

Version abrégée
1 Introduction
De multiples expériences montrent que la structure et la dynamique de l'eau près d'une interface ou dans un espace confiné sont fortement modifiées par rapport à l'eau liquide. De telles modifications sont d'une importance cruciale dans de très nombreuses disciplines scientifiques (biologie, physique, chimie, sciences de l'environnement, etc.) et doivent nécessairement être prises en compte pour comprendre les phénomènes d'adsorption aux interfaces solide–solution aqueuse.
2 Structure de l'eau
L'eau est un liquide tout à fait particulier (souvent appelé anormal) avec une constante diélectrique et un moment dipolaire élevés, qui lui confèrent ses propriétés solvantes, dissociantes et ionisantes [45,78]. Le comportement unique de l'eau par rapport à d'autres liquides tient principalement à l'association tridimensionnelle des molécules d'eau par liaisons hydrogène [79]. En effet, la molécule d'eau présente une configuration optimale pour la formation de telles liaisons [142], chaque molécule pouvant être à la fois donneuse et accepteuse de liaisons. Ce comportement est bien illustré par la structure de la glace I, qui présente un réseau tétraédrique de liaisons hydrogène (Fig. 1). L'application des techniques de diffraction des rayons X et de neutrons à l'eau liquide [19,30,102,106] révèle un ordre local tétraédrique et une forte dépendance vis-à-vis de la température, attribuable à une augmentation des corrélations angulaires à basse température. L'étude de la dynamique des liaisons hydrogène par des techniques diélectriques [12,69], Raman basse fréquence [101,108] et de diffusion neutronique [30,149] permet de proposer un modèle plausible de l'eau fondé sur les théories de la percolation (Fig. 2) [140,141,148]. L'eau peut alors être considérée comme un gel, dans lequel la connectivité des différentes régions est assurée par les liaisons hydrogène [42]. Ce réseau est modifié en permanence, les clusters ayant une durée de vie de l'ordre de la picoseconde, tout en conservant sa connectivité. Cette image est dans l'ensemble confirmée par les simulations informatiques de l'eau même si, à l'heure actuelle, aucun potentiel numérique ne permet de retrouver l'ensemble des propriétés de l'eau liquide [31,43]. Les questions qui restent ouvertes [42,91,162] tiennent principalement à la dynamique de fluctuation des liaisons hydrogène, qui est encore mal comprise.
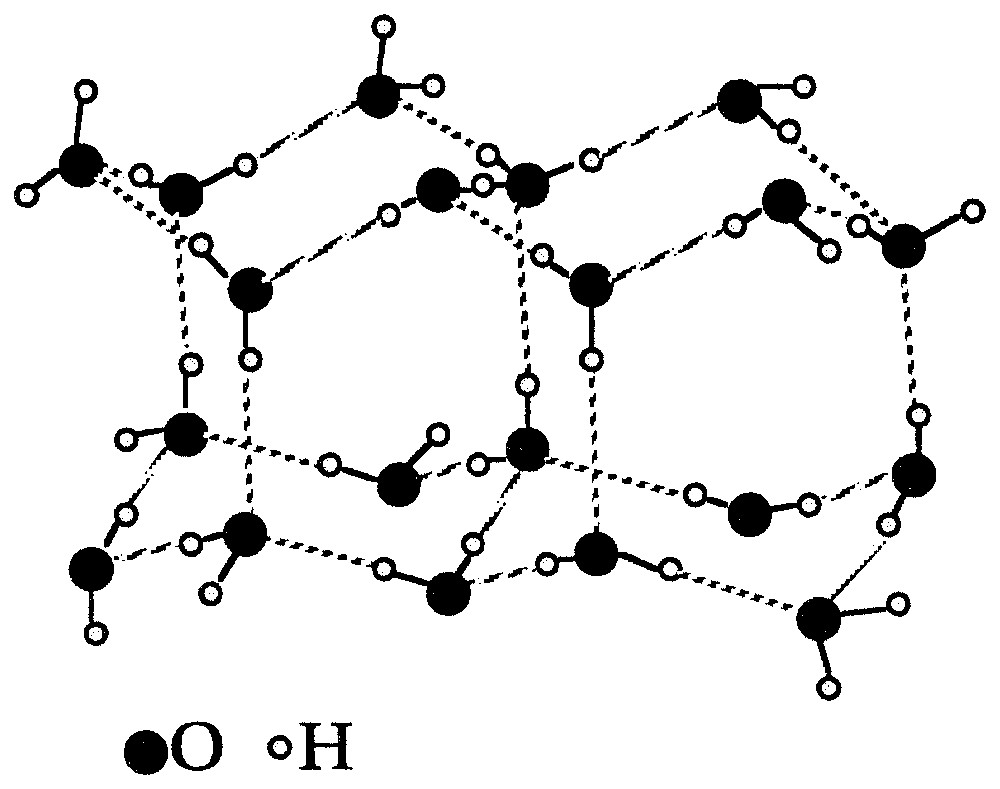
Structure of ice I (adapted from [45]).
Structure de la glace I (d'après [45]).

Picture of liquid water derived from the percolation model. The lines are drawn only for sites with four hydrogen bonds (adapted from [141]).
Image de l'eau liquide obtenue à partir du modèle de percolation. Les traits ne relient que les molécules d'eau engagées dans quatre liaisons hydrogène (d'après [141]).
3 Solutions aqueuses
La présence d'impuretés telles que des ions en solution modifie le réseau de liaisons hydrogène et par là-même les propriétés de l'eau liquide. Suivant le pouvoir polarisant des ions (défini comme le rapport z/r2), trois situations apparaissent : (i) les ions à fort pouvoir polarisant (Li+, Na+, Mg2+, F−, OH−) brisent les liaisons hydrogène et organisent de nombreuses couches d'eau autour d'eux ; (ii) les ions de pouvoir polarisant moyen (K+, Cs+, NO3−) modifient localement le réseau de liaisons hydrogène, mais n'ont que peu de molécules d'eau autour d'eux ; (iii) les ions de faible pouvoir polarisant (ions hydrophobes) ne brisent pas les liaisons. Ils s'incorporent au sein du liquide, dont ils modifient l'architecture locale [78,109]. De plus, la présence d'ions en solution modifie de façon significative les spectres SFG de l'interface eau–air [3,131,132] (Fig. 3).
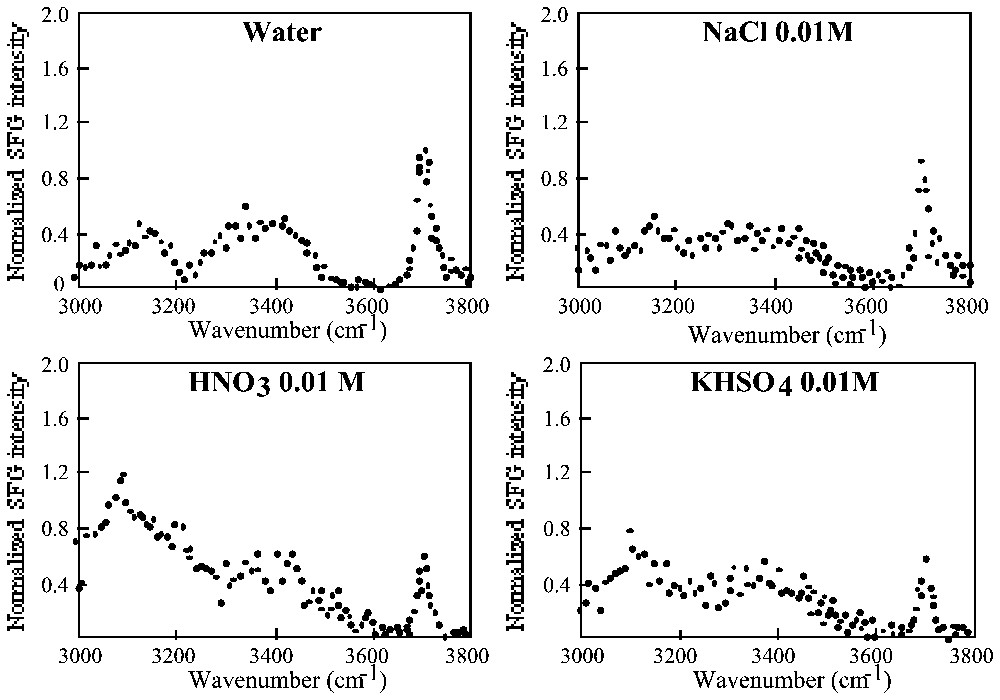
Sum frequency generation spectra of water and various solutes obtained at 277 K. Each spectrum is referenced to the free OH intensity at 3700 cm−1 (adapted from [3,131,132]).
Spectres obtenus à 277 K en génération de fréquence somme pour l'eau et différentes solutions aqueuses. Chaque spectre est normé par rapport aux OH libres à 3700 cm−1 (d'après [3,131,132]).
