

Abridged version
The thorium phosphate–diphosphate (TPD), Th4(PO4)2·P2O7, displays interesting properties that make it a good candidate as a potential host matrix for long-term storage of radioactive wastes. TPD structure can incorporate actinides like Th, U, Pu or Np. TPD can be easily synthesised either by solid-state or wet chemistry. In addition, leaching experiments performed on TPD yielded very low dissolution rates. It is therefore striking that, with such properties, TPD does not occur in nature. Apatite, Ca5(PO4)3(F,OH,Cl), is the most abundant phosphate in natural rocks. Possibly, in Th- and Ca-bearing phosphate systems, apatite crystallisation will sequester all the available phosphorus and will therefore preclude from the TPD formation. Since the long-term stability of TPD is of major concern for radioactive waste storage, we have undertaken an experimental study of TPD phase relations in a Ca-bearing chemical system. TPD was synthesised by mixing concentrated solutions of thorium nitrate (2.17 M) and phosphoric acid (5 M). The thorium nitrate solution was obtained by dissolution of Th(NO3)4·5 H2O (Fluka, 99%) in HNO3. After complete evaporation of the solution, the solid precipitate was held for 6 h at 400 °C and 18 h at 1250 °C in a platinum crucible. The X-ray powder pattern of the obtained powder product yielded single-phase of well-crystallised TPD. This product was used in hydrothermal experiments that were carried out under 50 MPa pressure and under a thermal gradient (temperature comprised between 400 and 320 °C, Fig. 1) in a horizontal externally heated pressure-vessel at the ENS. Forty-five milligrams of TPD were encapsulated together with 30 mg Portland cement (G class) in a 15 mm-long half-opened gold tube (3.8 mm outer diameter, A), which was placed at the cold end (320 to 350 °C) of a 150 mm-long sealed gold tube (4.8 mm outer diameter, called reaction tube, RT). A second half-opened container (B) with 60 mg of FoCa-7 (compacted benthonite) powder is placed at the hot end (390 to 400 °C) of the RT. In addition, the RT is filled with 1 ml de-ionised water. The temperature profile along the pressure vessel has been measured beforehand by introducing a series of three thermocouples under pressure within the vessel body. The role of the temperature profile in these experiments is to force mass transfer from one end of the RT towards the other, through aqueous species diffusion. Dissolution of the starting materials (TPD, benthonite and cement) and precipitation of newly formed phases along the temperature profile are therefore expected. This type of experiment is complementary to conventional leaching experiments and represents a valuable way to evaluate, experimentally, TPD reactivity and phase relations at relatively low experimental temperatures in a multi-component chemical system. Phases like apatite, Ca5(PO4)3OH, thorite, ThSiO4 or brabantite, CaTh(PO4)2, which are expected to share phase relations with TPD, belong to the investigated chemical system. Experiment was run during two months and then the system was cooled down at ca 50 °C min−1 under a compressed-air stream. The powder products recovered in the two half-tubes were X-rayed and the distribution of the newly formed phases along the RT was determined using the SEM. X-ray powder diffraction shows that, in the A tube, TPD has partly reacted with the cement to form hydroxylapatite, Ca5(PO4)3OH, and thorianite, ThO2 (Fig. 4). In the B tube, the FoCa-7 starting material has disappeared and, instead, a mixed-layer silicate of rectorite-type is recovered. Among the phases which formed outside the half-opened tubes A and B, xonotlite, thorianite, anorthite, garnet and a silica gel were identified using SEM (Fig. 3) combined with EDS analysis.
