

1 Introduction
Le colloque « From synthetic chemistry to synthetic biology » organisé au Collège de France le 5 mai 2009 a été l’occasion de réfléchir sur les enjeux de la synthèse chimique dans le cadre de la création de nouveaux médicaments. Plusieurs questions se posent : sommes nous à la fin d’une période florissante de la synthèse chimique de médicaments, le domaine des « petites molécules » selon l’expression actuelle de l’industrie pharmaceutique ou bien va-t-on vers un arsenal thérapeutique basé essentiellement sur des « biomolécules » comme les anticorps monoclonaux, les protéines recombinantes ou les fragments d’acides nucléiques ? Le développement exponentiel de la biologie moléculaire a permis de développer de nouveaux outils de diagnostic et de thérapie qui deviennent des éléments essentiels pour l’amélioration de la santé des personnes. Devant cette évolution que va devenir la chimie thérapeutique ?
2 Quel futur pour la création chimique de médicaments ?
La synthèse chimique de nouveaux médicaments va-t-elle disparaître dans les dix ou 20 prochaines années, les méthodes de synthèse issues de la biologie en utilisant des cellules de micro-organismes ? Nous pensons que ces méthodes de synthèses viennent s’ajouter à celles de la chimie classique. Par ailleurs, la conception de nouvelles méthodes chimiques dans la synthèse de médicaments est loin d’avoir atteinte ses limites. Après un bref aperçu sur les différentes stratégies pour créer de nouvelles molécules à visée thérapeutique, nous présenterons la conception de molécules hybrides comme stratégie pour la création de nouveaux agents anti-infectieux. Nous sommes là dans le domaine des « petites molécules », celui du médicament dit traditionnel et non dans celui des molécules d’origine biologique (protéines, anticorps, acides nucléiques, oligosaccharides,…) qui constituent depuis quelques années le domaine des produits « biopharmaceutiques ».
La création de nouveaux médicaments à base de molécules chimiques passe une période difficile : le temps entre le dépôt d’une demande de brevet et la mise sur le marché s’allonge de manière importante et certainement déraisonnable ; en 30 ans, cette durée est passée de 7 à 8 ans à plus de 12 à 15 ans. De nombreux facteurs sont responsables de cet allongement, les « précautions » prises à chaque étape de la mise au point de médicaments dans une société qui ne veut plus assumer le moindre risque au niveau collectif, les structures complexes des grands groupes pharmaceutiques, loin de la dynamique des petites entreprises et les relations difficiles entre investisseurs et créateurs. Les nouveaux médicaments sont moins nombreux et leur coût de développement s’est envolé, jusqu’à atteindre le milliard de dollars, ce qui nécessite une immobilisation de capitaux de plus en plus difficile à réunir. Rappelons que les coûts de création d’un médicament sont répartis de la manière suivante : environ 10 % pour la partie découverte et recherches initiales, 15 % pour la partie préclinique, 15 % pour la fabrication selon les méthodes dites « GMP » (Good Manufacturing Practices), 55 % pour les essais cliniques (cette part est en constante augmentation devant la demande croissante de sécurité) et enfin de l’ordre de 5 % pour les frais de commercialisation du médicament. La très forte variabilité inter-individu pose des problèmes difficiles à résoudre lors de la mise au point de nouveaux médicaments. Pour des médicaments à large prescription, il faut envisager des cohortes de patients allant de personnes obèses à des malades immunodéprimés, c’est-à-dire des extrêmes dans les paramètres de biodisponibilité et de risque de réactions secondaires. Il est possible d’envisager des approches thérapeutiques ciblées, à la carte, pour chaque individu ou sous-groupes d’individus, mais le corollaire de ces traitements ciblés sera l’augmentation inévitable du coût des traitements. Au cours des 30 dernières années, les coûts des budgets de santé publique ont fortement augmenté, souvent deux fois plus vite que la croissance économique des pays concernés. Dans les pays développés, la part du produit intérieur brut (PIB) consacrée aux dépenses de santé se situe entre 8 et 16 %, ce dernier chiffre étant celui des États-Unis [1], dont beaucoup s’accordent à reconnaître que la couverture sociale est loin d’être optimale avec des coûts intrinsèques très élevés en comparaison des autres pays développés. Même en tenant compte d’efforts de rationalisation dans les dépenses de santé publique, il faut noter que certains traitements thérapeutiques innovants posent de graves problèmes de coûts. Une étude suédoise faite entre 2000 et 2003 sur le coût annuel du traitement de l’arthrite rhumatoïde, une maladie très invalidante, est de l’ordre de 11 800 à 14 400 euros par an à l’aide d’anticorps spécifiques, très loin de la moyenne de 170 euros par an pour l’utilisation de médicaments classiques [2]. De telles augmentations de coûts de traitement pour des maladies chroniques posent de sérieux problèmes pour les budgets de santé publique, en dépassant très nettement les possibilités personnelles de financement de la quasi-totalité des malades et conduisant le financement collectif à une impasse budgétaire.