4 Eau aux interfaces
4.1 Surfaces métalliques
De nombreuses études se sont intéressées à la structuration des premières molécules d'eau sur des surfaces métalliques parfaitement définies. Le lecteur intéressé pourra se reporter à la revue de Thiel et Madey [152]. Les surfaces métalliques chargées sont d'une importance primordiale en électrochimie. Il apparaı̂t que, sur de telles surfaces, l'eau peut être très structurée sur deux à trois couches, avec des densités égales jusqu'à deux fois celle de l'eau liquide [154], et que la structuration dépend fortement de la charge de l'électrode. La localisation des ions dans la double couche électrochimique est également fonction de leur pouvoir polarisant et de la charge de l'électrode [136,137].
4.2 Surfaces minérales
Sur des surfaces cristallines modèles bien définies [50,51,95], l'eau s'adsorbe très souvent de façon associative [152], même sur des oxydes (Fig. 4), ce qui montre l'importance des défauts sur la plupart des surfaces « naturelles ». Dans ce dernier cas, les mesures de calorimétrie d'adsorption en fonction du pré-recouvrement [26,28,55,100,113,157] (Fig. 5), ainsi que des mesures du temps de relaxation en RMN du proton sur des suspensions à contenu solide croissant [54,55], montrent clairement que la structuration de l'eau interfaciale n'affecte que les trois premières couches adsorbées. L'existence de ces couches d'eau structurées entraı̂ne deux conséquences fondamentales pour les études d'adsorption à l'interface solide–liquide : (i) le plan de cisaillement auquel est mesuré le potentiel zêta (voir [153]) ne peut être assimilé au plan externe de Helmholtz. Il ne sera déplacé au-delà des couches d'eau structurée que dans le cas de l'adsorption de polymères pour lesquels des boucles peuvent s'étendre loin de l'interface ; (ii) à l'interface solide–liquide, les enthalpies d'adsorption mesurées correspondent en réalité à des enthalpies de déplacement de l'eau par l'adsorbat.
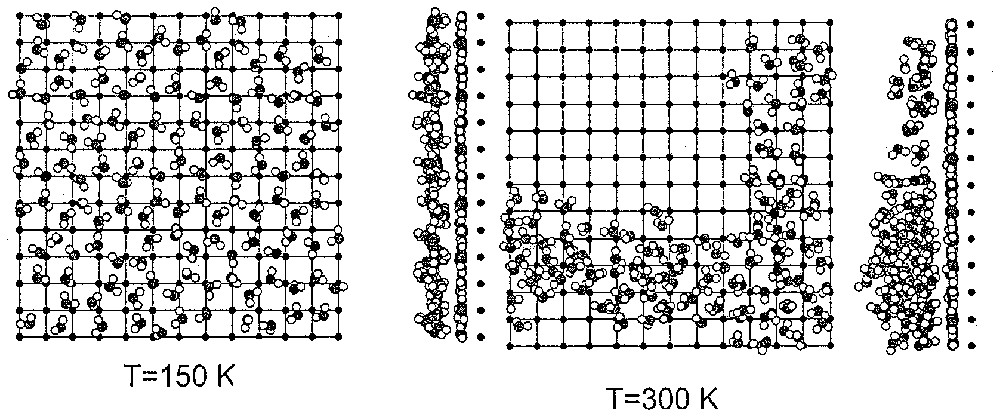
Molecular dynamics simulation of water adsorption on the (100) face of MgO (adapted from [95]).
Simulation par dynamique moléculaire de l'adsorption d'eau sur une face (100) de MgO (d'après [95]).
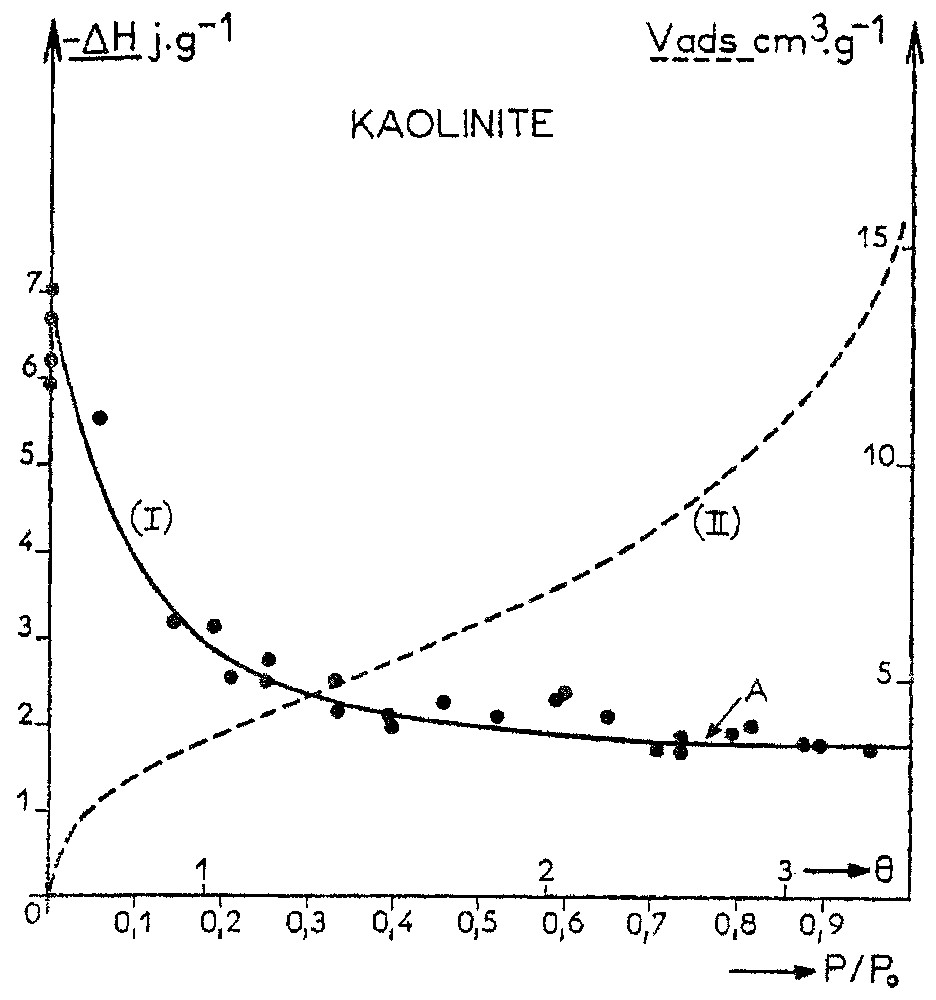
Evolution of the immersion enthalpy of Na-kaolinite at 30 °C vs pre-coverage. The dotted line relates to the corresponding water adsorption isotherm (adapted from [55]).
Évolution de l'enthalpie d'immersion de la kaolinite en fonction du pré-recouvrement à 30 °C. L'isotherme d'adsorption de vapeur d'eau correspondante est tracée en pointillés (d'après [55]).
4.2.1 Silice
De nombreuses études se sont attachées à l'étude de la structure de l'eau interfaciale et/ou de l'eau confinée sur des surfaces de silice. Les études de 1H RMN [18,23,34,85,110] révèlent une structure complexe, due à la variabilité du réseau de silanols de surface. La première « couche » d'eau est très fortement liée par des liaisons hydrogène. Dans le cas d'un verre siliceux poreux tel que le Vycor, des études Raman [32] et de diffraction des neutrons [7,22,135] montrent que, dans la première couche d'eau, le réseau de liaisons hydrogène est fortement distordu. Par diffusion inélastique et quasi élastique de neutrons [6] (Fig. 6), il apparaı̂t que les molécules d'eau adsorbées ont un comportement proche de celui de l'eau surfondue, avec des temps de résidence sur leurs sites supérieurs à ceux observés dans l'eau liquide. Des études en diéléctricité [62,129] et par relaxométrie RMN [58,86] permettent d'observer la dynamique des molécules d'eau à des échelles de temps supérieures à celles accessibles aux techniques de diffusion neutronique. On peut alors observer une diffusion ralentie des molécules d'eau à la surface des pores jusqu'à leur échange avec l'eau bulk. L'ensemble de ces informations est corroboré par les simulations informatiques [68,138].

Arrhenius diagrams of characteristic times τ1 and τ2 for water adsorbed in Vycor glass for various hydration states (adapted from [6]).
Diagrammes d'Arrhénius des temps caractéristiques τ1 et τ2 pour l'eau contenue dans le Vycor, à différents états d'hydratation (d'après [6]).
4.2.2 Matériaux carbonés
En ce qui concerne la structuration de l'eau au voisinage des surfaces « hydrophobes », la plupart des travaux se sont concentrés sur des matériaux carbonés. Les simulations informatiques [89,155] de la structuration de l'eau dans des pores à parois parallèles de graphite révèlent une couche de molécules d'eau orientées parallèlement à l'interface et de l'eau liquide au centre du pore (Fig. 7). Dans le cas des charbons actifs, la présence de groupes oxydés en surface [15,21,60,96] modifie complètement la structuration de l'eau, avec le développement de clusters d'eau liés par liaisons hydrogène sur les quelques sites hydrophiles de la surface (Fig. 8). La présence de tels clusters est confirmée par des mesures de diffraction et de diffusion des rayons X [8,72,73].