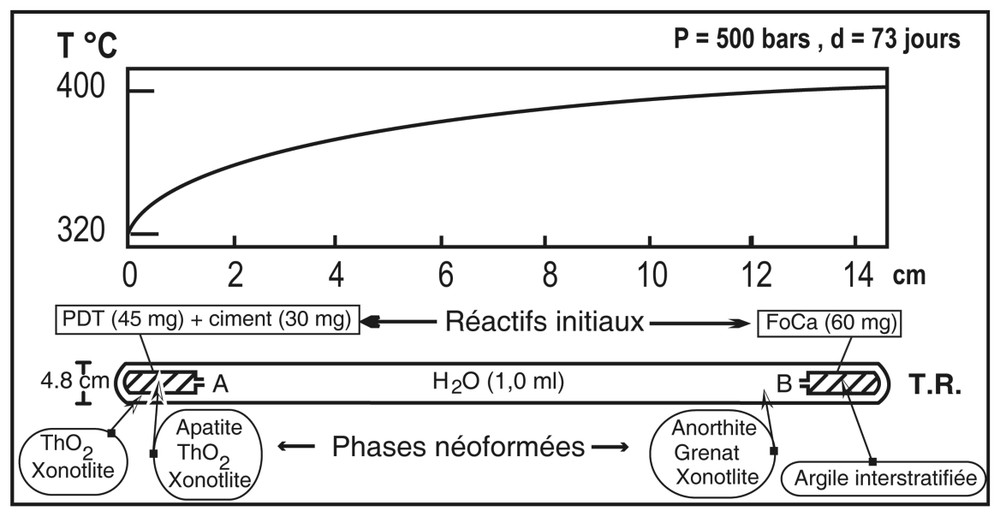
Configuration du TR et des capsules A et B. Sur ce schéma ont été reportés le gradient thermique appliqué (d'après [19]) ainsi que la distribution des réactifs initiaux et des phases néoformées après expérimentation.
Experimental set-up, location of the starting material in capsules A and B and of the run production.
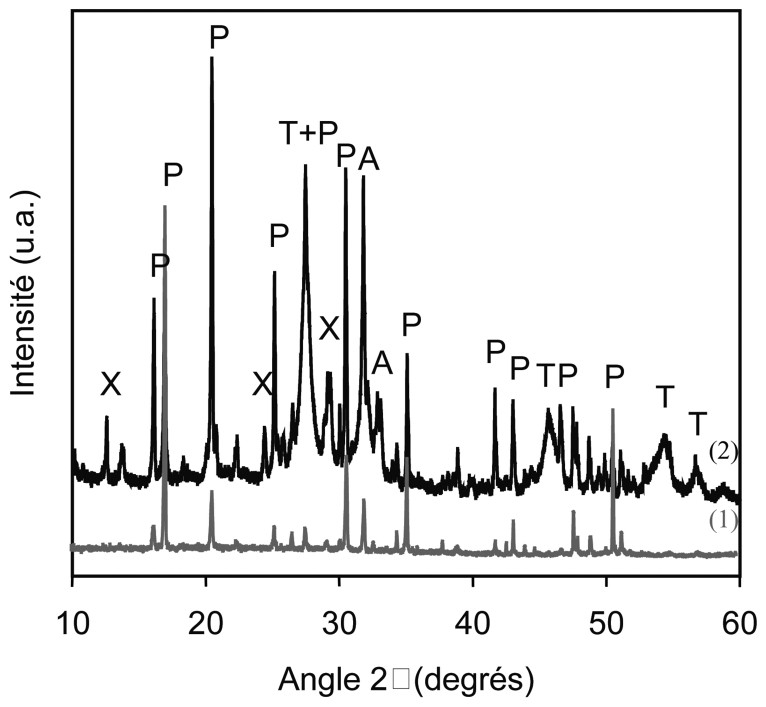
Diagrammes de diffraction RX (1) sur poudre du PDT (produit de départ) et (2) sur la poudre de tube A après expérimentation. Phases identifiées : PDT (P), apatite (A), thorianite (T), et xonotlite (X).
X-ray powder diffraction pattern obtained for (1) the TPD (starting material) and (2) the A tube after the experiment.

Microphotographies (MEB–électrons secondaires) des phases néoformées dans la capsule B entre 320 et 350 °C (A) et sur les parois internes du tube réactionnel autour de 320–330 °C (B, C). B : Cristal de thorianite isolé ; C : cristaux de thorianite enveloppant des fibres de xonotlite.