Le domaine des anti-infectieux a été en grande partie délaissé au cours des 20 dernières années pour plusieurs raisons. Les formidables avancées des années 1940 à 1960 dans la lutte contre les infections bactériennes ont laissé penser que l’arsenal des antibiotiques était largement suffisant pour faire face aux différentes situations pathologiques. La capacité d’adaptation des bactéries et la facilité avec laquelle elles échangent des gènes leur permet de s’adapter très vite aux molécules exogènes. C’est ainsi que de nombreuses souches bactériennes ont acquis des résistances à un ou plusieurs antibiotiques. La prolifération de ces bactéries en milieu hospitalier est devenue un problème majeur de santé publique. Les décès dûs aux infections nosocomiales aux États-Unis ont maintenant dépassé ceux liés au sida (plus de 19 000 morts par an en 2005). Dans le domaine des maladies parasitaires, tout le monde a en tête la perte d’activité de la chloroquine, un formidable médicament qui a permis de réduire les zones de paludisme endémique entre 1945 et 1960 avec l’aide d’un insecticide puissant, le DDT.
Que faire devant la montée des souches de pathogènes résistants aux molécules anciennes, c’est-à-dire celles qui sont utilisées depuis plus de 40 à 50 ans ? Tout d’abord, accepter l’idée que l’arsenal thérapeutique doit être renouvelé tous les 30 à 40 ans. Les parasites, comme les bactéries et les virus, ont de formidables capacités à s’adapter à des environnements changeants et contraignants. Cela leur permet de survivre grâce à des mutations spécifiques et de faire face à leurs nouvelles conditions de vie.
Depuis une vingtaine d’années les génomes de nombreuses espèces vivantes sont connues, y compris celui de l’homme. Dans le cas des maladies infectieuses, les génomes des virus, bactéries et parasites sont connus, ainsi que celui de vecteurs comme le moustique Anopheles gambiae. Connaître le génome ne signifie pas que la mise au point des médicaments ou des vaccins est facilitée. Les déconvenues des 25 dernières années sur la mise au point d’un vaccin contre le virus du VIH responsable du sida amènent les scientifiques à plus d’humilité en évitant des déclarations hâtives sous la pression des médias. Le rétrovirus du VIH a un génome de très petite taille, moins de 10 000 bases, bien en dessous de celui d’un parasite comme Plasmodium falciparum qui comporte 28 millions de bases. Dans les deux cas, la connaissance du génome n’a pas permis la mise au point rapide d’un vaccin. Il faut comprendre que la pénétration d’un virus ou d’un parasite dans la cellule humaine cible se fait par l’intermédiaire de nombreuses interactions à la fois très spécifiques et variées. Dans le cas du parasite du paludisme, plus de 58 protéines sont impliquées dans la reconnaissance et la pénétration d’un érythrocyte par un mérozoïte [3]. Cela explique en grande partie les échecs dans la mise au point d’un vaccin capable de protéger contre les différentes formes de Plasmodia transmissibles à l’homme : P. malariae, P. vivax, P. ovale et P. falciparum.
