



 CC-BY 4.0
CC-BY 4.0
1. Introduction
In a cell membrane, the stacked aliphatic chains of the lipid bilayer form an impermeable hydrophobic barrier. The simplest form of membrane transport, passive diffusion, refers thus to the diffusion of a chemical species by random processes across this lipid bilayer, from one side of the membrane to the other (and vice versa), independently of any metabolic energy. This passive diffusion is restricted to gas and essentially to apolar molecules and small (<200 Da) polar charged molecules.
Living organisms have evolved increasingly sophisticated transport systems to control exchanges across the membrane and enable the import of nutrients and energy-producing compounds, essential for survival, as well as the recognition of chemical or physical messengers. The transmission of these messages to the interior of the cell enables the exchange of information with cells and the surrounding environment. These transport processes are carried out by proteins integrated into the plasma membrane.
In addition to specialized proteins, endocytosis mechanisms are active processes that involve protein-assisted (and energy-dependent) deformations of the lipid bilayer, bringing extracellular material into the intracellular environment via closed vesicles [1] (Figure 1). The most studied process involves clathrin-coated endocytic vesicles. These vesicles are produced by complex modular protein machinery that assembles transiently on the plasma membrane. This machinery shapes/deforms the membrane to form inward-pointing buds, thus concentrating the molecules to be transported in enclosed vesicles. Parameters such as plasma membrane tension and rigidity control the dynamics of this endocytosis pathway. Various endocytic proteins bind to membrane phosphoinositides, which are essential for organizing the protein assembly steps throughout the endocytosis process.
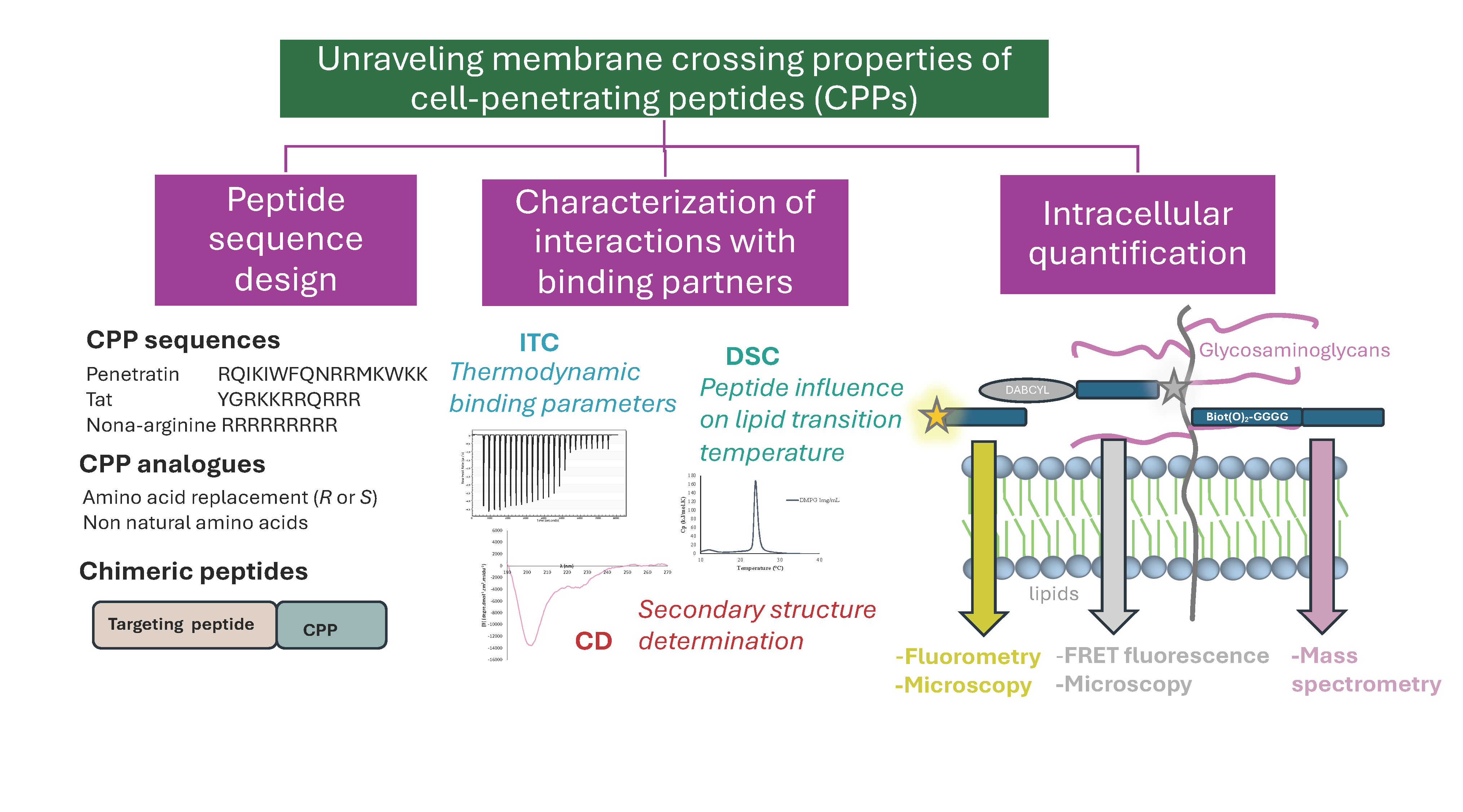
Schematic representation of the current mechanisms of internalization, endocytosis, and translocation of CPPs and the peptide sequences of the most studied ones. Cationic amino acids are highlighted in blue, aromatic in green, and conserved residues in HD-derived CPPs are in bold.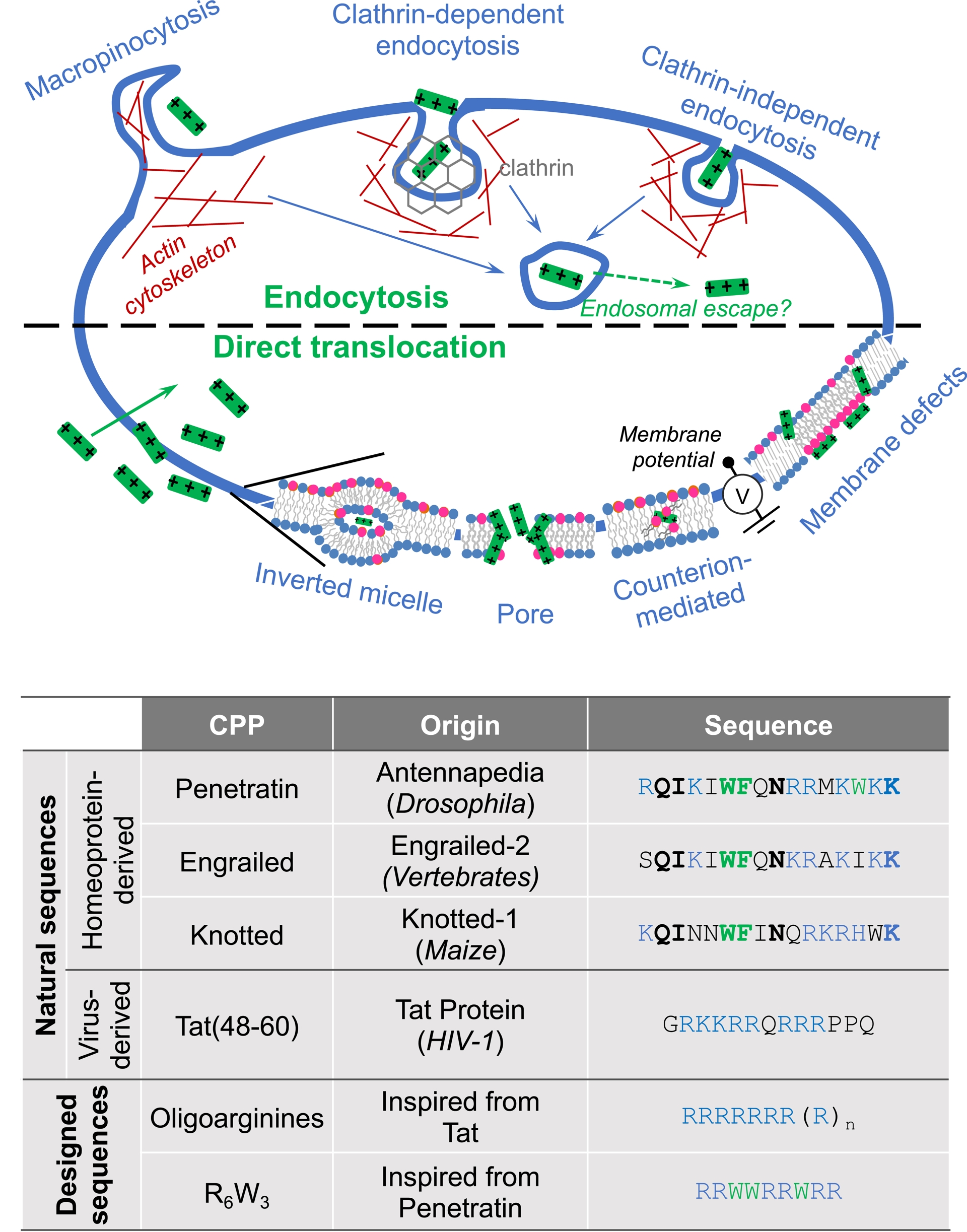
Macropinocytosis, on the other hand, is a regulated form of endocytosis that enables non-selective uptake of extracellular material. It involves the formation of protrusions from the plasma membrane to the outside of the cell (similar to ocean waves), which then fuse with themselves (or back to the plasma membrane), resulting in the uptake of all trapped extracellular components and their subsequent entry into the cell [1].
In the 1990s, it was described for the first time that polar cationic proteins were able to pass through plasma cell membranes in a non-conventional, non-endocytic process, a result that contrasted with the dogmatic view and general acceptance that a biological membrane is impermeable to hydrophilic molecules.
2. Emergence of the carrier peptides
