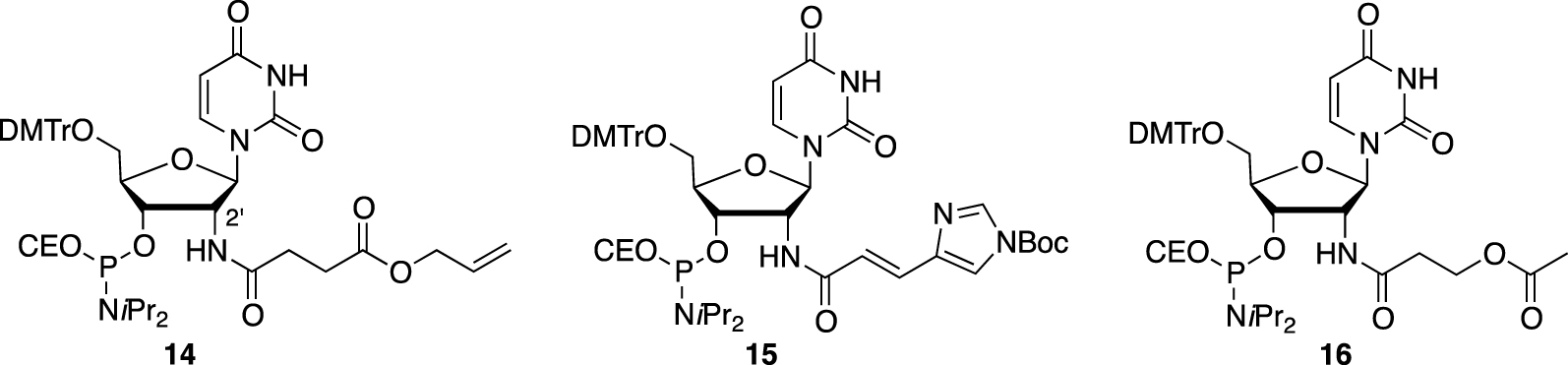
As the structuration properties of DNA have not been exploited to their full potential, we decided to build a 3 way junction (3WJ) from three oligonucleotides partially complementary to each other, bearing at its core the three functional groups involved in the catalytic triad [66].
First, we established our proof of concept by relying on a versatile convertible approach by introducing organic functionalities not at the usual 2′- or 5-position but at the underestimated 5′-position. As the 2′- or C5-modifications direct the chemical handle toward the major groove within a B-DNA duplex, we demonstrated that, depending on the stereochemistry of the newly generated 5′-C-stereogenic center, the modification could point either toward the solvent or the minor groove when the absolute configuration of the 5′-carbon is R-assigned or S-assigned, respectively [67]. Second, we decided to organize our potential DNA-based proteases into a flexible secondary structure such as a 3WJ. This assembly, bearing three unpaired thymidines at its core, was expected to bring the three added modifications in close proximity with sufficient flexibility to avoid steric hindrance. After the efficient incorporation of the convertible 5′-C-(S)-propargyl thymidine phosphoramidite 17 into three different oligonucleotides, each strand underwent a post-synthetic copper(I)-catalyzed alkyne-azide cycloaddition (CuAAC) “click” reaction with the three azide derivatives bearing either a carboxylic, alcohol, or imidazole functions (Scheme 5). The FuON were then assembled in an equimolar mixture, forming a DNA 3WJ with a triply modified core.
5′-convertible nucleotide for the synthesis and functionalization of a 3WJ structure with amino acid side chain mimics at its core.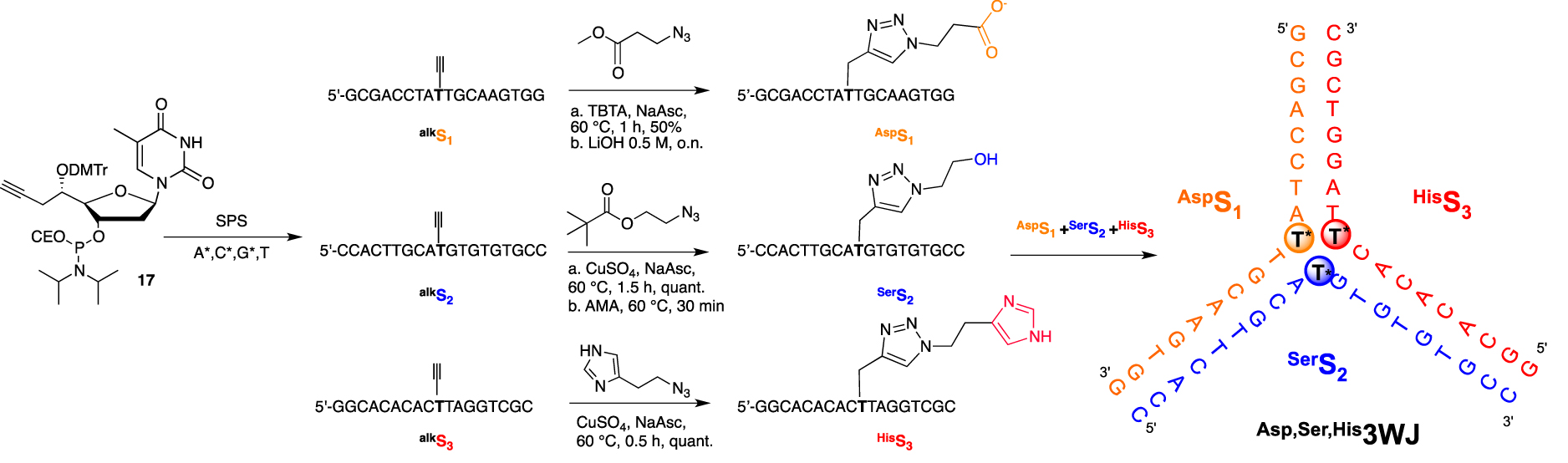
The 3WJ assembly was confirmed by the presence of only one slow migrated band during non-denaturing polyacrylamide gel electrophoresis analyses and was also corroborated by the characteristic B-DNA signature obtained by circular dichroïsm spectroscopy. Thermal denaturation data were obtained to evaluate the contribution of each amino acid side chain-like residue to the overall stability of the secondary structure. As expected in the context of a polyanionic DNA backbone and different pKa values for each residue, the presence of a carboxylate moiety was modestly destabilizing (−1 °C), neutral for the alcohol moiety, and slightly stabilizing for the (cationic) imidazole residue (+1 °C). Taken together, unmodified, triply propargyl- or Ser,His,Asp3WJs displayed the same Tm value of 43 °C, indicating that the size of the three added cycloaddition products did not destabilize the resulting secondary structure. This decorated three-way junction ultimately failed to provide any amide bond hydrolysis. This could be explained by the lack of cooperative interactions between the three appended functionalities, as an alleged over-stabilization (that could originate from a close proximity of the functional groups) was not observed. Moreover, the stabilization of the anionic tetrahedral intermediate in the proximity of the negatively charged DNA backbone must be considered. We are investigating new synthetic options to ensure cooperative interactions between the three functional groups to design a functional DNA-based protease that can cleave the amide bond.
