

1 Introduction
A soil is known as a hydromorphic or gley soil when some of its characteristics are due to an excess of water [4]. However some conditions with regard to hydric variations, biological and biogeochemical properties must be simultaneously fulfilled. They are: (i) an excess of water; (ii) the restriction of the oxygen sources; (iii) the presence of bio-available substrates; (iv) thermal conditions favourable to the microflora activity; (v) the presence of elements able to change their oxidation state and to record the variations more or less irreversibly, named geochemical markers of hydromorphic soils [49].
Among the elements whose oxidation state can change under earth-surface conditions, iron is the most abundant. The switching between the di- and trivalent redox state of iron initiates a number of significant geochemical reactions. Indeed, the large difference of mobility between Fe2+, soluble, and Fe3+, insoluble, leads to the segregation of iron in horizons and/or in soil sequences [29,40]. These variations are organized in landscapes and result in concretioning processes [29] and colour variations [44]. In virtually all soil classification systems, soil characteristics depending on iron dynamics are used to differentiate well-drained from poor-drained soils [5,41,51]. Thus soil colour is closely related with the nature of iron oxides, more specifically their degree of hydration and their amount [12,44,50]. Classically brown, red or yellow colours are associated with the occurrence of Fe(III) oxides, while grey, green, blue or black colours are associated with the occurrence of Fe(II) solids (oxides or sulphides), the white colour indicating the absence of any ‘free’ Fe oxides. Among these colours, the blue-green one is observed in moderately reduced waterlogged soils, and turns into ochre when soils are open to the outer atmosphere [33,50]. This colour has been ascribed to the occurrence in the milieu of mixed Fe(II)–Fe(III) compounds with a likely structure of green rusts (GRs). This assumption was formulated as early as in 1960, discussed largely in the literature (see reviews in [25] or [44]). GRs were first observed in corrosion products of steel [2,6,43], then in a waste sludge [22], but were only recognised as a mineral in soils since 1996 [47,48] (Fig. 1). The aims of this paper are to describe the different steps that led to the discovery of GR as a natural mineral, then to its homologation by the International Mineralogical Association as fougerite and to discuss eventually the questions that remain open today.

Gleysol of the site from Fougères (Brittany, France). The blue-green colour indicates the occurrence of Fougerite. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
Sol hydromorphe du site de Fougères (Bretagne, France). La couleur bleu-vert marque la présence de fougérite.
2 Field evidences
Field evidences for Fe mobility are mainly based upon morphological observations (colour), measurements of total Fe in soil and mineralogy by XRD. However, this gives only indirect evidences. No information is gained about the thermodynamic control and kinetics of the processes.
The study of the redox geochemistry of iron is rather difficult in natural environments due to the extreme sensitivity of iron towards aerial oxidation and to the lack of sensitive detection techniques. The study of Fe dynamics in situ is particularly pertinent in gleysols where iron is the major marker of the redox changes in soils. The originality of our field studies has been to investigate both the solution and the solid fraction, particularly containing iron, of soils, in order to check mineral–solution equilibriums. This was systematically made since 1987 [15,20,28,42].
2.1 Soil solutions
The in situ study of soil solutions in gleysols requires a specific procedure of sampling and measurement: soil waters are sampled without any contact with oxygen of air, to prevent oxidation, and in the dark, to prevent photoreduction, in chemically inert devices, placed at different depths, that act as zero-tension lysimeters [14,27]; this system allows us to sample free soil solutions. The non-conservative parameters are measured in the field: temperature, pH, redox potential, concentration by colorimetry with DPKBH (see [32], modified by [9,20]), nitrite and sulphide by colorimetry. In the laboratory, after filtration under nitrogen atmosphere, major cations and anions, metals, dissolved organic and inorganic carbon and alkalinity are measured. These data allow us, on the one hand, to establish the seasonal dynamics of the chemistry of soil waters, and, on the other hand, to check mineral–solution equilibriums in thermodynamic models providing for activity coefficients corrections and ion-pair formation, such as EQUIL(T) [17] or PHREEQC [31]. An originality of our approach is the following: total dissolved Fe(II) is used, with pH, major cations and anions concentrations to compute the activity of ; this value is not influenced by particulate or colloidal Fe(III), which would result in a large overestimation of , as dissolved Fe(III) cannot be separated from colloidal Fe. From the preceding value of the concentration of , the activity of is obtained. All mineral–solution equilibriums are checked by writing all reactions with the activity of , pH and pe as master variables [9] and by using a consistent set of thermodynamic data of the different iron species [9,36].