Quelles sont les voies principales pour la mise au point de nouveaux médicaments ? Sans être exhaustif, on notera :
- • les méthodes associant l’identification de nouvelles cibles grâce aux études de protéomique et leur criblage à l’aide de robots avec des centaines de milliers de molécules provenant de bibliothèques de composés chimiques (High-Throughput Screening [HTS]) ;
- • le retour vers la recherche de nouvelles molécules issues des produits naturels. Les produits naturels peuvent être actifs par eux-mêmes ou bien conduire à la conception de nouveaux pharmacophores par simplification des structures des molécules naturelles (démarche de la chimie « bio-inspirée »).
Actuellement, plus de 50 % des médicaments utilisés sont d’origine naturelle [36]. Cela devrait rassurer les personnes qui veulent utiliser des produits naturels en automédication : de nombreux médicaments sont effectivement des composés extraits de plantes, purifiés, identifiés et dosés de manière rigoureuse. La génétique chimique (chemical genetics) est également un moyen de mieux comprendre le rôle des gènes ou des produits de gènes à l’aide de petites molécules chimiques. Cette approche a été largement popularisé par Schreiber depuis 1998 [4].
La création de nouvelles structures chimiques a-t-elle atteint ses limites ? Cette question a souvent été posée au cours des années 1990 à 2000 en sous-entendant qu’il serait temps de réduire les budgets consacrés à la chimie thérapeutique dans les grands groupes pharmaceutiques. Cette attitude ne tient pas compte de deux phénomènes. En outre, la chimie de synthèse a été fortement renouvelée au cours des 20 dernières années. De nouvelles réactions, en particulier catalytiques, sont devenues des outils utilisables dans la fabrication de pharmacophores, permettant ainsi d’accéder à de nouveaux motifs structuraux considérés comme inaccessibles. L’exploration de l’espace chimique est encore très limitée. Fink et Reymond ont récemment créé une base de données structurales contenant plus de 26 millions de molécules pouvant être faites avec un jeu d’atomes allant jusqu’à 11 atomes de carbone, azote, oxygène et fluor et en utilisant les méthodes de synthèse actuellement connues [5]. De cet espace chimique, le nombre de molécules connues dans les banques de données existantes était seulement de 63 850 molécules, c’est-à-dire 0,24 % des possibilités offertes par la synthèse chimique. Nous sommes donc très loin d’avoir atteint les limites de la construction de nouvelles molécules.
Par ailleurs, nous allons également vers la diversification des synthèses mettant en jeu des micro-organismes modifiés, dans lesquels sont intégrés des gènes d’enzymes capables d’effectuer des étapes difficiles (oxygénation de liaisons carbone-hydrogène,…) comme cela a été réalisé récemment dans la synthèse d’un précurseur de l’antipaludique artémisinine en utilisant une levure modifiée [6]. La fermentation industrielle est à même d’intégrer rapidement ces méthodes de « synthèse biologique », car il s’agit simplement d’étendre la gamme des micro-organismes utilisables dans les fermenteurs de forte capacité comme utilisés depuis plus de 50 ans dans la synthèse des antibiotiques ou de vitamines.