An initial study showed indeed that the Tat protein, which controls the expression of the viral genome in cells infected with the human immunodeficiency virus, is capable of entering cells through a process that remained to be identified in order to exert its action [2] (Figure 1). A subsequent study highlighted endocytosis as the entry mechanism [3]. At the same time, another study described how the homeodomain from the Drosophila melanogaster homeoprotein (HP) Antennapedia, secreted by certain neurons, is able to enter cultured neurons by a non-endocytic route to induce their differentiation [4] (Figure 1). It was later shown by fluorescence imaging that a 16-amino-acid domain of this homeodomain retains the internalization capacity of the full-length protein [5]. Moreover, when incubated with cultured neurons, this fluorescence-labeled protein domain is detected inside cells (cytosol and nucleus) even when incubation is carried out at a temperature ⩽12–15 °C, at which endocytosis pathways are totally inhibited. Once in, the peptide is found intact in the cells, suggesting that it had not followed an entry route that leads to the lysosomal (endocytic) compartments that contain numerous proteolytic enzymes. These observations led Joliot and Prochiantz to propose an unconventional mechanism to explain this membrane passage, which was not linked to a process of endocytosis [6], henceforth referred to as translocation.
3. Molecular diversity of CPPs
Following those initial findings, studies of structure/function relationships identified penetratin (RQIKIWFQNRRMKWKK) [6] and Tat (described in the literature equally by the sequences YGRKKRRQRRR and GRKKRRQRRPQ) [7], which were the first vector peptides to be described in the 1990s. These peptides (Figure 1) are not only capable of crossing the biological membranes by a mechanism not fully understood yet but also of carrying different types of molecules with them [8]. These two inseparable criteria (membrane passage and transport) enable qualifying a peptide sequence as a cell-penetrating one. Over the past 30 years, numerous other cell-penetrating peptides (CPPs) have been described based on proteins [9] such as those that interact with heparin (such as superoxide dismutase, epidermal or platelet-derived growth factors, intestinal mucin, apolipoprotein B), with RNA (such as Tat and Rev from human immunodeficiency virus type 1), and with DNA (such as the HPs Engrailed, Fushi tarazu, Hoxa5, PDX1, etc.).
These peptides have in common a basic isoelectric point, being thus positively charged at pH 7.4, and are sometimes endowed with a more or less amphiphilic profile. Compared to antimicrobial peptides (AMPs), some of which exert their bacteriolytic activity directly by irreversibly disrupting the bacterial membrane, which are also generally positively charged sequences at pH 7.4, vector peptides are not cytotoxic. The CPPs can disrupt temporarily the membrane to enter cells, for example by forming transient pores, but are not cytotoxic. Transient pore formation in a cell membrane is for instance a biologically occurring process known as membrane repair [10, 11].