SEM micrographies of secondary phases precipitated in the capsule B between 320 and 350 °C (A) and along the reaction tube around 320–330 °C (B, C). B: Isolated thorianite crystal; C: xonotlite fibres surrounded by thorianite crystals.
The distribution of the newly formed phases shows that phosphorus and thorium did not migrate significantly and remained at the cold end of the RT. In contrast, calcium and silicon were mobile in this experiment, since Ca- and Si-bearing phases are widespread all over the RT (e.g., silica gel and xonotlite all along the TR, and grossular-rich garnet and anorthite at the hot end, Fig. 3). One of the most prominent results of this study is that TPD has reacted at the cold end of the TR (320–350 °C) with the cement, to form hydroxylapatite and thorianite according to the following simplified reaction:
This result shows that, at 320–330 °C, two months are sufficient to destabilise significant amounts of TPD in the presence of Ca-bearing phases (Fig. 4). Interestingly, TDP reactivity is comparable to that of the R7T7 glass under similar experimental conditions. The experimental breakdown of TDP in a Ca-bearing system into hydroxylapatite and a Th-rich phase (here thorianite) is in good agreement with natural data, since apatite is the most abundant phosphate in the Earth's crust and TPD has, so far, no natural equivalent. In very peculiar natural contexts of low-calcium environments where apatite (or REE phosphates) do not host the whole rock phosphorus, growth of TPD could then be envisaged. However, POP bonds in pyrophosphates are easily hydrolysed and pyrophosphates are expected to be less stable than the equivalent mineral assemblage containing orthophosphate(s). As a matter of fact, wooldridgeite, Na2CaCu2(P2O7)2·10 H2O is the only pyrophosphate reported so far from a natural occurrence. In addition to important consequences for long-term storage of actinides, the experimental results of the present study give an interesting clue to understand why TPD has no equivalent in nature.
1 Introduction
Dans l'optique d'un stockage de déchets nucléaires en formation géologique, les matrices cristallines phosphatées sont capables d'incorporer, au sein de leur structure cristallographique, différents radioéléments, tout en présentant des cinétiques de dissolution a priori relativement lentes et une relative inertie au rayonnement radioactif. C'est pourquoi ces matériaux sont étudiés comme matrice potentielle de confinement spécifique de radionucléides. La flexibilité structurale de certains phosphates cristallins permet l'incorporation de radionucléides de charges et de tailles variées, tels que des actinides tétravalents, comme le Pu et ses descendants radiogéniques, U et Np. Ces éléments se substituent alors au Th au sein de la structure cristalline du PDT. Dans le système ThO2MO2P2O5 avec M = U, Pu, Np, une série de composés de type Th4-xMx(PO4)4P2O7 a ainsi pu être obtenue pour des valeurs de x allant jusqu'à 3,0 (U4+), 2,09 (Np4+) et 1,67 (Pu4+) [9,10].
Des études récentes montrent également que, non seulement la structure du phosphate–diphosphate de thorium (PDT) [4,18], mais aussi celles d'autres phosphates, comme la monazite [17] ou les apatites silicatées [2], résistent remarquablement bien à l'altération et aux irradiations. Cependant, si des minéraux de structure monazite ou apatite sont présents dans le milieu géologique [5], le phosphate–diphosphate de Th n'a pas en revanche d'analogue naturel. Pourtant, le PDT semble présenter expérimentalement des signes d'une grande stabilité : (i) ce composé peut être facilement synthétisé, aussi bien par voie humide que par voie sèche, (ii) les tests de lixiviation révèlent des vitesses de dissolution très faibles (par exemple, à 90 °C et pH = 0 [19]), comparables à celles du verre nucléaire français R7T7 [1]. Cet apparent paradoxe pourrait trouver sa solution dans les relations de phases de ce composé dans les systèmes chimiques naturels, où l'apatite phosphocalcique est de loin le phosphate dominant [7,13]. Il nous est donc ainsi apparu judicieux d'étudier le comportement du PDT en milieu calcique, ce qui pourrait permettre d'expliquer l'absence d'analogue naturel du PDT. Remarquons dès à présent que l'existence d'une réaction de déstabilisation du PDT se traduirait par la libération, dans le milieu environnant, des actinides retenus dans sa structure. Comprendre les relations de phases que le PDT pourrait avoir avec les autres minéraux du système {P2O3CaOSiO2H2O} est donc important pour évaluer les propriétés de confinement du PDT vis-à-vis des radionucléides dans des conditions géologiques.