Two main patterns of seasonal dynamics can be distinguished, depending on the location of hydromorphic soils in landscapes (Fig. 2) [8]. The vertical movements of the watertable control the first pattern, which is generally observed in colluvio-alluvial systems, as follows: (i) when the level of the watertable is high, soils are waterlogged during a long time; reductive conditions occur and significant release of FeII in soil solutions is observed; (ii) when the level of the watertable gets lower, the environment becomes oxidant by entry of air, and FeII concentrations in soil solutions decrease drastically to zero (Fig. 3).
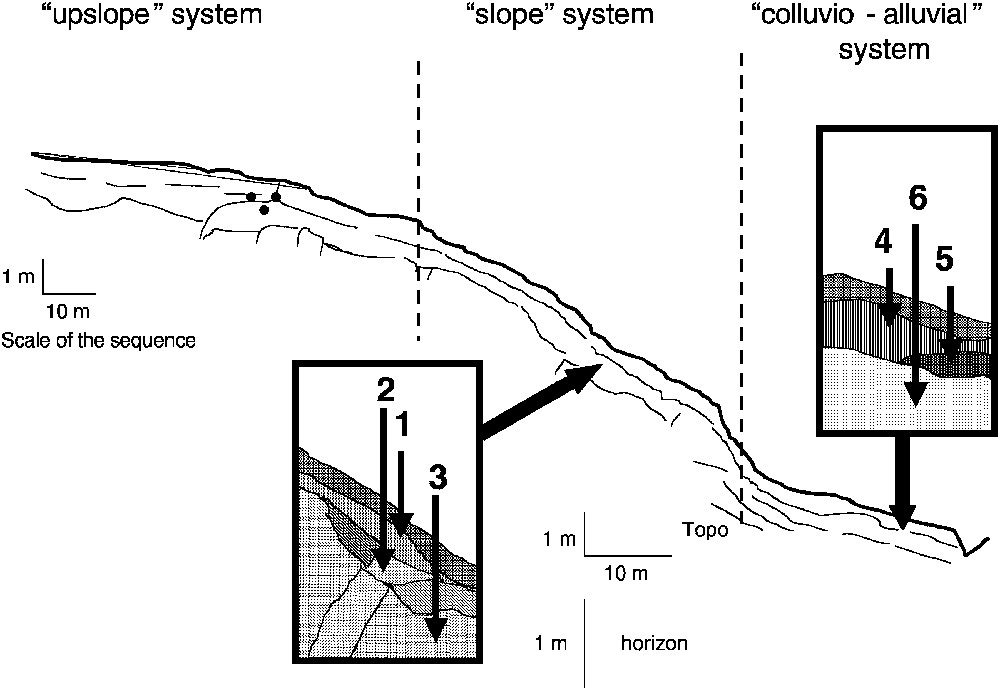
Location of sampling of soil waters in two different gleysol solums (from [8,28]).
Localisation des sites d'échantillonnage des solutions du sol dans deux solums hydromorphes différents (d'après [8,28]).
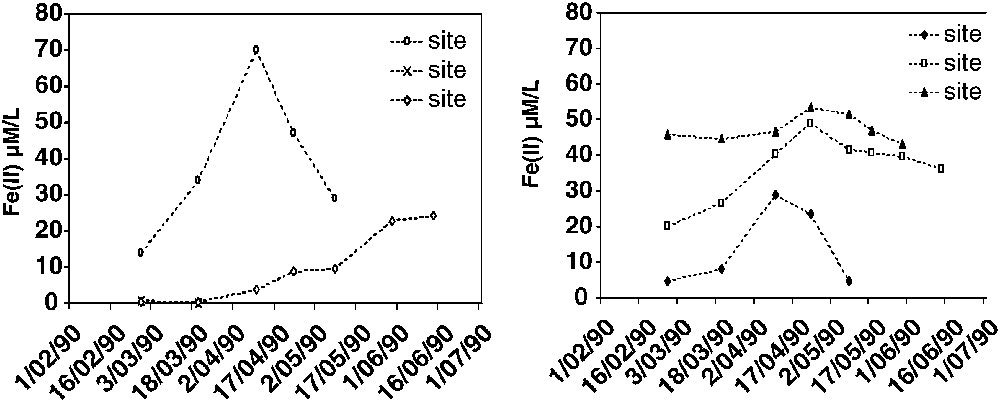
Seasonal dynamics of Fe(II) in soil solution in the different sites from Fig. 2 (from [8]).