4. An entry mechanism of CPPs still subjected to controversy
For years, many research groups working on the mechanism of internalization of CPPs, using multidisciplinary approaches (chemistry, biophysics, biochemistry, and biology), have contradicted each other article after article. Indeed, over the past 30 years, most publications have indicated that the route of entry of these peptides into cells is an endocytosis mechanism, implying that the peptides are able to exit endosomal vesicles so that the transported cargoes can exert the observed biological activity. Other studies, on the other hand, refer to an original mechanism known as translocation that can be assimilated as direct passage of the plasma membrane, even if it appears thermodynamically impossible for a heavily charged peptide to cross the hydrophobic core of the lipid bilayer. The controversy also mainly arose from the complexity and difficulties to observe translocation compared to endocytosis in living cells. Currently, it is quite accepted that the two main internalization processes, endocytosis and translocation (Figure 1), can coexist for one peptide used at one concentration [12, 13, 14], which makes the study of one pathway versus the other even more complicated. Various studies reporting translocation rely on model membrane systems that are less complex than living cells but allow observing peptides crossing a lipid bilayer without the requirement of an energy input. It is also possible to quantify a peptide entry through translocation by working with cells kept at low temperature (<12 °C), where all energy-dependent processes are inhibited. However, one should keep in mind that working with cells at low temperature not only inhibits endocytosis paths but can also affect membrane dynamics and fluidity since temperature modifies the phase behavior of lipids. Altogether, this translocation mechanism cannot be equated with passive diffusion in its admitted definition.
Indeed, quantitative studies to evaluate the intracellular concentrations of these peptides show that they do not diffuse through the lipid bilayer as can do small hydrophobic molecules such as benzene. Generally speaking, the ability of a peptide sequence to form intramolecular hydrogen bonds should promote more or less its passive membrane permeability by reducing the free energy cost of peptide desolvation as they insert into the membrane. Nevertheless, this prediction alone is not sufficient to fully design in silico efficient membrane-passing vector peptides as illustrated by studies using cyclic carrier peptides [15, 16, 17].
Cellular studies generally aim to qualitatively monitor the passage of the vector peptide and/or the transported cargo molecule. The methods used are thus indirect, generally based on monitoring fluorescence by imaging approaches or on detecting the biological activity of a transported cargo [18]. Other approaches are also used with cells, such as electron or Raman microscopy to study CPP secondary structure or CPP-induced membrane structures as they pass from the outside to the inside of cells [19, 20, 21].
5. Methods to quantify CPPs inside cells and study the mechanisms of entry
5.1. Mass spectrometry
In this context, we developed a method for quantifying CPPs inside cells or bound to cell membranes, using matrix-assisted laser desorption ionization–time-of-flight mass spectrometry (MALDI–TOF MS) [22]. This approach has been adapted from existing robust methods demonstrating in particular that MALDI–TOF MS can become quantitative, whatever the ionization yield of a peptide sequence [23, 24].
The method is based on the synthesis of peptides with an N-terminal tag consisting of a biotin and several glycines. The glycines are incorporated in one peptide sequence in hydrogen form (1H) and in the other in deuterated form (2H), which acts as an internal standard. Depending on the mass of the peptide, the number of glycines is adapted to ensure that the isotopic multiplets of the hydrogen and deuterated peptides do not overlap during MS analysis. For the sequences we have studied so far, the two peptides have generally an overall mass difference between 6 amu (3 glycines) and 10 amu (5 glycines). The biotin moiety enables selective “fishing” of the peptides out of the cell lysates using streptavidin-coated magnetic beads. At that point, it should be mentioned that for delivery issues, a biotin transporter is present and can be targeted in differentiated polarized animal cells and in dysfunctional cells such as some cancer cells. The biotin transporter can enhance the uptake of biotin-conjugated molecules in those cells [25]. However, in most other cell types, such as CHO-K1, the absence of biotin transporters makes the study of the cell internalization pathways of CPPs, endocytosis and translocation, much easier [26]. By contrast, in bacteria, it is well known that biotinylated peptides up to 31 amino acids in length can be taken up by the biotin transporter [27], making this transporter a good target for the development of novel antibiotic strategies.
Briefly, the quantification protocol consists first in the plating of a controlled number of cells. We found typically that one million adherent cells are convenient to get a detection signal sufficient for accurate quantification of CPP sequences. The cells are incubated with the peptide. At the end of incubation, the cells are washed and then treated (about 2 min at 37 °C) with trypsin, which both degrades the membrane-bound peptide and detaches the cells. After the addition of a trypsin inhibitor, the cells are transferred to microtubes. A known quantity of the 2H peptide is then added to the lysis buffer that is added to the cells. The cellular mixture is immediately boiled to destroy intracellular enzymes and any interactions of the internalized peptide with cellular biomolecules. Magnetic beads functionalized with streptavidin are then used to retrieve the peptide (in its 1H and 2H forms). These beads are stringently washed before releasing the peptide mixture directly with the acid matrix used for MALDI–TOF analysis. Once the mass spectrum has been obtained, the quantity of internalized 1H peptide is determined by the ratio of the areas of the isotopic masses corresponding to the 1H peptide and the standard 2H peptide introduced in known quantity.
The quantitative results obtained using this method show that translocation cannot be assimilated to passive diffusion since, depending on the experimental conditions and cell types, there is no equilibration of concentrations between extracellular and intracellular media [12]. In the absence of passive diffusion, a translocation mechanism implies that the peptide disrupts or disorganizes temporarily the lipid bilayer. Various modes of action have been proposed, most often based on observations made with membrane models (liposomes of different sizes and phospholipid composition) or proposed by analogy with antimicrobial peptides (formation of transient pores or reverse micelles).
5.2. Fluorescence imaging and spectroscopy
The use of fluorophore-labeled CPPs (Fluo-CPPs), to detect and/or quantify CPPs inside cells is a predominant method to study CPPs’ membrane-crossing activity. Although fluorescence is widely used because it allows quick, direct, and easy detection of Fluo-CPPs, mainly by fluorescence imaging and flow cytometry analyses, these two techniques neither reflect nor allow to determine quantities of those inside cells, when the main fluorophore generally used is carboxyfluorescein (CF). As the fluorescence quantum yield of fluorescein is pH-dependent, once linked to a CPP, some local membrane environment and cell compartmentalization prevent accurate detection of CF-CPPs. In addition, membrane insertion of the Fluo-CPP can lead to non-fluorescent aggregates or induce fluorescence fading. Quenching phenomenon of Fluo-CPPs can also affect fluorescence detection, potentially resulting in a tiny difference between the quantity of cell-associated peptide (cell-membrane-bound plus internalized peptides) versus internalized peptides only. As the membrane and intracellular behavior and fate strongly depend on the peptide sequence, all the above listed bottlenecks make difficult, if not impossible, a comparison between internalized species of different structurally unrelated CPPs.