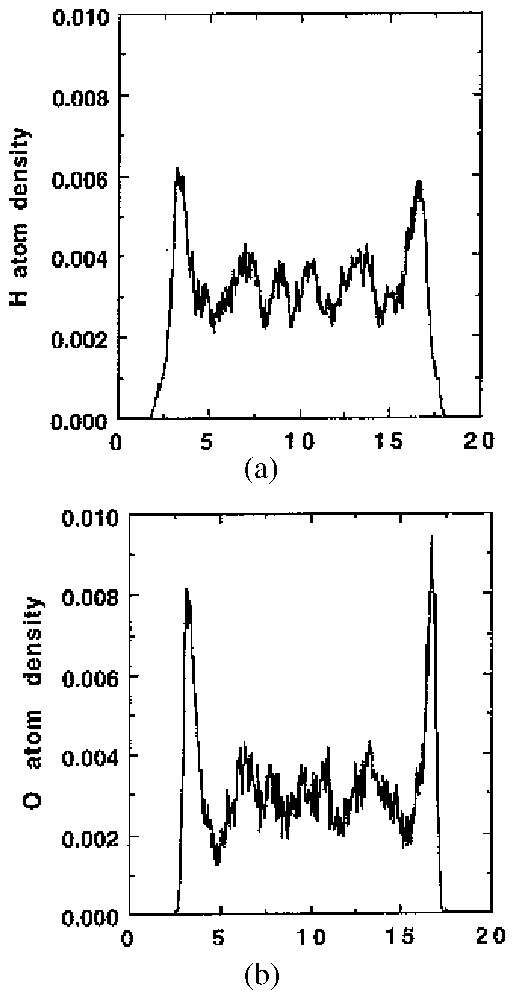
Distribution of (a) hydrogen and (b) oxygen at 298 K in a slit 20 Å graphitic pore (adapted from [155]).
Distribution (a) de l'hydrogène et (b) de l'oxygène à 298 K dans un pore parallèle de graphite de 20 Å (d'après [155]).

Configuration of water molecules in a simulated activated carbon slit (width 9.9 Å) for various relative pressures. The dark half-spheres represent oxidised surface sites. Top to bottom P/P0=0.01, 0.05 and 0.30 (adapted from [96]).
Arrangement des molécules eau dans un pore de graphite simulé (diamètre 9.9 Å) pour différentes pressions relatives. Les demi-sphères grisées représentent des sites de surface oxydés. De haut en bas P/P0=0,01, 0,05 et 0,30 (d'après [96]).
4.2.3 Solides lamellaires gonflants
Les solides lamellaires gonflants représentent une classe de matériaux particulièrement intéressants pour étudier la structuration de l'eau aux interfaces. La famille de minéraux la plus étudiée dans ce contexte est le groupe des phyllosilicates (Tableau 1) et notamment les argiles de type smectites. Les mesures d'adsorption d'eau couplées à l'étude de l'évolution des distances basales en fonction de la pression d'eau [10,24,25] (Fig. 9) révèlent l'importance primordiale de la nature du cation interfoliaire sur la « prise » d'eau. Ce rôle du cation est confirmé à la fois par spectroscopie infrarouge [14,49,77,115,118,121] et par les simulations informatiques [16,17,29,36–38]. Outre le cation interfoliaire, la localisation de la charge (tétraédrique ou octaédrique) et la quantité de charge jouent également un rôle important, bien que ces paramètres aient été moins étudiés [57,98,143,144]. La dynamique des molécules d'eau interfoliaire peut être observée par diffusion quasi élastique de neutrons [117,119]. Les molécules d'eau sont distribuées sur une superstructure et ont un temps de résidence d'environ 100 ps. Durant ce temps, elles tournent autour de leur axe C2 avec un temps de corrélation d'environ 4 ps. Les molécules d'eau dans la sphère d'hydratation du cation interfoliaire sont également impliquées dans la rotation lente (30 ps) de l'ensemble cation hydraté. Les temps plus longs peuvent être analysés en RMN [41] ou en résonance neutronique par écho de spin [145]. Il apparaı̂t alors une diffusion bidimensionnelle lente des molécules d'eau le long des surfaces.
Classification of phyllosilicates.
Classification des phyllosilicates .
| Charge/half unit cell | Dioctahedral | Trioctahedral |
| 0 | pyrophyllite Si4Al2O10(OH)2 | talc Si4Mg3O10(OH)2 |
| smectites | ||
| 0.2–0.6 | montmorillonite (octa. subst.) | hectorite (octa. subst.) |
| Si4(MgxAl2−x)O10(OH)2CEn+x/n | Si4(LixMg3−x)O10(OH)2CEn+x/n | |
| beidellite (tetra. subst) | saponite (tetra. subst) | |
| (Si4−xAlx)Al2O10(OH)2CEn+x/n | (Si4−xAlx)Mg3O10(OH)2CEn+x/n | |
| 0.6–0.9 | vermiculites | |
| micas | ||
| 1 | muscovite (Si3Al)Al2O10(OH)2K | muscovite (Si3Al)Mg3O10(OH)2K |
| 2 | margarite (Si2Al2)Al2O10(OH)2Ca | clintonite (SiAl3)(Mg,Al)3O10(OH)2Ca |
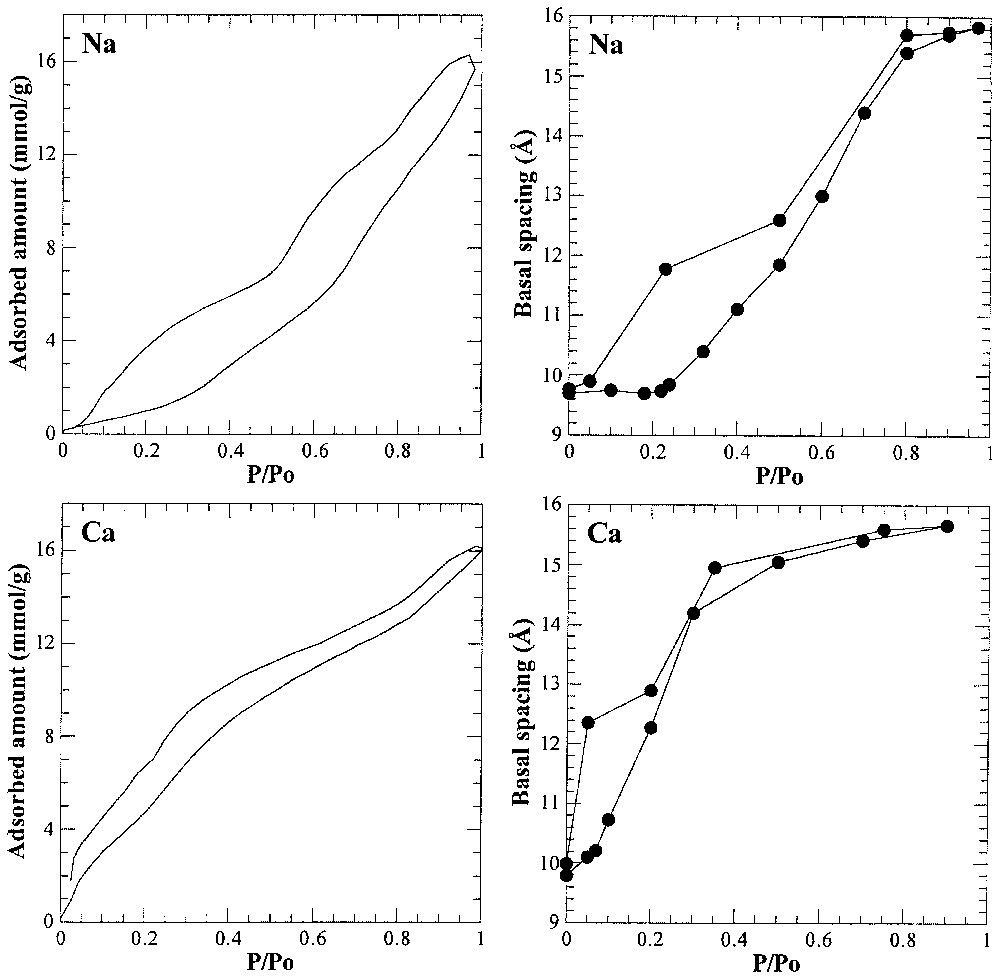
Water adsorption isotherms and evolution of basal distances with water pressure for Na- and Ca-montmorillonites (adapted from [10,24,25]).
Isothermes d'adsorption d'eau et évolution des distances interfoliaires avec la pression relative en eau pour les montmorillonites Na et Ca (d'après [10,24,25]).
5 Applications
Plusieurs applications dans le domaine des géosciences sont directement concernées par les phénomènes de structuration de l'eau aux interfaces.
- • Dans le cas du broyage humide des minéraux, la nature du solvant influence de manière importante la finesse et la structure des matériaux broyés [166]. Par exemple, dans le cas de la muscovite [111,126], l'addition de LiCl provoque à la fois un accroissement rapide de la surface spécifique et une amorphisation importante, ce qui peut s'expliquer par le fait que les ions Li+ peuvent perturber la structure de la couche d'eau liée au solide, dont l'élasticité protège le minéral lors du broyage.