Dynamique saisonnière du Fe(II) dans la solution du sol dans les différents sites de la Fig. 2 [8].
The recharge of oxygen by rainwater controls the second, generally observed in hydromorphic soils located at mid-slopes or over lithological discontinuities, as follows: (i) during the rainy season, enough dissolved oxygen is present, though the soils are completely waterlogged, so that the environment stays oxidant and no FeII is released in solution; (ii) when the rainflow decreases, the environment gets confined, becomes reductive and FeII concentrations increase strongly, until dissolved oxygen is supplied and Fe precipitates in situ.
These two patterns result in two different geochemical behaviours from a same initial condition, i.e. reductive and waterlogged. In the first case, there is a continuous flow of water when Fe is mobile. This results in the formation of bleached horizons, with exportation of Fe from the horizon. In the second case, there is no outflow when Fe is mobile, and Fe is globally conserved in the horizon, though locally it is segregated.
Check of Fe minerals–solutions equilibriums showed that all soil solutions are: (i) oversaturated with respect to Fe(III) oxides, haematite, goethite and lepidocrocite; (ii) undersaturated with respect to Fe(II) hydroxide; (iii) undersaturated with respect to GR1(Cl), GR1(CO3), GR2(SO4); (iv) close to equilibrium with a compound having the chemical formula , with [9]. This result is consistent with previous conclusions proposed in pioneering works by Ponnamperuma [33] in Philippines under rice crop or by Lindsay [26] in gleysols in Colorado. However, at this stage, the question remained whether this formula corresponds to a mean compartment of ferrosoferric complexes or to a specific mineral. Thus additional investigations of the Fe solid fraction of the soils were made.
2.2 The Fe-solid fractions in soils
To characterize the Fe solid fraction in soils, the principal experimental difficulties are the following: (i) the control of the redox state of soil, (ii) the heterogeneity of distribution of iron features, and (iii) the lability of the FeII–FeIII minerals.
Soil samples must be collected in a large volume with the surrounding soil solution and maintained in anoxic conditions in airtight box, without sieving and air-drying, the standard protocol in soil science. The permanence of the original blue or green-blue colours during all experiments is used as a preservation criterion. The physical separation of concretions or spots of Fe concentrations by microsampling under binocular in airtight box is preferred to a bulk analysis of the samples [45].
Kinetics by selective extractions [7] with citrate–bicarbonate [45] and dithionite–citrate–bicarbonate [21,30,46] define fractions of differential reactivity between microsites and complete the mineralogical and crystallographic analysis obtained by X-ray diffraction (XRD).
On a gleysol (Fig. 4) developed on granite in Brittany (France), seven microsampled soil features containing iron were distinguished from surface towards the depth. In the organo-mineral horizon, they are coatings with a reddish purple colour (2.5YR 3/6), noted LV, located around very fine roots and on the surfaces of aggregates. In the alluvial redoxic horizon, the BTO sample consists of diffuse ochre spots more clayey as the neighbouring white spots. In the structural redoxic horizon, the STO sample is made of diffuse ochre spots (7.5YR 5/8) in the matrix, with a similar texture as BTO sample. In the sandy-clayey saprolite, PO sample consists of large rusty spots (5YR 5/8) located in the most clayey part of the weathering zone, AR sample is the homogeneous bluish matrix (5GB 6/1), which turns into ochre when in contact with air and GRB and GRG are the rust coatings (7YR 5/8) developed directly on old root channels.