For the reasons mentioned above, we developed a method to quantify Fluo-CPPs’ cellular uptake by fluorometry after cell lysis [28]. By this method, the lysis of cells allows the release of Fluo-CPPs from confined cell organelles or membrane domains and hence the full recovery of fluorescence inside cells. Adapted from our MS quantification protocol, there are main drawbacks regarding the measured signal, and caution must be taken in the interpretation of data. First, as we observe a fluorescent signal, we have no guarantee about the molecular integrity or presence of the CPP behind the signal detection when, by MS, we only quantify an intact peptide. Second, it is now widely reported that the incorporation of a fluorophore into a peptide sequence impacts the physicochemical properties since it can enhance drastically the hydrophobicity of a small peptide as most fluorophores correspond to the molecular weight of 2–4 hydrophobic amino acids [29, 30, 31]. For instance, we have observed that N-terminally labeled peptides have generally higher internalization efficiency compared to biotinylated ones. This indicates that the hydrophobic/hydrophilic property of the N-terminal ending group in the sequence is crucial to the penetrating efficacy of the peptide.
Regarding the quantification of direct translocation, the fluorescent methods, as the MS one, describe the use of low temperature since the use of specific endocytic inhibitors leads to results difficult to interpret because of the existence of various biological side effects of those compounds in cell lines [32].
In collaboration with the group of Banoczi, we recently reported a new method that allows direct observation and evaluation of translocation in cells kept at 37 °C [33]. The protocol requires the use of 4-(dimethylaminoazo)benzene-4-carboxyl acid (DABCYL) and CF-labeled peptides. When the labeled peptide is intact, the fluorescence signal is weak thanks to the dark quenching property of DABCYL over CF. After complete enzyme hydrolysis of the peptide, we can measure a 10-fold higher fluorescence signal thanks to the release of CF. We used this “quenching and release” property to evaluate translocation at 37 °C and compare it to the 4 °C condition. The peptides can be internalized by endocytosis and then end up in lysosomal vesicles in which enzymes can degrade the peptide sequence. If the peptide translocates to reach the cytosol, it remains intact and non-fluorescent. By harvesting cells and systematically analyzing them intact or after full lysis, we have access to the quantity of peptide internalized either by the two paths or by endocytosis only. Subtracting the two conditions allows us to quantify translocation at 37 °C and compare it to the 4 °C one. This method, based on Förster resonance energy transfer (FRET) quenching and release upon exposure of peptides to enzyme degradation, is easy, does not require any cell engineering, can be adapted very quickly to any cell type, and allows quantifying the internalization of CPPs [34, 35]. However, the major drawback of this method is that it requires the incorporation of DABCYL and CF moieties that impact the hydrophobicity of the studied peptide sequences.
6. Key amino acids for cell penetration of CPPs