3 Exemples de molécules hybrides à activité antipaludique
Dans cas particulier, nous nous sommes orientés vers la synthèse de molécules hybrides comme stratégie de création de nouvelles molécules antipaludiques. Pourquoi faire des molécules contenant deux pharmacophores liés par un lien covalent en lieu et place de deux pharmacophores donnés indépendamment ? Une partie de la réponse est liée à des travaux avec mon équipe sur la Bléomycine® [7,8], un médicament antitumoral. Cette molécule hybride comporte trois domaines différents ayant chacun une fonction très précise : un disaccharide permettant la pénétration dans les cellules tumorales, un bithiazole comme l’intercalant de l’ADN et une partie peptidique capable de complexer fortement un ion ferrique, site de la formation d’une espèce métal-oxo à l’origine des coupures d’ADN [8]. L’autre partie de la réponse est liée à la nécessité de rompre avec la monothérapie dans la lutte contre le parasite du paludisme. L’utilisation de la seule chloroquine pendant plus de 30 ans a conduit à la mutation du parasite et à l’émergence dès 1970 à 1980 de souches résistantes de Plasmodium falciparum. L’Organisation mondiale de la santé (OMS) recommande l’utilisation de bithérapie afin d’éviter la sélection de nouvelles souches résistantes [9]. La résistance d’un parasite provient de mutations. Selon White et al. [10], la chance d’apparition d’un parasite capable de résister simultanément à deux antipaludiques est le produit du risque de résistance pour chacune de ces molécules. Par exemple, si un parasite sur 109 est résistant à un médicament A et un parasite sur 1013 est résistant à un second médicament B, et si les deux mutations génétiques sont indépendantes, alors un parasite seulement sur 1022 sera résistant aux deux médicaments. Sachant qu’une personne ne peut être infectée par plus de 1012 parasites (la charge parasitaire par individu est comprise généralement entre 108 et 1012), la probabilité d’avoir un parasite résistant aux deux médicaments est donc très faible. La démonstration de l’efficacité d’une bithérapie a été faite dès 1950 avec l’association entre la Streptomycine® et le Rimifon® (isoniazide) dans le traitement de la tuberculose. Les polychimiothérapies ne sont imposées depuis longtemps dans le traitement des cancers et plus récemment dans celui du sida.
Dans le traitement du paludisme, pour créer des bithérapies, il y a d’un côté de nombreux dérivés de quinoléines avec des demi-vies plasmatiques assez longues et de l’autre des dérivés de l’artémisinine ayant une très courte durée de vie plasmatique de l’ordre de 30 minutes (il est possible que ces courtes demi-vies soient en partie dues à la décomposition de la partie péroxydique de ces molécules dans les échantillons de sang ex vivo, après prélèvement). Pour éviter d’associer deux molécules ayant des pharmacocinétiques très différentes, nous avons eu l’idée de créer des molécules antipaludiques hybrides dont nous pouvions espérer qu’elles présentent un mode d’action dual [11]. Il s’agissait d’associer par un lien covalent un motif aminoquinoléine (existant dans la chloroquine) à un trioxane (pharmacophore de l’artémisinine). Il était également possible de regarder la partie aminoquinoléine comme un excellent vecteur de la partie trioxane. En effet, la capacité des aminoquinoléines à se protoner ou se déprotoner lors des passages de membranes et ensuite celle de s’accumuler dans la vacuole digestive acide des plasmodiums permettaient de drainer des trioxanes vers l’intérieur d’érythrocytes infectés. Ces idées simples ont été à l’origine de la conception des trioxaquines. Cette conception de nouveaux antipaludiques fait suite à des études sur le mécanisme d’action de l’artémisinine et de ses dérivés [12–17]. L’artémisinine est extraite des feuilles d’une armoise courante en Chine, Artemisia annua, et utilisée dans la médecine traditionnelle depuis plus de 1500 ans [17–19]. Il est remarquable de constater que tous les dérivés actifs de l’artémisinine sont capables d’alkyler l’hème, non seulement in vitro, mais également in vivo. En effet, les adduits hème–artémisinine ont été identifiés chez les souris infectées par Plasmodium et traitées avec l’artémisinine à des concentrations thérapeutiques et en aucun cas chez les souris saines exposées à l’artémisinine [15]. Cette capacité d’alkylation des dérivés de l’artémisinine a été corrélée avec leur activité antipaludique in vitro ou in vivo pour plus d’une dizaine de ces composés. Toutefois, cette proposition de mécanisme est parfois remise en cause par d’autres auteurs qui préfèrent voir l’artémisinine comme un élément perturbateur de l’équilibre redox des érythrocytes infectés « .. the intimate nature or early steps in the process that is initiated by electron transfer to the peroxide from an endogeneous reductant, not necessarily containing iron(II), and how these early steps may affect the redox balance within the parasite » [20].