- • La flottation des sels solubles, tels que les halogénures d'alcalins ou les sels doubles de potassium [65,66], par des tensioactifs anioniques ou cationiques est parfaitement corrélée au caractère structurant ou déstructurant des ions (Tableau 2). Ceci peut s'expliquer en considérant l'influence de ces ions sur l'eau superficielle. Si les couches d'eau superficielles sont en forte liaison hydrogène en raison de la présence d'ions structurants, les molécules de collecteur ne peuvent accéder à la surface du minéral pour la rendre hydrophobe. Au contraire, des ions déstructurants permettent l'adsorption des tensioactifs et donc la flottation.
- • La rhéologie des suspensions minérales concentrées, utilisées en céramique et dans l'industrie minière, peut être contrôlée à partir de la compréhension des forces interparticulaires [53,74–76]. La contrainte seuil d'une suspension d'alumine est ainsi maximale pour un pH correspondant au point isoéléctrique, c'est-à-dire lorsque les répulsions électrostatiques sont minimales (Fig. 10a). Lorsque la concentration en électrolyte de la suspension augmente, les courbes contraintes–pH s'élargissent en accord avec l'évolution du potentiel zêta [153]. De plus, la nature de l'électrolyte modifie les contraintes seuil (Fig. 10b), ce qui est en accord avec le caractère structurant–déstructurant des ions de l'électrolyte [13].
- • L'eau d'hydratation des argiles gonflantes joue un rôle important dans de très nombreux phénomènes. Nombre de réactions chimiques entre molécules organiques et argiles gonflantes se produisent par l'intermédiaire des molécules d'eau liées au cation [103,125,163]. De ce fait, la nature du cation interfoliaire et la quantité d'eau adsorbée influencent de manière significative nombre de ces réactions, qui participent au contrôle des phénomènes de migration des polluants organiques dans les sols et les sédiments. Dans les opérations de forage pétrolier et en géotechnique, les boues à base d'argiles gonflantes sont très utilisées, en raison de leurs propriétés rhéologiques [93]. De plus, le gonflement des shales argileux peut provoquer de graves incidents lors des forages. Dans le cas des smectites échangées par des cations monovalents, le potassium se comporte comme un inhibiteur de ces phénomènes de gonflement, ce qui est directement lié à la structure et à la dynamique des molécules d'eau autour du potassium [10,17,112]. Le non-gonflement osmotique des argiles échangées par des cations divalents résulte, en revanche, d'attractions électrostatiques dues à des corrélations inter-ioniques [39,114]. Les mêmes phénomènes de gonflement–dégonflement sont également en jeu dans la stabilité, la porosité et l'utilisation agricole des sols argileux [1,123,167]. La présence d'ions sodium diminue la porosité et la stabilité des sols, ce qui peut être corrigé par l'utilisation de différents cations inhibiteurs de gonflement, tels que le potassium, le calcium ou des polycations d'aluminium ou de fer [123,167]. La présence d'eau liée est également un paramètre crucial pour comprendre les phénomènes de transport dans les barrières argileuses utilisées dans la gestion des déchets municipaux, industriels et radioactifs (par exemple, [87,92,122,151]). Il apparaı̂t toutefois que la question du rôle spécifique de l'eau vicinale dans le transport reste mal comprise [44,82,105,133,164] dans les barrières argileuses et même dans les milieux poreux en général. Vu l'importance de ces phénomènes, il semble donc que des recherches nouvelles dans ces directions devraient être amenées à se développer dans les années futures. Une compréhension complète du comportement dynamique de l'eau liée ne pourra être atteinte que par le couplage de techniques expérimentales donnant accès à différentes échelles de temps. Dans ce contexte, l'étude de différents matériaux lamellaires (argiles, hydroxydes lamellaires doubles, acides siliciques...) semble être une voie d'étude prometteuse, en raison de la versatilité de ces matériaux. Par ailleurs, relativement peu d'études se sont intéressées à l'influence des propriétés physico-chimiques des solutions aqueuses sur la structuration de l'eau aux interfaces, bien que l'on puisse montrer que ces paramètres jouent un rôle non négligeable [84] (Fig. 11).
‘Floatability’ of alkali halide salts using either dodecyl ammoniumhydrochloride or sodium dodecyl sulfate (adapted from [65]).
Flottabilité des sels d'halogénures d'alcalins par le chlorure de dodécyl ammonium ou le dodécylsulfate de sodium (d'après [65]).

Shear yield stress properties of concentrated α-alumina suspensions (volume concentration = 0.250) as a function of (A) pH and KCl concentration, (B) pH and nature of the electrolyte at a concentration of (adapted from [75]).
Évolution de la contrainte seuil de suspensions concentrées d'alumine α (concentration volumique 0,250) en fonction de : (A) pH et concentration en KCl, (B) pH et nature de l'électrolyte à une concentration de (d'après [75]).
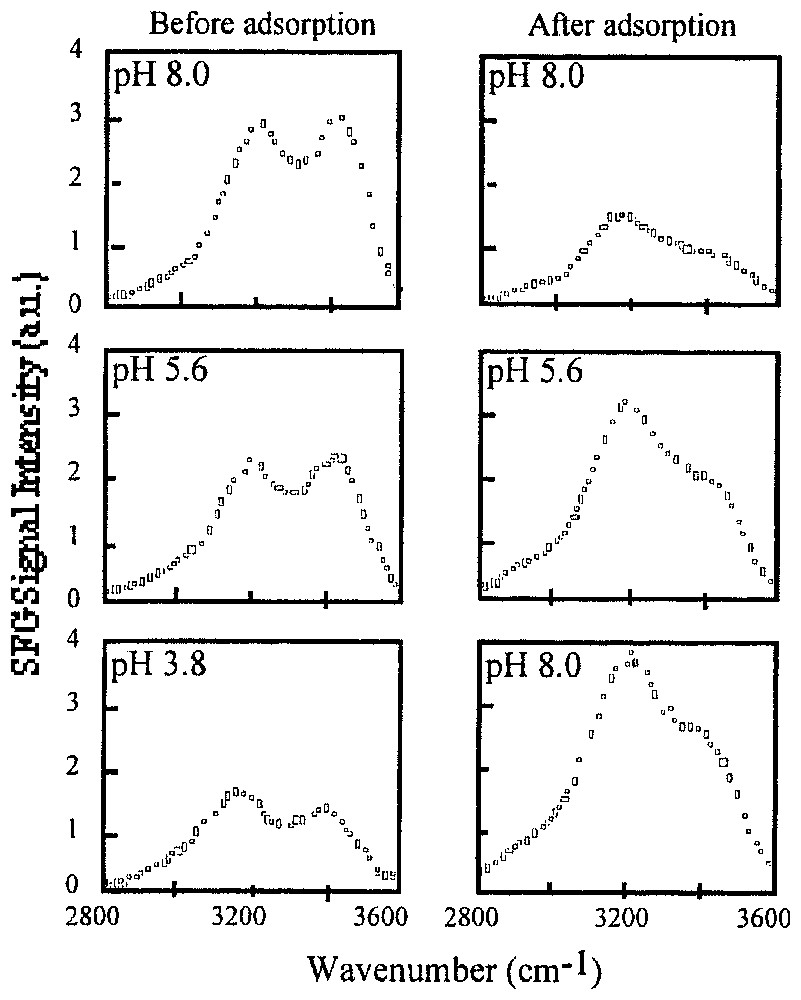
SFG spectra of the quartz/water interface in the OH stretching region at pH 8.0, 5.6 and 3.8 before and after adsorption of polydiallydimethylammonium chloride (adapted from [84]).
Spectres SFG de l'interface quartz/eau dans la région des vibrations d'élongation des OH à pH 8,0, 5,6 et 3,8, avant et après adsorption de chlorure de polydiallydiméthylammonium (d'après [84]).
1 Introduction
Due to its ubiquitous presence on the Earth's surface and to its crucial role in biological processes, water has been a major object for scientific investigation since the very early times of scientific research. For instance, the first report describing the maximum density of water at 4 °C dates back to 1667. Despite the huge amount of work devoted to the molecular level understanding of the structure of water, there exists at present no comprehensive model that can describe all its microscopic and macroscopic properties [162]. Still, there is clear evidence that the structure and dynamics of water are modified when water molecules are close to interfaces or confined in porous spaces. Understanding the interaction of water with surfaces is of prime importance for many scientific disciplines. In electronics, the relative humidity and thus the structure of adsorbed water layers modifies the durability of hard disk drives by changing the media tribology [80]. In biology, the physicochemical properties of interfacial water profoundly influence the biological response to materials [159]. For instance, using organised surfaces of different crystals, containing a controlled and known amount of surface water, it was shown [64] that the protein fibronectin is strongly adsorbed on water-free surfaces, whereas it does not adsorb on faces containing a continuous water layer. Furthermore, the effects of hydration on equilibrium protein structure and dynamics are fundamental to the relationship between structure and biological function [5]. In chemistry, surface water is crucial in electrochemistry, in corrosion and passivation chemistry and in catalysis [152]. In environmental sciences, the presence of interfacial and confined water certainly plays a role in transport phenomena, as illustrated by a recent study showing that water saturation modifies the retardation of volatile organic compounds in unsaturated model soils [83]. Adsorbed water molecules also appear to play an important role in the chemistry of aerosols, both organic [127] and inorganic [165]. Finally, in all the studies dealing with adsorption phenomena at solid–aqueous solution interfaces, the presence of structured water layers must be taken into account as under those conditions, the adsorbent is always covered by water molecules.