In addition to the development of reliable quantification methods, the work of our group focuses on the molecular understanding of uptake mechanisms, one aspect of which is studying the precise role of some amino acids in CPP sequences. As already described as the magic arginine and tryptophan power [36], these two amino acids are crucial to cell penetration thanks to the unique non-covalent bonds they can establish, which we detail (vide infra). Other amino acids such as cysteine can significantly impact internalization, in particular through disulfide cross-exchange with thiol-containing cell membrane proteins [37, 38].
6.1. Magic arginine
Arginine contains a guanidinium group on its side chain. The ability of guanidinium groups to form bidentate hydrogen bonds easily endows oligo-arginines with a hydrophilic or hydrophobic character, depending on the associated counter-anion (Figure 2). For example, in an octanol/water mixture, all fluorescent octa-arginine (R8), octa-lysine (K8), octa-ornithine (Orn8), and Tat(49-57), distribute in water only [39]. When free fatty acids (⩾C10) are added to the mixture, the peptide partition shifts to the octanol phase.
Non-covalent and covalent bonds between guanidinium (Arg), indole (Trp), and thiol (Cys) groups and biological molecules at the cell surface, which are important for cell entry of CPP sequences. CPP linked by disulfide bridges to membrane proteins can be internalized after endocytosis of the given protein or transbilayer disulfide exchange.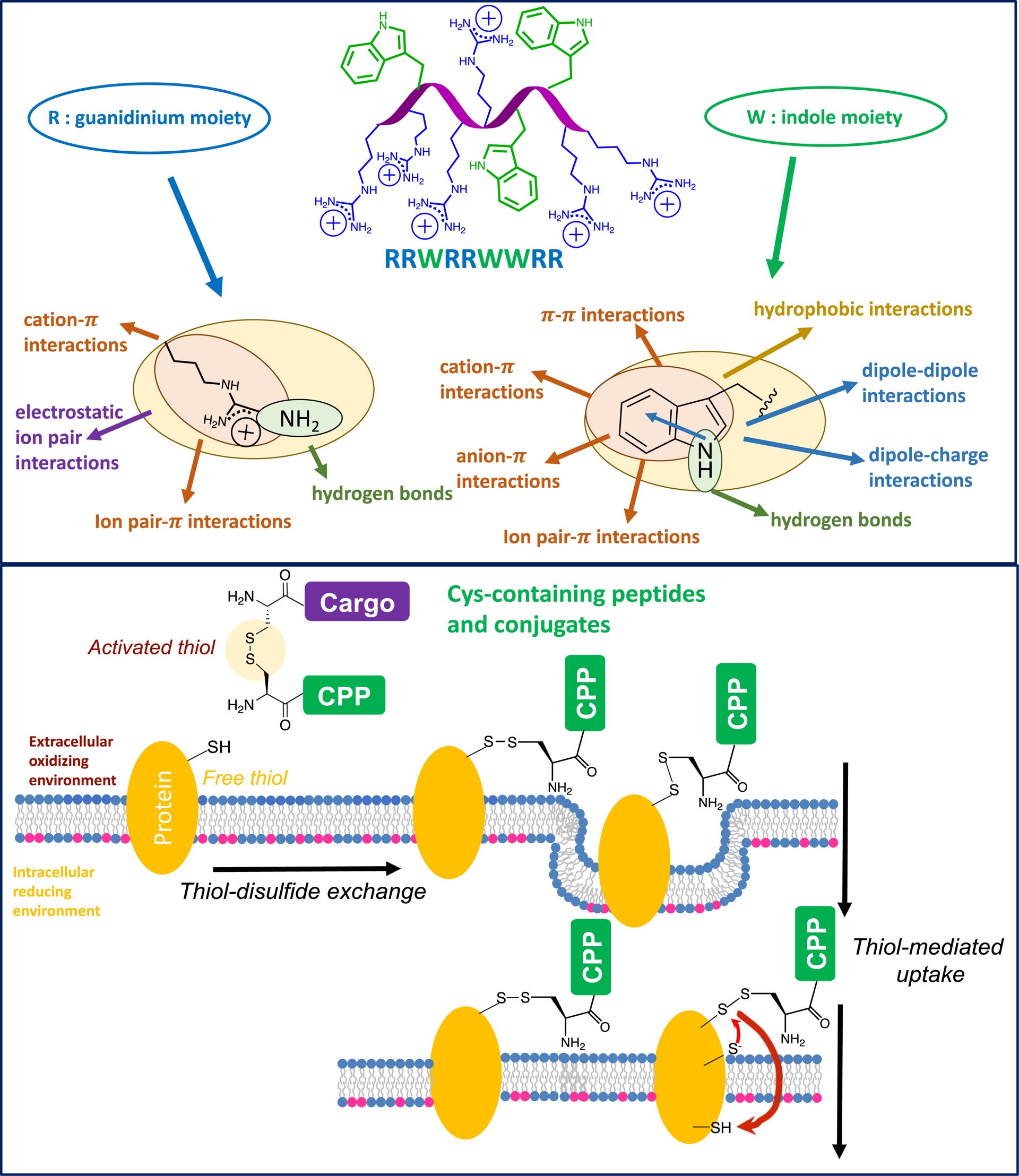
The order of peptide partition in octanol is R8 > Tat > K8 ≈Orn8. The octa-arginine peptide localizes 100% in octanol with 2 equiv of sodium laurate (C12), while Tat (6 Arg) does with 4 equiv. In sharp contrast, octa-lysine (K8) and octa-ornithine (Orn8) are equally distributed between water and octanol with the addition of 5 equiv of sodium laurate. These guanidinium-rich peptides are only weakly associated with counter-ions in the extracellular medium, and can readily exchange these counter-ions to form hydrogen bonds with phosphates, sulfates, and/or carboxylates present on the cell surface [40]. These hydrogen bonds transform the polar cationic peptides into lipophilic ion pairs whose hydrogen bonds become stronger as they penetrate the non-polar lipid bilayer. This way, the interaction between these arginine-rich peptides and the phosphate groups of the lipid heads masks the peptide charge, attenuating its polarity and enabling adaptive diffusion across the plasma membrane [41].
6.2. Powerful tryptophan
As for tryptophan (Trp), it is an aromatic amino acid with unique physicochemical properties. It is often found in membrane proteins, notably at the interface between the lipid bilayer and the surrounding aqueous environment. It plays an essential role in the stability, anchoring, and orientation of membrane proteins in the lipid bilayer. It is generally considered a hydrophobic amino acid. However, this hydrophobicity varies greatly depending on the method used to determine it. Tryptophan is considered either a very hydrophobic (Wimley and White scale, 1996), moderately hydrophobic (Kyte and Doolittle scale, 1982), or not very hydrophobic (Moon and Fleming scale, 2011) amino acid. Whatever the case, this amino acid can be engaged in many types of non-covalent bonds (π–cation, π–π, π–anion, and hydrogen bonds) and thanks to its dipole moment, in dipole–dipole or dipole–charge type interactions [42, 43].