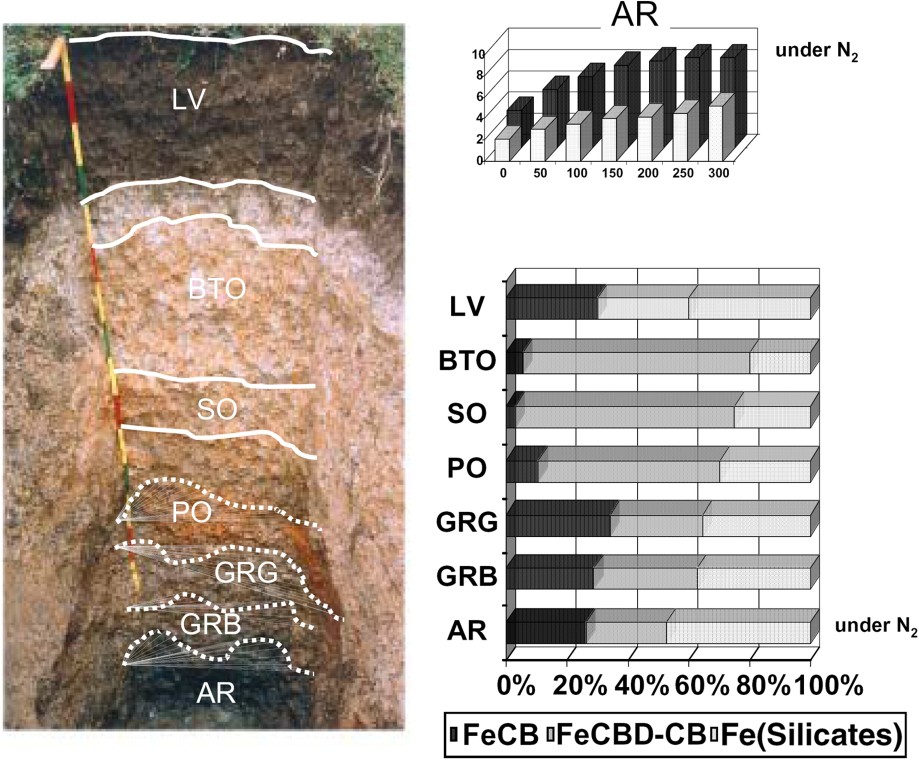
Sampling in a typical gleysol developed on granite and iron distribution in the extractible fractions by citrate–bicarbonate and dithionite–citrate–bicarbonate: for the AR sample, occurrence of a Fe fraction available without reduction under nitrogen atmosphere (from [49]).
Échantillonnage dans un sol hydromorphe développé sur granite et distribution du fer dans les compartiments extraits par les réactifs : citrate–bicarbonate et citrate–bicarbonate–dithionite : pour l'échantillon AR, mise en évidence de l'effet conservatoire d'une fraction du fer disponible sans réduction par manipulation sous atmosphère d'azote (d'après [49]).
Selective extractions [49] show the occurrence of a Fe labile fraction available without reduction whose size and reactivity can be determined by citrate–bicarbonate extractions. This fraction depends quantitatively on the waterlogged and oxido-reduction states of the milieu. In fact, even a short contact (a few hours) with air of the AR sample before CB extraction resulted in the immobilization of a large Fe fraction, e.g., up to 40% for AR sample (Fig. 4), with respect to the amount extractible by CB under N2 atmosphere. By calibration of the CB extraction method on synthetic minerals, it can be shown that mixed FeII–FeIII compounds, such as green rusts may contribute to the signal [46]. However, these extractions, though selective, are not specific of a mineral. As the total Fe content is small, about 5%, X-ray diffraction is not sensitive enough, and moreover the main GRs' line is very close to the main line of kaolinite. The question merges with the preceding, whether this Fe labile fraction corresponds to a mixed compartment of ferrosoferric complexes or to a specific mineral.
3 Fougerite discovery
From the preceding, it was clear that Mössbauer spectroscopy was the adequate technique to obtain an answer to the preceding questions. I proposed to choose the Fougères site for the following reasons: (i) an analogous study had shown that in a gleysol, 95 to 98% Fe was extractible by CB [15,47]; (ii) soil solutions had been studied since several decades; (iii) detailed soil maps and mineralogical data were available, showing that the clay fraction mainly consists of kaolinite and Al-vermiculite, with practically no silicated Fe, which is not the case on schists; (iv) the forested environment ensures no sludge application has been made so that the compounds determined are natural minerals.