The aim of this review is to try to provide a quick state of the art of the present knowledge concerning interfacial and confined water. We will first summarise the most important features of liquid water and then examine the structural modifications brought by the vicinity of an interface. We will finish by a few examples taken from the field of geosciences where water structure near interfaces plays a major role.
2 Water structure
The structure of an isolated water molecule was precisely determined by infrared spectroscopy in 1956 [9]. The molecule is symmetrical with regard to the bisector of the HOH angle equal to 104.523°. The length of the OH bond is 0.9572 Å. The dipole moment of the molecule points from the oxygen end toward the hydrogen atoms and has a relatively large value of [45], which explains the solvating power of water. Due to its Lewis base character, water bears ionising properties and can easily coordinate with Lewis acids. Because of its high dielectric constant (ε=78.5), water is also a dissociating liquid [78].
The properties of liquid water are mainly due to the association of water molecules through hydrogen bonding. Such an interaction occurs when a hydrogen atom linked to a strongly electronegative atom (F, O, N) associates with another electronegative atom [11]. The energy of the hydrogen bond is around 20–40 kJ/mole, i.e., an intermediate value between Van Der Waals interactions () and covalent bonds () [108]. Hydrogen bonding is highly directional. In comparison with other associative liquids with hydrogen bonds, water is clearly unique due to the three-dimensional character of hydrogen bonds [90]. For instance, the behaviour of liquid methanol is markedly distinct [79]. The water molecule, with its two hydrogen atoms and its only oxygen atoms arranged in a non-linear fashion, presents an ideal configuration for forming hydrogen bonds [142]. The molecules can both accept and donate hydrogen atoms. Such a dual behaviour is perfectly illustrated by the structure of ice I (stable polymorph at atmospheric pressure). In that case, the three-dimensional network of hydrogen bonds leads to a tetrahedral structure in which each oxygen atom is located at the centre of a tetrahedron formed by four equidistant oxygen atoms (Fig. 1). In such an arrangement, the length of the OH and O⋯H bonds are 1.01 and 1.75 Å, respectively, whereas the hydrogen bonds O⋯HO have a length of 2.76 Å. Such a structure exhibits lacunae, which explains the lower density of ice with regard to liquid water.
Numerous experimental and computer modelling techniques have been used for studying the network and dynamics of hydrogen bonds in liquid water and their evolution with temperature. In terms of structure, water was first studied with X-rays (which are mainly sensitive to oxygen atoms) [102,106]. The main findings from these works were that water was tetrahedrally bonded in a manner not dissimilar to that occurring in hexagonal ice. Further studies [19,30] have shown that the number of first neighbours is almost insensitive to a decrease in temperature and that the rearrangement of oxygen atoms with decreasing temperature occurs in the second neighbour shell, which tends to move further apart (adopting a more tetrahedral configuration), thus revealing the influence of angular correlations. SAXS experiments have also been used to discard the presence of clustered aggregates in liquid water [20,71]. Neutron diffraction experiments on heavy water have revealed [6,30] that the structure factor of D2O is strongly temperature-dependent. Its evolution at low temperature suggests that the structure of supercooled water is close to that of amorphous ice. The strong temperature dependence must be assigned to an increase in angular correlations at low temperature, i.e., the more hydrogen bonds are intact, the more tetrahedral is the symmetry.
The dynamics of hydrogen bonds can be studied using dielectric spectroscopy [12,69] low frequency Raman spectra [101,108] and neutron scattering techniques [30,149]. These latter experiments suggest a possible mechanism for the molecular dynamics of liquid water. Large-amplitude vibration movements are the main mechanism of hydrogen bond breaking. The breaking of an individual hydrogen bond enables a molecular reorientation to occur. This process is thermally activated and the associated activation energy () is comparable to the hydrogen-bond energy. Molecular diffusion is possible only when several hydrogen bonds are broken simultaneously. As the number of bonds increases with decreasing temperature, the diffusion process is strongly temperature-dependent.
All these results provide a picture of liquid water that can be described in a first approximation on the basis of the percolation model (Fig. 2) [140,141,148]. Water can be thought of as a gel structure where extensive connectivity of different regions is established by the hydrogen bonds [42]. There are changes in time with a constantly varying local structure arising from the re-arrangement of the local network while still retaining the essential connectivity within the network. Transient hydrogen bonded clusters (mainly hexagons and pentagons with a lifetime less than one picosecond) are permanently formed and broken and very few free water molecules are present, as confirmed by recent IR experiments [94]. Most computer simulations tend to confirm this picture of water, even if at the moment no numerical potential is able to correctly predict all the thermodynamics of water [31,43]. In most simulations, water appears as a three-dimensional network of randomly distributed hydrogen bonds with a local tetrahedral geometry. One of the main questions still open regarding water structure is the way in which hydrogen bonds fluctuate to enable the structural changes to occur. Indeed, hydrogen bond dynamics is not characterised by single relaxation times. This was explained in terms of cooperation between different hydrogen bonds [108,142], as initially suggested by Frank and Wen [52]. However, recent molecular dynamics simulation [91] would tend to discard this notion of cooperation. It then seems, as stated by Dore [42], that “our fundamental understanding of the disordered hydrogen-bonded network is still at a fairly rudimentary stage”.
3 Aqueous solutions and aqueous solution interfaces
As revealed from the previous section, water structure is due to a delicate balance between the ordering effect of hydrogen bonds and the disordering effect of thermal motion. It then seems logical that the presence of any foreign molecule will have important effects on water structure by modifying the hydrogen bond network. Indeed, most of the common substances dissolved in water tend to reduce or even suppress most of the abnormal behaviours of liquid water. Due to the polarity and high dielectric constant of water, ionic or polar compounds are solvated in the solution, cations being associated to water molecules through the oxygen atom, anions being associated through the hydrogen atoms. The more polarising the ion (high charge, small radius, the polarising power being defined as z/r2), the more water molecules are associated to it. Depending on the polarising power of the ion, three situations are encountered: (i) ions with a strong polarising power (Li+, Na+, Mg2+, F−, OH−) break hydrogen bonds and organise numerous water layers around them (they are commonly referred to as structure-making ions); (ii) ions with a medium polarising power (K+, Cs+, NO3−) locally modify the hydrogen bond network, but do not have numerous water molecules around them; they are commonly referred to as structure-breaking ions; (iii) ions with a weak polarising power (hydrophobic ions) do not break hydrogen bonds; they are incorporated in the liquid and strongly modify the local architecture. In some cases, clathrate type structure can develop, in which the hydrogen bond network forms a cage around the element [78].
The difference between those three classes of ions translates in their influence on some macroscopic water properties. For instance, a LiCl solution is more viscous than pure water, whereas a CsCl solution is less viscous. These ions also affect the dynamics of water molecules in aqueous solutions. The rotation of water molecules is slowed down in solutions of structure-making ions and accelerated in solutions of structure-breaking ions [109]. Low frequency Raman spectra of NaCl solutions [101] also suggest that the hydrogen bond network is more distorted than in pure water, and that the reorganisation of the network is somewhat slowed down. It must also be noted that the presence of various ions in the solutions modifies the infrared–visible sum frequency generation (SFG) spectrum of the air–water interface (Fig. 3) [3,131,132]. In the spectrum of pure water, the peak around 3700 cm−1 is assigned to free OH groups of water, i.e., OH groups projecting out of the bulk solution into the vapour phase, free of H bonding. They seem to correspond to approximately 0.25 monolayer. The two other features around 3100 and 3400 cm−1 have not been assigned definitively yet, but may measure ordering of hydrogen-bonded water in the surface and the disordered environment at the interface, respectively. Acid solutions provoke a strong increase of the SFG intensity in the hydrogen bonded region, which reveals a greater alignment of water molecules near the interface and/or a larger number of affected water layers, due to the formation of an electrical double layer at the interface. The effect of salt solutions is marked only by a decrease in the band corresponding to free OH groups, which may be due to a displacement of water by contact ion-pair complexes that disrupt the hydrogen-bonded region.
4 Interfacial water and confined water
The presence of a solid interface in water clearly represents a major perturbation to the hydrogen-bond network. We will try in the following sections to analyse how the nature of the surface influence the structure of water and to what extent this actually propagates into the solution.
4.1 Metallic surfaces
A vast body of literature is concerned with the interaction of water molecules with clean metallic surfaces (e.g., [152] and references herein) at low temperature in ultra-high vacuum. As this is not directly in the scope of the present review, such studies will just be briefly mentioned. Depending on the metal used, the adsorption of water at surfaces can be either associative or dissociative. In the former case, long-range three-dimensional ice-like structures can be observed, in which the first-layer H2O molecules tend to occupy specific adsorption sites and the second layer molecules are hydrogen-bonded to the first layer.