We reported that Trp-containing CPPs have enhanced internalization efficacy compared with analogues that do not contain this unique amino acid [44, 45, 46]. We have recently added a new type of interaction to the already long list of interactions that the indole group of Trp can establish: π–ion pairs [47, 43]. We used a series of nona-peptides composed only of Trp (from 0 to 5) and Arg (from 9 to 4), which we compared to a series of oligo-arginines (5 to 9), for their capacity to internalize (Figure 3). Results of the quantification of the internalization efficacy of the peptides indicate that R8 and R8W internalize with the same efficacy in cells [47]. So do R7 and R7W2, but R6W3 and R5W4 are better internalized in cells, respectively, than R6 and R5 and even than R9. These results indicate that Trp can advantageously replace Arg to maintain or enhance internalization efficacy. In addition, the number of Trp essentially impacts internalization only in cells expressing negatively charged glycosaminoglycans (GAGs), showing the importance of these cell-surface molecules for the internalization process. In parallel, we could show by isothermal calorimetry (ITC), to analyze the thermodynamics of peptide/heparin, a favorable enthalpy in the formation of the complexes. This enthalpy is balanced by an unfavorable entropy resulting from the loss of flexibility of the peptides bound to heparin, overtaking the favorable counter-ion release. Increasing the number of Trp leads to enhanced favorable binding enthalpies. To explain these results, a DFT analysis by Bauzá and Frontera highlights the crucial role of π–ion pairs involving sulfates from glycosaminoglycans and Arg and Trp in the peptide sequence, which contribute to the binding enthalpy of the peptides to heparin [47]. Formation of heparin/peptide complexes is thus more stable in the presence of ion pair–π interactions (Figure 3).
Impact of Trp on thermodynamics of interaction and internalization efficacy of arginine-rich CPPs thanks to its unique ability to form π–ion pair interactions with guanidinium and carboxylate/sulfate/phosphate groups [47].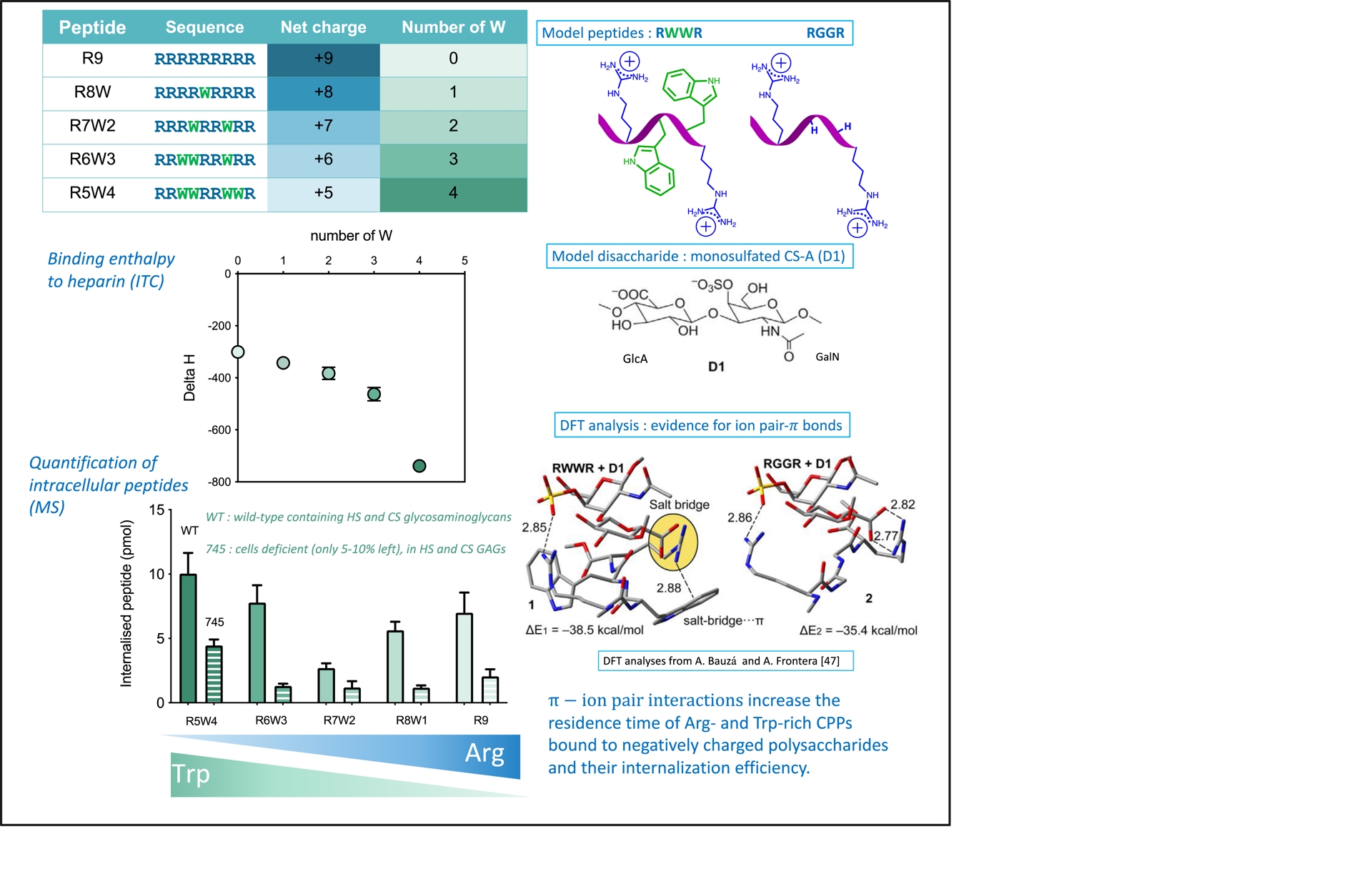
Interestingly, in the hundred of HPs so far identified, a tryptophan can be found at position 48 in the sequence, and is over 90% conserved. Tryptophan 48 is found in the third helix of the homeodomain, which corresponds to the so-called penetratin sequence (Figure 1). This tryptophan is essential for the peptide translocation across the membrane [48], notably due to its ability to interact through π–cation with choline-type polar heads (quaternary ammonium) found in phosphatidylcholine or sphingomyelin (Figure 4).
The different types of molecules (heparan and chondroitin sulfate glycosaminoglycans, sialic acids, cysteine-containing proteins, lipid polar heads) that interact with CPPs in the plasma membrane.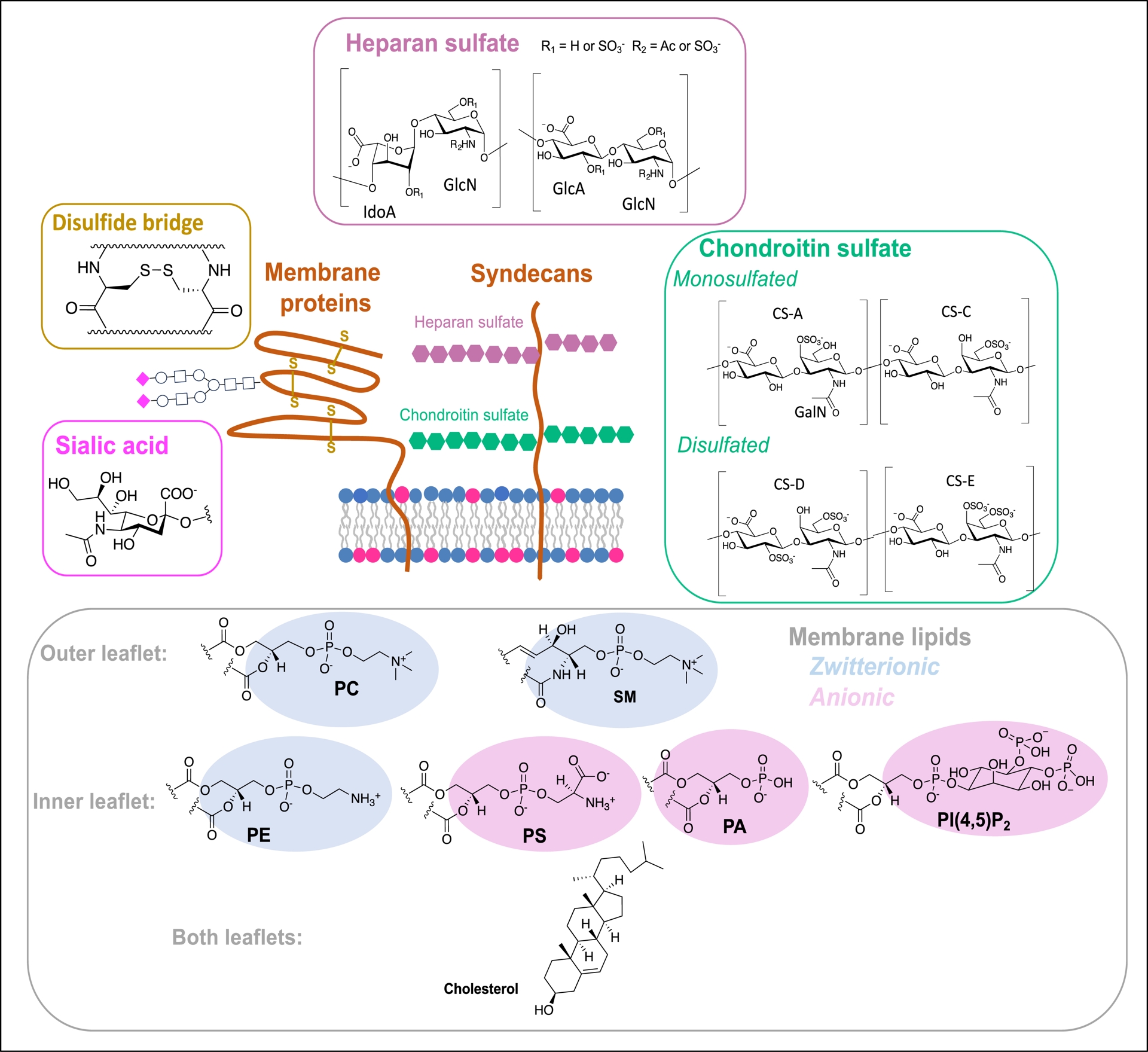
In addition to these interactions with membrane lipids, when present in an arginine-rich peptide sequence, tryptophan can establish π–ion pair interactions with anionic cell-surface glycosaminoglycans. These π–ion pair interactions increase the enthalpy of interaction of these peptides with glycosaminoglycans. This increase in enthalpy implies a longer residence time or interaction of the peptides with negatively charged glycosaminoglycans. This longer residence time helps to explain greater internalization, either by endocytosis or translocation of these peptides directly to the membrane [47]. We are currently developing Trp analogues to tune these crucial π–ion pair properties, and we intend to design more efficient CPPs.