Mössbauer and Raman spectroscopies have confirmed the occurrence of a green rust mineral, fougerite [47,48]. It was tentatively assumed that the anion present in the interlayer was OH−, and the global formula, , was proposed with [9,16]. Thermodynamic calculations on mineral–solution equilibriums gave values of , −1244.1 and −1944.3 kJ mol−1 for Fe3(OH)7, Fe2(OH)5, and Fe3(OH)8 respectively at 282.15 K [9]. These values were fitted to soil solution data on granite (Fougères and Quintin) and schists (Naizin). A more detailed study of the mineral was then necessary to establish more firmly the structure of the mineral, to meet the requirements of International Mineralogical Association (IMA).
4 Mineral characterizations
Fougerite is difficult to analyse because it is dilute in soil (total Fe2O3 content is ca. 4% only), is labile with a particle size less than 500 nm (Fig. 5) and cannot be separated from the other minerals. Thus its structure was established by X-ray absorption spectroscopy in addition to data obtained by Mössbauer spectroscopy [37] and microprobe analysis.

Fougerite, observed by electron microscopy, shows hexagonal plates with a mean size of 500 nm.
La fougérite, observée par microscopie électronique à balayage, montre une structure en plaquettes hexagonales de l'ordre de 500 nm.
4.1 Structure of the green-rust mineral fougerite and presence of Mg
The structure of the mineral was proposed to be GR1, analogous to GR1(Cl) [34], with a symmetry group type and lattice parameters and (Fig. 6). The structure of the mineral was studied by X-ray absorption spectroscopy (XAS), both by recording the Extended X-ray absorption fine structure signal (EXAFS) and the X-ray absorption near edge structure signal (XANES) at the Fe K-edge, along with transmission Mössbauer spectroscopy (TMS). The same techniques were applied to synthetic GR1(Cl), GR1(CO3), and to a synthetic pyroaurite with approximate formula . Two samples of hydroxycarbonates obtained by mixing Fe2+, Fe3+ and Mg2+ salts and coprecipitating hydroxides by addition of NaOH in the presence of Na2CO3 were also prepared and analysed by TMS. The x mole ratios were measured by Mössbauer spectroscopy. Powder X-ray diffractograms were performed on the synthetic samples. The global formulae of the compounds obtained were Fe(II)2Mg(II)2Fe(III)2(OH)12 CO3 and Fe(II)4Mg(II)2Fe(III)2(OH)16CO3. The main results obtained [37] are briefly summarized.
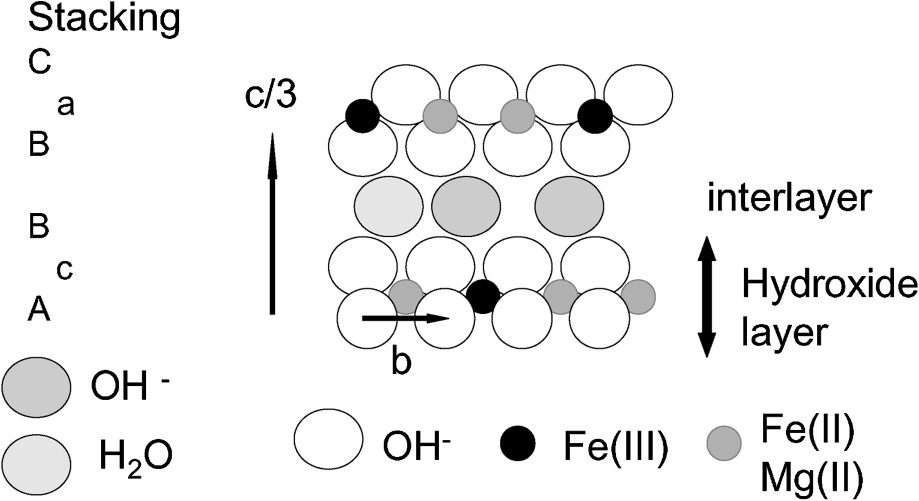
Proposed crystal structure of (OH) fougerite (from [18]).
Structure cristallographique proposée pour la fougérite (OH) (d'après [18]).