2 Stratégie expérimentale
Le PDT a déjà fait l'objet d'études détaillées de lixiviation, qui ont permis de mettre en évidence l'influence de différents paramètres tels que la surface spécifique, la température, le taux de renouvellement, le pH et la concentration en espèces phosphatées de la solution. Ces résultats ont permis de déterminer les lois cinétiques de dissolution du PDT [18]. Dans cette étude, nous avons développé une approche expérimentale complémentaire visant à encadrer le champ thermodynamique de stabilité du PDT, en le forçant à réagir en présence de matériaux susceptibles d'exister dans le champ proche d'un stockage de déchets nucléaires.
Bien que les températures attendues en stockage au sein du colis ne devraient pas dépasser 200 °C [14], nous avons volontairement choisi, dans un premier temps, une gamme de températures plus élevées, comprises entre 320 et 400 °C, pour obtenir des cinétiques de réaction compatibles avec des durées d'expériences raisonnables. De plus, ces expériences ont été conduites sous conditions hydrothermales en présence d'un fort gradient thermique (de l'ordre de 5 °C cm−1). Des études antérieures ont, en effet, montré que la présence d'un gradient de température dans l'axe du tube réactionnel horizontal engendre des transferts de matière en solution le long du gradient de température via des processus complexes de dissolution–transport–précipitation (voir, par exemple, [11,16,19]). En particulier, il a été montré que certains éléments ont tendance à migrer et à précipiter vers le point chaud du système, alors que d'autres se concentrent préférentiellement dans les phases précipitant dans la zone froide [11,15,16]. Ces expériences permettent donc de réaliser des lixiviations accélérées, où le moteur de dissolution n'est plus le degré de sous-saturation de la solution régulièrement renouvelée, mais l'existence d'un gradient d'activité induit par le gradient de température. Ce gradient d'activité est source d'un transport diffusif, sans que l'on puisse exclure aujourd'hui l'intervention de processus de transport non diagonaux, comme la thermodiffusion (voir, par exemple, [8]). Enfin, le PDT a été associé dans le tube réactionnel, d'une part, (i) à l'argile FoCa-7 (argile de Fourges-Cahaignes), largement étudiée dans le passé comme constituant potentiel de barrière ouvragée [15], et, d'autre part, (ii) à un ciment assurant un milieu calcique réactif. Cet ensemble définit un système chimique suffisamment large pour permettre l'expression de plusieurs phases minérales susceptibles de partager des relations de phase avec le PDT, comme l'hydroxyapatite, Ca5(PO4)3OH, la thorite, ThSiO4 ou la brabantite, CaTh(PO4)2.
3 Produits de départ, dispositif expérimental et caractérisation
3.1 Produits de départ : PDT, ciment et FoCa-7
Le PDT a été synthétisé par voie humide à partir du mélange de solutions concentrées de nitrate de thorium (2,17 M) et d'acide phosphorique (5 M) [4]. La solution de nitrate de Th est obtenue par dissolution de Th(NO3)4·5 H2O (Fluka, 99 %) dans l'acide nitrique. Le résidu obtenu après évaporation du mélange est chauffé sous atmosphère ambiante successivement à 400 °C pendant 6 h, puis à 1250 °C pendant 18 h. Le diagramme de diffraction sur poudre du produit obtenu correspond bien au diagramme attendu pour le PDT monophasé (fiche ICDD 86–669). Le ciment utilisé est un ciment conventionnel Portland de classe G, tandis que l'argile FoCa-7 est constituée à 80 % d'un interstratifié smectite/illite, associé à de la kaolinite. La composition chimique de ces deux matériaux est reportée dans le Tableau 1.