5. Conclusion
Despite the exquisite evolutionary optimization of DNA to perform reactions thought to be only devoted to enzymes, organic chemists came into play to increase the DNA catalytic repertoire. As unmodified DNAzymes were first developed to cleave the phosphodiester bond of RNA, mimicking the catalytic activity of Ribonucleoase A, chemists rapidly took inspiration from the active site of such enzyme to functionalize oligonucleotides with organic groups reminiscent of the side chain of the amino acids involved in catalysis. Although a reduced catalytic efficiency was observed on non-nucleic acid substrates, FuON were also modified with metal complex ligands to confer either peroxidase or phosphatase properties or organic functions to mimic the amidolytic cleavage of the peptide bond, as exemplified in this review. Moreover, the recognition properties of DNA were also exploited to develop DNA catalysts capable of recognizing their target and performing the desired reaction in the so-called nucleoaptazyme approach.
Despite these promising results, more challenges lie in front of (bio)chemists in terms of matching the efficiency and selectivity of enzymes, with DNA catalysts being less potent than their enzymatic counterparts. Within the nucleoaptazyme approach, improving the binding properties would consequently increase the effective concentration of the substrate, leading to more efficient catalysis. Moreover, guidance could also be provided by in silico design [68] and algorithms [69] to rationally design functional FuON bearing, at the right positions, catalytic modifications or even to reach new stable secondary structures. Finally, advances must be made to improve each step of the process by developing new SELEX protocols and engineering new enzymes to overcome the limitations of functionalized nucleotides that are sometimes poorly recognized by polymerases. Evolution could also be directed toward reactions not yet known to be catalyzed by DNA, for example, by modulating the modification status of RNA, as already explored with RNA-catalyzed RNA bio-orthogonal labeling reactions [70]. However, the catalog of enzymatic reactions remains underexploited, as exemplified by the challenging site-specific cleavage of DNA by DNAzymes [60, 71]. Organic chemistry is associated with this ongoing optimization process with recent developments toward the expansion of the genetic alphabet [72], original orthogonal bioconjugation [73], or revisiting the solid phase synthesis process using P(V) chemistry to give ODN with optically pure phosphorothioate linkages [74]. Finally, starting from a single oligonucleotide displaying catalytic properties, recent advances in their supramolecular assembly in 3D organizations such as DNA tetrahedral [75] or origami nanostructures [76] or their capacity to mimic the enzymatic cascade process [77] will undoubtedly widen the scope and success of their applications in the foreseeable future.
Declaration of interests
The authors do not work for, advise, own shares in, or receive funds from any organization that could benefit from this article, and have declared no affiliations other than their research organizations.