The case of charged metallic surfaces has been extensively studied by electrochemists [88]. In the case of an Ag(111) electrode, recent X-ray reflectivity measurements [154] reveal that water is ordered in layers extending to about three molecular diameters from the electrode. The orientation of water molecules at the interface depends on the applied potential. For a negative potential of −0.23 V, the water molecules are oriented on average with the oxygen atoms pointing away from the surface, and the water layers exhibit a density 1.3 times higher than that of bulk water. For a positive potential of 0.52 V, the orientation is reverse and the density reaches more than two times that of bulk water. Molecular dynamics simulation [2,56] is in agreement with such data and show structured interfacial water layers extending over 10 to 15 Å from the surface, with an ice-like structure. More recent computer simulation of the electrochemical double layer near metal interfaces show that for concentrated NaCl solutions, whatever the electrode potential, Na+ cations are located in between the first and second adsorbed layer. In contrast, on positively charged electrodes, Cl− anions are contact adsorbed, whereas they are shifted into and beyond the second water layer for negative surface charge densities [137]. The orientations of water molecules in the first layer appear rather broad, but clearly depend on the nature of the electrolyte, CsF being different from NaCl [136].
4.2 Mineral surfaces
4.2.1 Model well-defined surfaces
Many experimental and simulation studies deal with water structure at well-defined equilibrium faces of various ionic crystals. Various recent results dealing with both non-oxide and oxide minerals can be quoted.
For the (111) faces of CaF2 or BaF2, water molecules adsorb associatively with their dipole nearly parallel to the surface [35,95]. The same situation is encountered for the (100) faces of NaCl and KCl [95,97]. In the case of the BaSO4 (001) and (210)-water interfaces, X-ray reflectivity data reveal [51] that the location and the number of adsorbed water molecules are consistent with the saturation of broken metal-oxygen bonds. A similar situation appears for the calcite (104)-water interface [50]. In all these cases, it then appears that the adsorbed water structure can be understood in the context of water–cation interactions.
For the (100) faces of MgO, one observes a first water layer strongly structured by Mg2+ cations. This first layer is covered with water layers interacting weakly with the monolayer and with a temperature-dependent structure (Fig. 4). At 300 K, this subsequent layer is only weakly structured and close to bulk water.
The situation here encountered is common to many well-ordered oxide single crystals, i.e., samples are often non-reactive for H2O dissociation [152]. This is at first sight rather surprising, as most work on powders of common oxides indicates that H2O is generally very efficient in hydroxylating the surface, thus resulting in the development of a pH-dependent surface charge (on this last topic, see [153]). This strongly suggests that in most powders H2O dissociation is dominated by reaction at defect sites and facet edges. This once again emphasises the importance of the concept of surface heterogeneity (see [158]) in studying natural samples.
4.2.2 ‘Natural’ surfaces
The first question that needs to be answered is the range to which natural surfaces influence the structure of adsorbed water molecules, i.e., how far from the surface is the water structure the same as in the bulk. This problem was extensively studied in our laboratory in the 1980's by combining water adsorption gravimetric measurements and immersion enthalpy determination at increasing water pre-coverage. Such an approach was based on the work carried out by Partyka et al. [113], who were the first to apply a modified Harkins and Jura procedure to measure the external surface area of solids from immersion calorimetry measurements. For all the studied solids, such as kaolinite, hectorite, montmorillonite [26,55], talc [100], chlorite [157] calcite [28,113], aerosil [113], ground quartz [113], gibbsite [113] etc., it was clearly revealed that at most two to four water layers are influenced by the surface field of the solid, i.e., an approximate thickness around 10 Å. Fig. 5 illustrates this conclusion in the case of a Na-kaolinite. A plateau corresponding to an immersion enthalpy independent of water pre-coverage (the heat of immersion is equal to the internal energy of the bulk water/water vapour interface) is reached at P/P0=0.7, which corresponds to a statistical coverage of 2.5 water layers. Parallel 1H and 2H NMR measurements in suspensions with increasing solid concentrations [54,55] yielded an average number of bound water layers around 3.
As will be seen in the following sections, this conclusion on the short-range (<15 Å) influence on the surface field is consistently confirmed in nearly all the experimental and computer simulation studies dealing with interfacial and confined water. This has two immediate consequences in terms of adsorption phenomena at the solid–liquid interface: (i) the shearing plane at which the zeta potential is measured (see [153]) cannot be assimilated with the outer Helmholtz plane (it will be displaced past the structured water layers only upon polymer adsorption when loops can extend into the solution); (ii) at the solid–liquid interface, the measured adsorption enthalpies correspond in fact to enthalpies of displacement of water by the adsorbate.
In the following sections, we will mainly focus on the description of the structural and dynamic behaviour of vicinal water and how it is modified with comparison with bulk properties. In order to try to bring clarity to the general discussion, we will present the data according to the various adsorbents used.
4.2.2.1 Silica-based materials
Many experiments dealing with the effect of confinement on water structure use various types of silica based materials that can be silica gels, pyrogenic silicas, sol–gel silicas, glasses, and mesoporous silicas of the MCM type.
1H MAS NMR or 1H CRAMPS have been implemented on silica samples under various hydration levels [18,34,85,110]. In the case of silicagel, one observes a first water layer strongly hydrogen-bonded adsorbed on the surface silanols. In such a system, the measurements of longitudinal and transverse relaxation times could be described by assuming three proton populations: silanols, protons from the first water layer, protons from ‘free’ water [110]. For pyrogenic silicas, the situation is similar though slightly more complicated as five proton signals can be assigned [34]: (i) isolated internal silanols, (ii) weakly coupled external silanols, (iii) strongly hydrogen bonded external silanols, (iv) first adsorbed layers bonded to silanols, (v) adsorbed water close to bulk water. Variable temperature NMR studies reveal that the first water layer exhibits a freezing point that is shifted towards lower temperatures. Furthermore, the freezing point of a binary mixture of H2O/HNO3 adsorbed at the surface of pyrogenic silica is 10 °C lower than that of pure water [18]. As this shift is much stronger than in solution (≈1 °C), it is suggested that the presence of nitric acid slightly modifies the adsorbed layer.
Porous silica glasses represent useful models for studying confinement, as their pore size is well defined. In a saturated Vycor glass with pores around 40 Å, by coupling DSC, IR and NMR measurements, it is possible to show the presence of a first water layer structured by surface silanols, which represents approximately 25% of the total water contained in the material [23]. Detailed Raman and Rayleigh wing investigation of water confined in nanoporous sol–gel glasses clearly reveal that the interaction of water molecules with the silanol surface thoroughly modifies the tetrahedral H-bond network of water destroying its intermolecular network [32]. Neutron scattering experiments on water confined in Vycor glass [7] showed that the structure factors are modified only for low hydration levels, i.e., for the water layer adsorbed at the pore surface. More recent neutron diffraction experiments with isotopic substitution [22,135] tend to show that water in Vycor is still hydrogen-bonded, but that the bond network is strongly distorted. When the sample is cooled down, the surface water does not crystallise and even at very low temperature, exhibits a neutron diffraction pattern close to that obtained for low-density amorphous ice [7].
Quasi-elastic and inelastic neutron scattering experiments can also be used in that case to study the dynamics of water confined in Vycor. By applying the same formalism as that used for the study of liquid water [149], the times characteristic of a jump diffusion mechanism (τ1) and of reorientational correlations (τ2) can be calculated and compared with the case of liquid water [6] (Fig. 6). The residence times (τ2) are significantly increased for medium and low hydration levels, whereas (τ1), which can be assimilated to the lifetime of hydrogen bonds, is modified only for the lowest hydration level, i.e., for the water layer adsorbed at the pore surface. The picture that can be derived from these experiments suggests that adsorbed water molecules remain in their sites for longer times than in the bulk, local reorientations being rather similar. In terms of water dynamics at the time scale probed by neutrons, interfacial water exhibits a behaviour that mimics that of the supercooled state. It must be noted that such a behaviour can be modelled in the framework of the percolation model by assuming that the lifetime of hydrogen bonds formed between surface silanols and adsorbed water molecules is much longer than that in liquid water [150].
Water dynamics on longer time scales can be analysed by dielectric relaxation [62,129]. Dielectric relaxation experiments on controlled pore size sol–gel glasses reveal four distributed relaxation processes: a first one assigned to the reorientation of water molecules in ice-like water cluster structures, a second one assigned to water molecule re-orientating in the vicinity of a defect, the third one assigned to the self diffusion at the surface of the pores between hydration centres and the fourth one due to interfacial polarisation. Nuclear Magnetic Relaxation Dispersion (NMRD) is also an extremely useful tool for analysing the dynamics of interfacial and confined water [86]. NMRD experiments on a controlled pore size chromatographic glass (mean pore diameter 75 Å) reveal that water molecules diffuse near the pore surface until such a process is interrupted by chemical exchange with the bulk phase. A translational diffusion coefficient is obtained, i.e., around one third of the value obtained in bulk water [86]. The situation is markedly different in granular packings of silicon carbide and limestone. In that case, the levelling of T1 at low frequency allows determining a surface residence time for water molecules of 1.6 μs for SiC and 5.3 μs for CaCO3, thus revealing the higher affinity of CaCO3 towards water. In that latter case, the slow diffusion behaviour observed may also correspond to the diffusion of hydrated calcium anions [58].