Composition partielle en poids d'oxydes (%) du ciment conventionnel Portland de classe G et de l'argile FoCa-7 [15], utilisés comme produits de départ.
Composition (weight %) of the reactants: the Portland cement (G class) and the FoCa-7 clay [15].
| Oxydes | FoCa-7 | Ciment |
| SiO2 | 44,08 | 19,4 |
| Al2O3 | 24,31 | 4,4 |
| CaO | 2,66 | 64,2 |
| FeO | 7,53 | 5,5 |
| MgO | – | 2 |
| TiO2 | 1,77 | – |
| Na2O | 0,32 | 0,1 |
| SO3 | – | 2,8 |
3.2 Dispositif expérimental
Le dispositif expérimental est identique à celui décrit par Poinssot et al. [15,16] pour des expériences similaires effectuées sur des verres. Il s'agit d'un tube réactionnel (TR) en or (150 mm de longueur et 4,8 mm de diamètre), soudé à ses deux extrémités et contenant deux capsules expérimentales semi-ouvertes (15 mm × 3,8 mm) contenant elles-mêmes les réactifs. Le TR est placé dans un autoclave à joint froid. La température est mesurée par un thermocouple chromel–alumel et la pression par une jauge électronique. Le profil de température le long de l'axe de l'autoclave a été calibré par une série de thermocouples introduits dans le corps de l'autoclave [11]. La différence de température entre les deux extrémités du TR est de 80 °C (entre 400 et 320 °C).
Les réactifs finement broyés sont 45 mg de PDT et 30 mg de ciment, introduits dans la capsule (A) disposée à l'extrémité froide du TR, entre 320 et 350 °C, et 60 mg de FoCa-7 introduits dans la seconde capsule (B) disposée à l'extrémité chaude, entre 390 et 400 °C, (Fig. 1). Le TR est rempli de 1 ml d'eau milliQ à . En fin d'expérience, l'autoclave est refroidi sous un flux d'air comprimé.
3.3 Caractérisation des produits expérimentaux
Les réactifs et les produits expérimentaux néoformés, ont été caractérisés par diffraction de rayons X sur poudre, grâce à un diffractomètre INEL CPS 120 (radiation Kα du cobalt, 30 kV, 25 mA) ou un diffractomètre Philips PW 1720 (radiation Kα du cuivre, 40 kV, 30 mA). Les produits néoformés ont été caractérisés directement sur les parois du tube découpé dans le sens de la longueur, par microscopie électronique à balayage (MEB, Hitachi S-2500, ENS, Paris) couplée à un système de spectrométrie de rayons X par dispersion d'énergie (EDS, Kevex Sigma).
4 Résultats
4.1 Phase néoformée après 73 j à 500 bar et 400–320 °C
4.1.1 Sur les parois du TR
Deux phases principales tapissent la paroi interne du TR dans toute sa longueur : il s'agit (i) d'un gel de silice localement associé, vers le point froid, à de rares cristaux de thorianite (Fig. 2C) et (ii) de la xonotlite, Ca5,7Si6,3O17,7(OH)2 (Fig. 3A). Au point chaud entre 380–390 °C du TR, à proximité de la capsule B, cristallisent l'anorthite (Si1,99Al2,08Ca0,93O8,03) et le grenat (grossulaire–andradite, Si3,09Al1,21Fe0,61Ca3,09·O11,99) (Figs. 2A et 2B). À l'extrémité la plus froide du TR, entre 320 et 330 °C derrière la capsule A, des cristaux de thorianite (ThO2) croissent isolément, ou bien recouvrent les cristaux de xonotlite (Figs. 3B et 3C).


Microphotographies (MEB–électrons secondaires) des phases néoformées sur les parois internes du tube réactionnel entre 380 et 390 °C (A, B) et autour de 360 °C (C).
SEM micrographies of secondary phases precipitated along the reaction tube between 380 and 390 °C (A, B) and around 360 °C (C).