Toutes ces études sur les propriétés alkylantes des dérivés ou des modèles de l’artémisinine [21] nous ont permis de concevoir la synthèse de molécules hybrides contenant un motif 1,2,4-trioxane comme dans l’artémisinine et une entité aminoquinoléine comme dans la chloroquine [11]. La conception de molécules hybrides à activité duale s’inscrit dans le contexte du développement de la polypharmacologie, c’est-à-dire d’une pharmacologie qui va au-delà du concept fructueux « clé–serrure » (ou « une molécule–une cible biologique ») qui est la base classique de la pharmacologie. Il est possible d’imaginer des molécules hybrides dont les deux entités constitutives sont capables d’agir sur la même cible (cas A de la Fig. 1), ou bien chacun des deux pharmacophores est capable d’agir sur deux cibles indépendantes (cas B de la Fig. 1) ou dernier cas, celui d’une molécule hybride dont les deux entités agissent sur deux sites distincts (indépendants ou non) d’une même macromolécule biologique (cas C de la Fig. 1).

Représentation schématique de trois modes d’interaction possibles pour des molécules hybrides : (A) une seule cible avec des molécules du type épée à double tranchant, (B) deux cibles indépendantes (chaque entité de la molécule hybride interagit avec sa cible) et (C) deux cibles reliées (les deux parties de la molécule hybride agissent en même temps sur les deux cibles connectées).
Schematic representation of three possible modes of interaction for hybrid molecules: (A) one single target for double-edge sword molecules, (B) two independent targets (each entity of the hybrid molecule interacts with its target), and (C) two related targets (both entities of the hybrid molecule act at the same time with two connected targets).
Les trioxaquines ont été conçues en associant deux éléments capables d’agir sur l’hème libérée lors de la dégradation de l’hémoglobine par Plasmodium dans les érythrocytes infectés. D’un côté une 4-aminoquinoléine capable de s’associer fortement avec l’hème (par stacking) et, en outre, un trioxane capable d’alkyler l’hème et de former ainsi des adduits capables, entre autre propriété, d’inhiber la synthèse de l’hémozoïne. Les premières trioxaquines se sont avérées très actives sur les souches de Plasmodium falciparum résistantes à la chloroquine, ce qui a conduit à la synthèse de plusieurs séries de molécules actives en cherchant au fur et à mesure du développement des synthèses à prendre en compte les différents critères entrant dans la sélection d’un candidat-médicament (molécule PA1103/SAR116242, Fig. 2) pouvant être développée pour aller vers les premières phases cliniques [22–28]. La synthèse à l’échelle du kilogramme de cette nouvelle entité chimique contenant un trioxane a été réalisé en 2008. Il est intéressant de noter que la stabilité thermique de ces trioxanes est bien meilleure que celles de molécules de masse moléculaire du même ordre ayant un simple motif endopéroxyde. Cela souligne le rôle essentiel du troisième atome d’oxygène en position 4 du trioxane comme élément de stabilisation de la liaison OO du péroxyde. Une étude théorique en DFT devrait permettre de mieux comprendre cette stabilité qui est un élément important pour une molécule qui doit être distribuée dans des pays chauds avec des moyens de transport et de stockage dont les températures sont peu ou pas contrôlées.
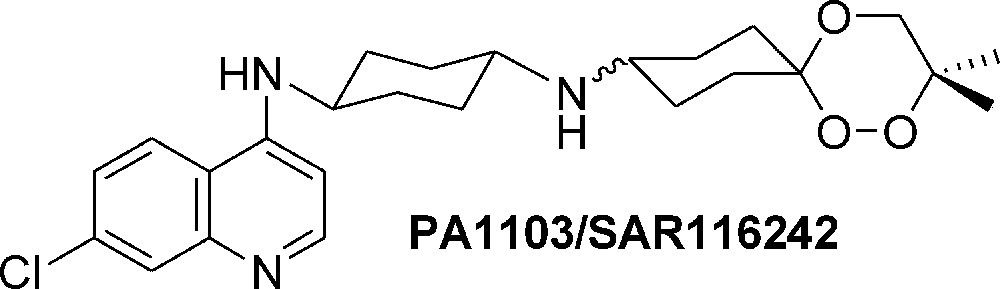
Structure de l’antipaludique trioxaquine PA1103/SAR116242.