Some of the above-described experiments are currently being implemented on a new family of mesoporous siliceous materials with monomodal adjustable pore sizes called MCM [4]. The evolution of 1H NMR signals with temperature reveals that some water molecules freeze at very low temperature only [67]. Assuming that such water family corresponds to interfacial water, the thickness of the adsorbed layer would be around 5.5 Å, i.e., in good agreement with what is observed for other siliceous materials and other solids (see Fig. 5). X-ray diffraction measurements [134] and neutron scattering experiments [42,147] reveal features that are rather similar to those observed in porous glasses. Decreasing pore size induces an increase distortion and/or breaking down of hydrogen bonds. When hydration is limited to the monolayer, neutron spectra can be analysed with a model in which the motions of the monolayer water molecules are rotations only.
Molecular dynamics simulations performed for water in modelled Vycor glass for various hydration degrees [68,138] reveal various features agreeing with the experiments. Close to the surface, two layers of water molecules are formed. In this region, the hydrogen bond network is distorted as hydrogen bonds between water molecules are partially substituted by bonds between water and surface. A mobility profile is observed across the pore. The mobility of molecules directly adsorbed on the surface is reduced by more than one order of magnitude, whereas for full hydration water molecules in the central part of the pores exhibit short time diffusion coefficients close to that of the bulk.
All these experiments and simulation results seem to provide a rather coherent picture of the behaviour of interfacial and confined water near silica-type surfaces. Water molecules in the first and maybe second layer exhibit properties significantly different from that of the bulk with a distorted hydrogen bond network, and reduced diffusion properties. It must be noted however, that the situation encountered for those types of surfaces may not be easily extended to other hydrophilic surfaces. Indeed, the presence of various types of silanols at the silica surface generates diverse water adsorption behaviours and the concept of adsorbed layer may not be fully applicable to those materials.
4.2.2.2 Carbon-based materials
Whereas silica-based materials are often used as representative of so-called ‘hydrophilic’ surfaces, most of the work devoted to water structuration near ‘hydrophobic’ surfaces has focused on carbon-based materials, mainly graphite and porous carbons. In this technologically important class of materials, the presence of water can severely handicap the intended process [21]. In fact, relatively few experimental studies have studied in detail, the structure and dynamics of adsorbed water near hydrophobic interfaces. Graphitised carbon blacks adsorb virtually no water molecules for relative pressures less than 0.8 [60], as the fluid–fluid attractive interaction is much stronger than the fluid–wall interaction. Computer simulation studies have been performed to try to deduce the orientation and structure of adsorbed layer, mostly in graphitic slits [89,155]. Using a Monte-Carlo type simulation, it can be shown that, for graphitic slits ⩾10 Å, two regions can be distinguished in the pore: a weakly hydrogen-bonded layer near the pore surfaces and liquid-like layers. As shown by the density profiles of oxygen and hydrogen (Fig. 7), water molecules near the pore surface exhibit an orientation parallel to the interface. Such preferential orientation does not extend into the pore centre.
In the case of porous carbons, the situation is markedly different, due to the presence of various oxidised surface groups [15] that can act as anchoring sites for water molecules; pore filling then occurs through adsorption of water on previously adsorbed water molecules and subsequent cluster formation. The importance of surface oxidised centres can clearly be deduced from experiments where oxidation conditions are varied in a systematic way (e.g., [21,60] and references quoted). The increase in adsorbed amount observed on water adsorption isotherms shifts towards lower relative pressure with increasing oxidation. Molecular dynamics simulation provide coherent pictures of the evolution of water structure in porous carbons (Fig. 8) [96]. Such simulation studies also clearly show the importance of the site density of oxidised groups that thoroughly modifies water adsorption [96]. Structural studies of water confined in porous carbons have been based on X-ray diffraction [8,72], neutron diffraction [8] and Small Angle X-ray Scattering (SAXS) [73]. The main results evidenced a more extensive distorted hydrogen bonded network near the surfaces, and the formation of ice-like water, the structure of which depends strongly on pore size. SAXS experiments reveal cluster formation in agreement with the previous discussion. Once again, cluster size depends on pore size.
The behaviour of interfacial water in carbon-based materials then appear as rather difficult to generalise, as a rather complex surface chemistry is nearly always involved with a mixture of hydrophilic and hydrophobic sites or patches at the interface. It still clearly deserves further study in view of the technological importance of such materials; moreover, the observed complexity of hydrophilicity/hydrophobicity could somewhat be reminiscent of some of the situations encountered in biological materials.
4.2.2.3 Swelling layered materials
In several classes of layered solids, such as smectite clay minerals, layered double hydroxides, Group 4 metal phosphates, calcium silicate hydrates or layered silicic acids, the solid layers carry a net electrical charge. This electrical charge is compensated by counter-ions occupying the gallery region between layers. Due to the presence of these charge-balancing ions, the interlayer space is generally accessible to water molecules, which can then provoke a swelling of the structure in the direction perpendicular to the layers. Due to the structural diversity of these classes of material, they clearly represent very relevant models for studying the structure of interfacial and 2D confined water.
Due to their ubiquitous presence in soils and their widespread industrial use, the most extensively studied family of charged layered solids is by far swelling clay minerals. In that family of materials, charge arises from substitutions in the crystalline network yielding an overall negative charge compensated by interlayer cations. Numerous experimental works and computer simulation studies have been devoted to understand the arrangement of water molecules in the interlayer spaces of clay minerals and its evolution with increasing water content. Only the main results will be mentioned here and the reader interested in that topic can find supplementary information in various reviews (e.g., [63,107,139]). The classification of phyllosilicates (two tetrahedral layer sandwiching an octahedral one) is presented in Table 1. The criteria used are (i) the charge of a half unit cell; (ii) the filling of the octahedral layer; (iii) the localisation of isomorphous substitution. These criteria and the nature of the interlayer cation appear as the relevant parameters whose influence on water structure should be followed.
Cases and co-workers [10,24,25] carried out an extensive study on the influence of the nature of the interlayer cation on water uptake in montmorillonite by combining water adsorption gravimetry, X-ray diffraction under controlled water vapour pressure and immersion enthalpy measurements. They showed that the nature of the interlayer cation plays a prominent role in defining water uptake (Fig. 9). In the case of the sodium cation, the two-dimensional pressure of the water film adsorbed on the external clay surfaces also was important in opening up the interlayer spaces [10,24]. Furthermore, the hydration states observed exhibit some degree of layering, but can never be considered as truly homogeneous, which renders the precise definition of one-layer, two-layer and three-layer hydrates rather difficult. IR spectroscopic studies [14,49,77,115,118,121] also reveal the crucial role of the cation, as an IR signal assignable to water molecules linked to the cations and depending on the nature of the exchangeable cation dominates the spectra for low water contents. This crucial role of the cation is also revealed in computer simulation of clay hydration [16,17,29,36–38] that show parallel layers of water molecules oriented at the clay surface in agreement with 2H NMR experiments, which exhibit a splitting of the resonance line of D2O [40,59,161]. For cations with a low hydration energy such as potassium, water/surface interactions start playing a significant role [17].
Charge localisation in the tetrahedral layer, i.e., closer to the cation, appears to homogenise the hydrated states [57,143,144]. Preliminary experiments on the influence of charge density in synthetic saponite samples [98] also suggest significant changes in the molecular arrangement of interlayer water.
Neutron diffraction experiments [27,70,116,120] reveal that water in the interlayer of montmorillonite is rather strongly hydrogen-bonded. The dynamics of water molecules confined in the interlayer of various clay minerals (hectorite, montmorillonite and vermiculite) were studied by quasi-elastic neutron scattering [19,117]. The dynamics of the clay water interface could then be described as follows. Water molecules are distributed over a superlattice and remain on average 10−10 s on a site. During this time, they spin around their C2 axis with a correlation time around 4 ps. If they are in the hydration shell of an exchangeable cation, they are also involved in the slow rotation of the entire hydrate with a correlation time around 30 ps. Both these times appear to be sensitive to the charge density of the clay layer, higher charges leading to an increase in correlation times. For high water contents, the dynamics observed are closer to that of bulk water.
Longer time scales can be probed from a precise 1H NMR analysis of the evolution of the longitudinal relaxation time with frequency [41] at the clay water interface. Such experiments revealed a two-dimensional diffusion of adsorbed water molecules with a residence time at the surface (before exchange with the bulk) of around 10−6 s. Such a value appears extremely long, but is in agreement with the adsorption energy derived from Monte-Carlo simulations of the clay/water interface [41]. Long time scales were also probed recently by neutron resonance spin echo measurements in Na-vermiculite [145]. Those experiment that probe times up to 3000 ps reveal slow dynamics assigned to the diffusional motion of water molecules interacting with the clay surfaces, confirming the above-mentioned NMR results.
The water structure in the interlayer of swelling clay minerals appears to be mainly controlled by the nature of the exchangeable cations that, depending on their hydration energy, structure water molecules around them. Still, the exact influence of tetrahedral charge and charge density is not clearly defined yet and obviously deserves further investigation. We hope to participate in that research effort in the coming years.
In comparison with clay minerals, very little is known on the detailed hydration behaviour of other classes of charged layered solids, except for a few studies [81,130,146,156,160]. This appears rather surprising, as such information could shed new light on the role of charge amount, charge localisation and layer structure on the arrangement of water molecules in confined interlayer spaces. For instance, we recently showed [46,47] that in a layered sodium silicate, such as Na-magadiite, the balance of surface/water/cation interactions is very different from that observed in swelling clay minerals, thus resulting in significantly different water structure in the interlayer. In addition, the family of layered double hydroxides (LDH, also referred to as anionic clays) certainly represents a useful model, as they can be easily synthesised with variable interlayer anions and charge densities [99]. Preliminary QENS measurements in carbonate and terephtalate MgAl LDH reveal water diffusion coefficients significantly lower than in bulk water.