4.1.2 À l'intérieur des capsules A et B
Dans la capsule B, au point chaud, entre 390 et 400 °C, l'argile de type FoCa-7 a réagi pour former un composé dont la raie de diffraction principale d001 est à 23,75 Å. Après traitement à l'éthylène glycol, cette raie de diffraction est déplacée à 27,17 Å, valeur caractéristique du gonflement des feuillets, suggérant un interstratifié régulier de type rectorite [3,19].
Dans lacapsule A, au point froid, entre 320 et 350 °C, en plus de la xonotlite, le phosphate–diphosphate de Th coexiste maintenant avec l'apatite et la thorianite (Figs. 3A et 4).
5 Discussion
5.1 Stabilité du PDT dans le milieu naturel ?
Nos résultats mettent en évidence la déstabilisation du PDT pour former de l'hydroxyapatite et de la thorianite, selon une réaction du type : 10 [CaO]ciment + Th4(PO4)4P2O7 + H2O → Ca10(PO4)6(OH)2 + 4 ThO2, où le CaO provient du ciment. Cette réaction est accompagnée d'une quasi-absence de mobilité du phosphore, qui reste cantonné au point froid dans la capsule B, d'une faible mobilité du Th vers le point froid, en contraste avec la forte mobilité de Si et Ca, qui participent à la cristallisation du grenat, et l'anorthite au point chaud [Figs. 2A et 2B]. Ces expériences démontrent que la réactivité du PDT n'est pas fortement différente de celle d'autres phases, comme les silicates ou les verres, placées dans des conditions expérimentales équivalentes [15]. Sa déstabilisation, en milieu calcique, pour former de l'apatite phosphocalcique et une phase riche en thorium (ici, la thorianite) est bien en accord avec la prédominance de l'apatite Ca5(PO4)3(F,OH,Cl) parmi les phosphates naturels, alors que le PDT n'a, à ce jour, pas d'équivalent naturel. Dans certaines conditions de chimie particulières rencontrées dans la nature (par exemple, un fort appauvrissement en CaO), on pourrait imaginer que le PDT puisse être exprimé [6]. Remarquons cependant que le PDT présente des liaisons POP, qui résistent mal à l'hydrolyse, ce qui limite la présence de pyrophosphates dans le milieu naturel, où le seul représentant reconnu est la wooldridgéite, Na2CaCu2(P2O7)2(H2O)10 [12]. Cette caractéristique cristallochimique est donc un obstacle supplémentaire à la cristallisation d'un équivalent du PDT dans la nature.
5.2 Implication pour le stockage
En l'absence de propriétés thermodynamiques pour le PDT, il est difficile de conclure définitivement sur la stabilité et le devenir de cette phase dans un système ouvert sur le milieu géologique. Cependant, le fait que le PDT ne cristallise pas dans le milieu naturel, doit conduire à envisager une déstabilisation du PDT au contact d'eaux naturelles souterraines calciques, à la faveur d'apatite et d'un composé thorifère. Nos expériences montrent que, dans les conditions de hautes températures (320–350 °C), avec une durée relativement courte (deux mois), ce sont bien ces phases qui cristallisent. Dans un système où les phases sont potentiellement stables dans une large gamme de température, il est classique de considérer que les expériences menées aux températures les plus hautes sont équivalentes à celles menées aux températures les plus basses, mais avec des cinétiques plus élevées. L'apatite étant une phase parfaitement stable à basse température, il n'est pas déraisonnable de considérer que le même type de réaction intervienne dans des conditions moins extrêmes, mais avec des cinétiques plus lentes. Le PDT apparaı̂t donc comme une phase synthétique, qui, dans le milieu naturel, se transformerait à terme en apatite, et une phase thorifère, qui pourrait être la thorianite.
Remerciements
Ce travail est soutenu par le GDR Nomade (CNRS–CEA–EDF–Cogema). Nous remercions Michel Genet pour l'intérêt qu'il a porté à ce travail. Nous tenons aussi à remercier François Gauthier-Lafaye et Pierre Costesèque pour leurs relectures attentives et leurs avis encourageants.