Structure of the antimalarial trioxaquine PA1103/SAR116242.
Très récemment, nous avons pu montrer que certaines de ces trioxaquines sont capables d’éliminer, de détruire les schistosomules responsables de la bilharziose (ou schistosomiase), maladie tropicale touchant plus de 250 millions de personnes et étant à l’origine de plus de 200 000 décès par an [29].
4 Que devient la recherche de nouveaux antibiotiques ?
La découverte et le développement des antibiotiques est une des plus belles pages de l’histoire des médicaments bien connue de tous, peut-être même, trop connu. Le nombre important de nouveaux antibiotiques efficaces mis à la disposition du corps médical et leur utilisation larga manu pendant plus de 50 ans ont induit l’idée que le traitement des infections bactériennes est une bataille gagnée. Cela a conduit à de fortes régressions dans le respect des règles d’hygiène pasteuriennes et l’apparition de souches bactériennes multirésistantes est venue nous rappeler que la mise au point de nouveaux antibiotiques doit rester une des grandes priorités de la recherche thérapeutique. Les grands groupes pharmaceutiques ont largement délaissé le domaine des anti-infectieux dans les années 1990 pour se focaliser sur des maladies dites prioritaires (cancers, maladies cardiovasculaires, affections du système nerveux central ,…). À titre d’illustration, seulement six candidats-médicaments étaient en développement en 2004 sur un total de 506 molécules dont 67 antitumoraux, 34 entités pour lutter contre les désordres métaboliques ou 33 anti-inflammatoires [30]. Le nombre de décès aux États-Unis liés à des infections nosocomiales, 19 000 morts par an en 2005, a maintenant dépassé les cas de décès liés au sida [31]. Cette donnée épidémiologique largement diffusée par les médias nord-américains donne la mesure de l’insuffisance dans l’innovation thérapeutique dans le domaine des anti-infectieux. Beaucoup d’efforts de recherche ont porté sur la biologie et la génétique des bactéries, peu a été fait sur la recherche de nouveaux antibiotiques. Le groupe GlaxoSmithKline a travaillé pendant sept ans sur des études de génomiques bactériennes (1995 à 2001) en examinant les produits d’expression de 300 gènes différents. Sur ces cibles potentielles, plus de 70 campagnes de criblage à haut débit ont été effectuées avec des collections de 260 000 à 530 000 composés. Le petit nombre de molécules identifiées comme point de départ n’a pas conduit à des médicaments et cette approche a été considérée comme un échec en regard des sommes importantes investies [32]. Ces travaux de génomiques confirment que la « voie royale du gène au médicament » n’existe pas [33].
À partir de ce constat d’échec, il est important d’étendre le champ des recherches à la protéomique structurale (détermination des structures des protéines exprimables par un génome), chimie combinatoire avec des synthèses permettant des diversités structurales importantes, retour vers la chimie des produits naturels ou des produits « bio-inspirés ». N’oublions pas que plus de la moitié des médicaments actuels est d’origine naturelle [34]. Ajoutons à cette liste, l’utilisation de méthodes de calculs de chimie théorique qui vont continuer à s’adapter à la pharmacologie et de la résonance magnétique nucléaire (RMN) pour l’optimisation de molécules actives [35]. La « chimie génétique » vient s’ajouter depuis peu à ces différentes approches, il s’agit d’associer à chaque gène, une ou plusieurs molécules capables d’en modifier l’expression et permettant d’observer les modifications phénotypiques associées à un ou plusieurs gènes [36].
Dans ce contexte, nous avons entrepris la synthèse de molécules hybrides en associant de manière covalente une entité ayant une activité antibiotique (ou potentiellement antibiotique) à une 4-aminoquinoléine qui permet :
- • de moduler l’hydrophobie de la molécule ;
- • de favoriser l’interaction avec les membranes des pathogènes et souvent leur franchissement ;
- • de protéger la partie antibiotique en milieu acide grâce à la protonation aisée de la partie aminoquinoléine.