5 Applications
5.1 Wet grinding of minerals
Around 3% of the world energy consumption is dedicated to grinding operations [61]. Improving grinding efficiency therefore clearly represents a major challenge both on an industrial and fundamental point of view. In the case of wet grinding, it has been shown that the nature of the solution in which the solids are suspended has a strong influence on the size distribution and structure of the obtained powders [166]. Such a tendency is particularly well illustrated in the case of muscovite [111,126], for which grinding was carried out under controlled conditions in various media. In that case, the influence of the nature of ions in the solution was clearly evidenced. Notably, the addition of LiCl in the grinding medium leads to a quicker initial increase of the specific surface area of the ground products. However, a limiting value of specific surface area is obtained for long grinding times, which is accompanied by an important ‘amorphisation’ of the structure of ground muscovite. The observed features can be explained by considering the presence of structured surface water layers at the surface of muscovite particles which elasticity may protect the mineral and distribute evenly the stresses applied. In that context, the influence of Li+ ions can be understood through their ability to break the hydrogen bond network. Further support for this assumption can be found in the study of wet grinding in various organic media [166]. Indeed, hydrogen-bonded associative liquids such as methanol tend to protect the structure from ‘amorphisation’. The addition of LiCl in methanol provokes similar effects as those observed in water.
5.2 Minerals flotation
In minerals flotation, surfactant molecules are adsorbed at the solid/aqueous solution interface to create a hydrophobic surface state. This can only be achieved if the surfactant molecules penetrate through and/or displace the interfacial water film initially present at the mineral surface. In that context, flotation of soluble salts such as alkali halides [65] or potassium double salts [66] represents a specific case. Indeed, flotation can be achieved independently of the surface charge with both cationic and anionic surfactants for some salts whereas other salts are totally non floatable (Table 2). Except in the case of LiF, there is a strong correlation between the floatability of the salts and the structure-breaker/structure-maker character of the ions, which can be explained as follows [66]. If the structure of surface water layers is strongly hydrogen-bonded, due to the presence of structure-making anions and cations, then collector molecules cannot reach the surface and be adsorbed. Ions that have a tendency to destroy the structure of water can create a condition for the adsorption of collector molecules and subsequently allow for the flotation of soluble salt minerals.
5.3 Rheological behaviour of concentrated mineral suspensions
The behaviour of concentrated mineral suspensions is of prime importance in various industrial fields and particularly in the ceramics and mineral industry where concentrated suspensions have to be either processed for further use or disposed of with minimal water content. In a series of recent papers [53,74–76], it was shown that the rheological behaviour of concentrated mineral suspensions could be controlled by understanding and regulating the net inter-particle force. As shown in Fig. 10a, the shear yield stress of a model alumina suspension is maximal at the pH corresponding to the isoelectric point due to a decrease in the electrical double layer force and thus, to an increase in the net inter-particle interaction. An increase of the ionic strength results in a broadening of the curves, in agreement with the observed evolution of zeta potential with ionic strength (see [153]). In addition, Fig. 10a also reveals that the yield stress values obtained at the vicinity of the isoelectric point decrease as the electrolyte concentration rises. This latter behaviour is indicative of an ion-based structural repulsion; that is, a short range barrier to inter-particle approach generated by a layer of adsorbed ions on each surface which prevents the particle from coming into physical contact [74]. Furthermore, in that case, some ion specificity can be observed (Fig. 10b) the yield stress varying in the order Li+>Na+>K+ >Cs+ across the entire pH range. Such a tendency is consistent with the structure making-structure breaking model initially proposed in [13]. In that scheme, structure-making species such as Li+ or Na+ can approach the surface more closely that structure breaking ions, resulting in smaller inter-surface separation and thus higher inter-particle attraction.
5.4 Swelling clay hydration
The structure of water molecules in the interlayer of swelling clay minerals is of prime importance in various applied problems.
- – In order to forecast the migration of hazardous organic pollutants through soils and sediments, it is essential to study the adsorption of these molecules by clay minerals. Indeed, it is now generally agreed that the clay fraction is most important in influencing the reactivity of organic molecules [168]. Among the various mechanisms by which organic species react with clay minerals surfaces, association via water through acid–base reactions to form a protonated organic base has been shown to be one of the major processes [103,125,163]. The amount of base that is protonated depends upon (i) the field strength of the interlayer cation, (ii) the alkalinity of the adsorbed molecules, (iii) the water content, (iv) the charge localisation in the clay layer. For instance, ammonia can be partially protonated by all cations including monovalent ones [128], whereas urea, a weaker base is protonated only by Al3+, Fe3+ and H+ interlayer cations [104]. In the case of pyridine, adsorption on a hydrated Mg-montmorillonite does not provoke any protonation, whereas pyridinium ions are formed under partial dehydration [48,125], thus revealing the strong acidic character of water molecules bound to the interlayer Mg cations.
- – Swelling clay minerals are extensively used in drilling muds in civil engineering and in the oil industry. In well construction, the use of water-based drilling fluids is increasingly favoured due to environmental constraints [93]. Most of these fluids are then based on smectite clay colloids that form gels at low volume fractions (). Furthermore, shale rocks that contain a reasonable proportion of swelling clays can swell dramatically upon exposure to water, which can create major drilling problems [93,124]. A proper understanding on the role of various physicochemical parameters in controlling the rheology and swelling behaviour of clay based materials is therefore clearly of prime importance. In that context, the role of potassium as a swelling inhibitor directly depends on the structure and dynamics of water molecules around potassium ions in the interlayer space of montmorillonite [10,17,112]. In contrast, the rheological behaviour of divalent exchanged clays results from electrostatic attractions induced by inter-ionic correlations that are stronger than in the case of monovalent cations [39,114]. It must be pointed out that the problems encountered in drilling fluids and related to swelling and to the influence of the interlayer cation on inter-particle forces are also relevant to the stability, structure and management of clayey soils [123,167]. Due to the swelling behaviour of Na-smectites, sodic soils are prone to mechanical instability [1,167] and their low porosity can be detrimental for agricultural practices [33,123]. Porosity correction and/or recovery of sodic affected soils can then be obtained through the addition of swelling inhibitors such as potassium brines, gypsum, or aluminium or iron polycations [123,167].
- – Clay hydration is also an important factor affecting the usability of smectites as a component in clay barriers (clay liners, slurry walls, compacted clay walls, etc.) used to minimise the transport of contaminants from municipal, industrial or nuclear waste sites [87,92,122,151, e.g.]. The hydration level of swelling clays influences the retention of elements and thus their transport in the barriers [44]. As stated in [105], ‘a macroscopic model for smectic clays requires an accurate description of the anomalous behaviour of the vicinal fluid. Hence, the treatment of the adsorbed water as a separate phase from the bulk water is mandatory’. Still, it appears that the general problem of taking into account the specific behaviour of vicinal water in transport phenomena in clay barriers remains largely unsolved [44,82,105,133,164]. In fact, this latter statement can be applied to porous media in general as there is, to the author's knowledge, no clear understanding of the role of vicinal water in transport phenomena inside porous networks. Considering the importance of such phenomena for environmental sciences, research along those directions should certainly be implemented.
6 Conclusions and perspectives
This brief review reveals the complex structure and dynamics of interfacial and confined water, which exhibit strongly modified properties when compared to bulk water. Part of the difficulty arises from the fact that no theory is able at the moment to precisely predict water interaction with a surface, even when the surface structure is well characterised. Still, it appears that, whatever the substrate, the spatial range at which water structure is perturbed is rather limited, less than 15 Å from the surface. In terms of dynamics, the puzzle of interfacial water can only be approached using various techniques, sensitive to different time scales, as primarily suggested by Eisenberg and Kauzman [45]. In that regard, the development of dielectric techniques, NMR relaxometry and spin echo neutron resonance techniques opens promising perspectives for studying slow dynamics. In comparison with silica-based and carbon-based materials whose surface chemistry is complex, highly variable, and sometimes ill-defined, lamellar materials could certainly represent very useful models for studying the behaviour of vicinal water as their structure and charge can be adequately controlled. Finally, relatively few studies have focused on the influence of solution properties (pH, ionic strength...) on water structure. In fact, as shown by studies on metallic electrodes, these parameters should have a significant influence on water structure. This was recently demonstrated by SFG studies at the quartz/water interface (Fig. 11) [84]. For the bare quartz/water interface, the SFG intensity of both peaks increases with increasing pH showing a more ordered structure of interfacial water resulting from the increased charge density at the quartz surface. The tendency is reverse after adsorption of a positively charged polyelectrolyte polydiallydimethylammonium chloride, as its adsorption overcompensates for the initial surface charge. It then seems that much information could be extracted by studying water structure in the electrical double layer developing at the surface of charged minerals, also taking into account ion specificity. This could also help in assessing the role of structured water in transport phenomena in porous media.