6.3. Versatile cysteine
As mentioned above, these vector peptides are molecules with a strong avidity for membranes and can interact with various membrane molecules or partners. Any modification of the physicochemical parameters, such as amino acid sequence modification of these peptides, impacts their ability to interact with the membrane components and thus their internalization efficiency and entry pathways. In this context, we have first shown that cysteine-containing peptide vectors are capable of cross-reaction with thiol groups present in the membrane (cysteine-containing proteins) and that this reactivity strongly influences their internalization into cells (Figure 2) [49]. More generally, the link between a carrier peptide and a cargo molecule to be transported into cells influences the internalization efficiency, the mechanism of conjugate entry, and the final intracellular localization of the conjugate (Figure 2). The disulfide bridge is often used to enable peptide vectors to transport bioactive molecules into cells. The disulfide bridge is in fact relatively stable in blood plasma, and it is only cleaved once it has entered reducing intracellular compartments, in particular the cytosol, allowing the transported molecule to be released and exert its biological action. Thiol/disulfide bridge exchanges at the cell surface have thus been studied for their impact on the entry of carrier peptide/transported molecule conjugates or of carrier peptides cyclized by a disulfide bridge (Figure 2). Thiol/disulfide exchanges at the cell surface can lead to the reduction of disulfide bridges or to covalent coupling to membrane proteins. Conjugated species then remain trapped on the surface, or allow peptides (vector peptides and originally non-permeable molecules) to enter the cell.
These specific thiol–disulfide exchanges and abilities to internalize thiol-containing compounds are now being widely exploited in a biomimetic way to transport biologically active molecules into cells [50, 51, 52, 53, 54].
7. Reaching cell-specific penetration of CPPs through membrane partners (Figure 4)
7.1. Membrane protein targeting
Besides the poor understanding of their mechanisms of entry, the fact that CPPs can enter any type of cell somehow prevents or impedes their easy use in therapeutic or biotechnological applications. Some attempts have been made to endow these peptides with more specific cell entry. The more classical approach is to conjugate the cell-penetrating sequence with a targeted one. In most cases, the CPP is conjugated to a peptide sequence that recognizes an overexpressed membrane receptor in specific cells, for example in cancer cells [55]. These targeted sequences are referred to as cell-targeting peptides [56], tumor-targeting peptides, or tumor-homing peptides [55].
The receptors that are overexpressed on tumor cells are various, such as folate receptors, integrins, somatostatin receptors, transferrin receptors, and epidermal growth factor receptors. It is interesting to note that natural ligands retain their specific recognition of these overexpressed receptors and can be conjugated to CPPs for tumor targeting and entry. In terms of therapeutics, targeting specific cells or tissues decreases unwanted side effects.
7.2. Targeting cell-surface glycosaminoglycans to attain specific cell internalization
Besides this protein-targeted approach, the group of Prochiantz reported the existence of GAG-specific recognition motifs within the sequence of the HP Otx2, involved not only in the development during the embryonic stage but also at postnatal stages and in adulthood [57, 58]. The mouse binocular vision usually develops from postnatal 20 to 40 days and is marked by the maturation of the inhibitory interneurons expressing parvalbumin (PV-cells) in the visual cortex. During that period, plasticity is progressively opened and closed as the inhibitory/excitatory balance shifts with enhanced inhibition [59]. If the eye of the mouse is closed before postnatal 20 or after postnatal 40 days, it leads to amblyopia of the closed eye. Otx2 is a key regulator that is transported into the PV-cells during the critical period of binocular vision. This non-autonomous activity of Otx2 implies that specific binding sites for Otx2 must be expressed on the PV-cell membrane surface at the onset of the plastic period. Injection of exogenous Otx2 in the visual cortex leads indeed to the accumulation of the protein preferentially into PV-cells thanks to its interaction with disulfated chondroitin glycosaminoglycans expressed in the perineuronal nets: (2S, 6S)CS or CS-D and (4S, 6S)CS or CS-E. A short peptide motif partially overlapping the Otx2 homeodomain was found as the CS-binding sequence [60].
We extend this study to another HP, Engrailed2 (En2) [61]. This HP controls the patterning of vertebrate embryos, in particular through the regulation of boundary formation in the developing brain. By contrast to Otx2, extracellular En2 poorly accumulates in PV-cells, the amino acid sequence preceding the homeodomain strongly differing from that of Otx2 [60]. The En2 sequence is indeed highly enriched in basic amino acids with homology with nuclear localization signals. Using in particular, chemical shift deviations by NMR analyses, we found a high-affinity GAG-binding sequence (RKPKKKNPNKEDKRPR), upstream of the homeodomain [61]. We could determine that this sequence controls En2 internalization through selective interactions with highly sulfated heparan sulfate GAGs. We are currently developing chimera of conjugated CPPs and GAG-targeting sequences to evaluate with cells varying in their plasma membrane GAG content, whether these mini-HPs can enter better in cells containing the targeted sulfated polysaccharides. In addition, other HPs will be studied to identify other natural GAG-targeting peptides.
7.3. Other important partners in the cell membrane: sialic acids
As most CPPs are positively charged peptides at pH 7.5, it seems obvious that they can interact with anionic molecules present in the plasma membrane of cells. We have already mentioned GAGs that are long, linear polysaccharides consisting of repeating disaccharide units of the extracellular matrix. In addition, sialic acid rich oligosaccharides are found on glycoconjugates (glycolipids, glycoproteins, proteoglycans) present in the outer layer of the plasma membrane. These glycoconjugates contribute to the net negative charge of the cell surface. We have identified sialic acids as binding receptors for penetratin [62] as previously described for Antennapedia HP [63].
Using plasmon waveguide resonance (PWR), we have shown that penetratin has better affinity for cell membranes containing heparan sulfates, chondroitin sulfates, and sialic acids (wild-type [WT] CHO-K1 cells) over those losing sialic acids (CHO-lec2) when cell membranes lacking 85–90% of heparan and chondroitin sulfates (CHO-pgsA745) are poor binders of penetratin [62]. These sialic acid rich membrane components retain the peptide at the cell surface but prevent its internalization as we demonstrated that the peptide is less internalized in WT than in lec2 cells. These results imply that sialic acids can act as passive binders that maintain and retain the peptide at the cell surface until the peptide is captured by higher-affinity partners, such as glycans and lipids.