C’est ainsi que des antibioquines ont été préparées et leur activité antibactérienne évaluée [37] (Fig. 3 pour une représentation schématique de ces antibioquines). Les efforts se sont portés actuellement portés sur la série des Vancomyquines® dérivés de la Vancomycine®, un glycopeptide très actif qui a longtemps été le dernier recours dans le traitement des infections bactériennes dues à des souches de Staphylococcus aureus multirésistant.
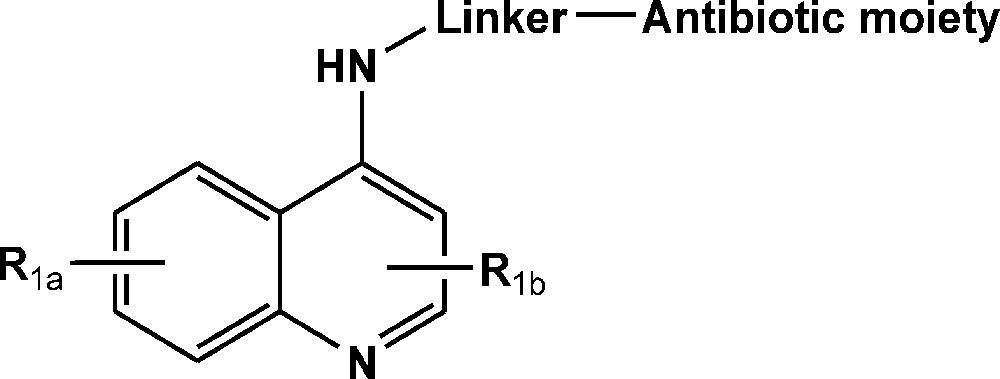
Représentation schématique des structures de type antibioquine.
Schematic representation of antibioquine structures.
La Vancomyquine® PA1409 a été sélectionnée en 2008 pour un développement préclinique réglementaire et les principaux résultats ont été présentés lors du dernier congrès International Conference on Antimicrobial Agents and Chemotherapy (ICAAC) en septembre 2009 aux États-Unis [38–40]. Des propriétés de cette molécule nous retiendrons essentiellement les points suivants :
- • une très forte bactéricidie à faible concentration, y compris sur des inocula de forte concentration ;
- • des activités sur des souches d’entérocoques résistants à la Vancomycine® et des paramètres de pharmacocinétiques très favorables (la valeur du rapport aire-sous-la courbe/concentration minimale inhibitrice chez le chien est de 60 fois celle de ce même rapport pour la Vancomycine®).
5 Conclusion
La conception de nouveaux médicaments devra intégrer de nombreuses nouvelles méthodes de synthèse qui devront être efficaces et avoir un minimum d’impact sur l’environnement. L’époque de la synthèse multi-étapes s’éloigne et sera progressivement remplacée par des méthodes de synthèse utilisant des micro-organismes génétiquement modifiés permettant des synthèses de squelettes chimiques compliqués (synthetic biology). En revanche, les chimistes pourront toujours imaginer de nouvelles structures simples et hors du champ d’application des synthèses utilisant des micro-organismes modifiés. Les molécules hybrides à activité duale sont un de ces exemples.
Remerciements
B.M. exprime ses sincères remerciements à tous les coauteurs (CNRS, Inserm et Palumed) des articles sur les antipaludiques (dérivés de l’artémisinine et trioxaquines dont la PA1103) publiés depuis 1997, ainsi que toute l’équipe de Palumed qui a mis au point les antibioquines et en particulier la Vancomyquine® (PA1409). Il faut également remercier le petit groupe de capitaux-risqueurs (IRDI-SOCRI, CDC-FCJE et Vivéris) ayant permis la création de la seule société privée dédiée initialement à la mise au point de nouveaux antipaludiques. L’auteur tient à remercier un des deux rapporteurs pour une lecture très attentive du manuscrit.