Interestingly, the same kind of result has been observed for HPs. Joliot and collaborators early on showed that high polysialic acid expression traps Otx2, leading to a reduction in HP internalization [63]. However, the coinfusion of polysialic acid and Otx2 in the cortex is mandatory for the protein activity in the brain. Polysialic acid increases indeed the diffusion of the protein in the brain [60]. In the absence of polysialic acid, Otx2 is taken up by cells around the infusion site, when its presence allows the protein to reach neurons embedded in highly negatively charged perineuronal nets, leading to internalization of the protein. These results suggest that during its physiological travel in the brain, Otx2 binds low-affinity glycans and, once in the cortex, transfers to higher-affinity ones found in perineuronal nets such as disulfated chondroitins [59]. It is thus important to keep in mind that the affinity of peptides for cell plasma membrane components does not reflect their internalization capacity into the cells.
7.4. Other important partners in the cell membrane: phosphatidylinositol bisphosphate
Besides glycans, we have studied the role of specific phospholipids in the plasma membrane by a combination of biophysical and cell biology approaches. In contrast to the general and well-admitted thought, positively charged CPPs can interact more widely than only negatively charged molecules (Figure 4). We have shown using PWR analysis that penetratin can bind to various sets of phospholipid bilayers [62]: egg L-a-phosphatidylcholine (PC, zwitterion), egg L-a-phosphatidylglycerol (PG, anionic), egg PC/POPG (3:1; anionic, POPG being 16:0–18:1 or 1-palmitoyl-2-oleoyl-sn-glycero-3-phospho-(1′-rac-glycerol)), and egg PC/DOPE (1:1; zwitterion, DOPE being 1,2-di-(9Z-octadecenoyl)-sn-glycero-3-phosphoethanolamine or PE(18:1(9Z)/18:1(9Z)) (Figure 4). The peptide binds first the phospholipid polar heads before eventually inserting in between the alkyl chains. The binding affinity of penetratin for these bilayers reflects the interaction with the polar heads, being either zwitterions, anions, or cations. Our results show that penetratin has 100 times higher affinity for egg PG compared to egg PC bilayers. However, when incorporating PE into PC bilayer (Egg PC/DOPE), although still zwitterionic, the bilayer binds penetratin with a 60-fold increased affinity, similar to egg PG alone. Due to its small headgroup, the critical packing parameter of PE is above 1 [64], making it an “inverted-cone” shaped phospholipid, PE-rich domains being known to exert bending fluctuations of the plasma membrane. These results indicate that the dynamics and membrane curvature properties of the bilayer are crucial parameters for the interaction of CPPs with the cell membrane bilayer.
We also demonstrated with photolabeling coupled to MS analyses [65, 66] that penetratin showed a preference for negatively charged (versus zwitterionic) polar heads and for unsaturated (versus saturated) and short (versus long) saturated phospholipids. These studies highlight the potential of using benzophenone to probe the environment and insertion depth of membranotropic peptides in membranes [65, 67].
More recently, we analyzed the role of PI(4,5)P2, a minor lipid in the cell plasma membrane but bearing three negative charges at pH 7.4, for the internalization of penetratin [68]. Penetratin partitioned more favorably in PC/PI(4,5)P2 than in PC/PS liposomes of similar global negative charge. Interestingly, this preference for PI(4,5)P2 over PS is also shared by En2 [69]. Regarding penetratin, PI(4,5)P2 of the outer leaflet of the bilayer seems to act as sialic acid, being a binder but not an active player in the internalization process. Blocking this pool of PI(4,5)P2 by a selective PIP2-binding protein domain results in an increase of penetratin internalization. An opposite effect was found for the PI(4,5)P2 in the inner leaflet. Occupancy of this lipid present in the inner leaflet by its binding to a selective PI(4,5)P2-binding domain enhances penetratin internalization. Altogether, these results show that depending on its location on either side of a plasma membrane, the same molecule can inhibit or enhance the internalization efficacy of a CPP, adding more complexity to the full understanding of the process.
8. Conclusion and perspectives
The existence of cell-penetrating peptides or protein-transduction domains was described about 35 years ago. It is obvious that such peptides are of major interest for delivery purposes, in particular therapeutics since most drugs in the market target extracellular membrane proteins. The field of CPPs is much younger than that of AMPs. The first (rather large) AMP, lysozyme, was reported by Alexander Fleming, shortly after the first World War. AMPs can act at the membrane level or inside bacteria and address a specific target. These two families that have been studied separately share at least a strong avidity for biological, although different, membranes. AMPs acting at the membrane level can destabilize the architecture of the bacterial cell wall (such as the lipopolysaccharide) or create stable pores in the lipid bilayer, which are deleterious to bacterial survival. Many AMPs have been approved by the US Food and drug Administration (FDA) [70].
By contrast, successful applications of CPPs as conveyors of therapeutic molecules inside cells are lacking and no CPP–drug conjugate except a CPP excipient that binds non-covalently to its cargo [71] has been so far approved by the FDA, unlike other peptides [72]. Many issues should be indeed addressed before translating CPPs into clinics, such as immunogenicity, stability, or bioavailability questions.
Apart from these expected applications, the field of CPPs is totally interdisciplinary, including synthesis, analytical and theoretical chemistry, biophysics, biochemistry, and cell biology. In our laboratory, we try to design potent penetrating sequences to identify membrane partners and to understand their mechanism of action as well as to study drug–peptide conjugates for biotechnological or therapeutic applications. The field definitely benefits from the close proximity of HPs, which naturally travel from one cell to another. Understanding the mechanism by which the peptides internalize into cells, in particular by direct translocation of the plasma membrane, permits us to exploit the path as for the ring tension applied to thiol-mediated cellular uptake or incorporation of more hydrophobic moieties such as DABCYL [34, 35, 73, 74, 75]. In this direction, we are currently developing highly efficient CPPs incorporating Trp analogues.
Declaration of interests
The authors do not work for, advise, own shares in, or receive funds from any organization that could benefit from this article, and have declared no affiliations other than their research organizations.
Funding
This work was supported by projects ANR-20-CE44-0018, ANR-17-CE11-0050 and the Arc Foundation for Cancer Research (PJA20181208198). FT is supported for her PhD by the ANRT - CIFRE Sanofi.
Acknowledgments
The authors would like to express their warmest thanks to all the colleagues at the Laboratoire des Biomolécules who contributed to some of the work cited in the manuscript, as well as to the MS3U mass spectrometry platform at Sorbonne University.