Both Fe2+ and Fe3+ valence states occur in close neighbourhood, as evidenced from the XANES part of spectrum, which is sensitive to the oxidation state of Fe and to its local symmetry; there are no clusters of Fe2+ or Fe3+. The mineral is confirmed to be a green rust, a double-layered hydroxide, because its pseudo-radial distribution functions (PRDF) display the same set of peaks (P1–P6) and are in agreement with previous studies on similar compounds [38] (Fig. 7). The lattice parameter a, is obtained at about 0.31–0.32 nm, as earlier predicted, from the second P2 peak of PDRF, i.e. the FeFe distance. The positions of farther – and smaller – peaks of the PRDF correspond to the hexagonal array of cations, second, third…neighbours of Fe, in the sequence (Fig. 8).
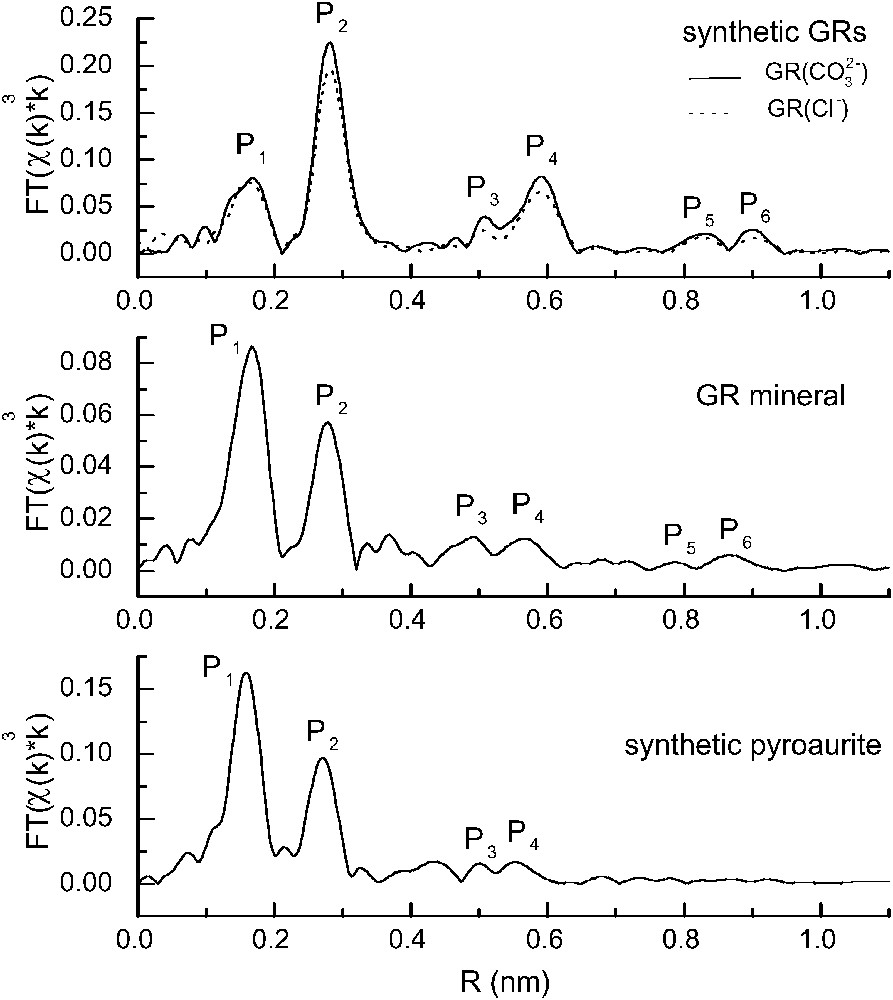
FeK pseudo-radial distribution functions (PRDF) for synthetic carbonate and chloride green rusts, fougerite mineral and synthetic pyroaurite. The real interatomic distances can be found approximately by adding about 0.04 nm to the distances given here (from [37]).
Distribution pseudo-radiale (PRDF) FeK des rouilles vertes de synthèse carbonatée et chlorurée, du minéral fougérite et d'une pyroaurite de synthèse. Les distances interatomiques réelles peuvent être obtenues approximativement en ajoutant 0,04 nm aux distances données en abscisse (d'après [37]).
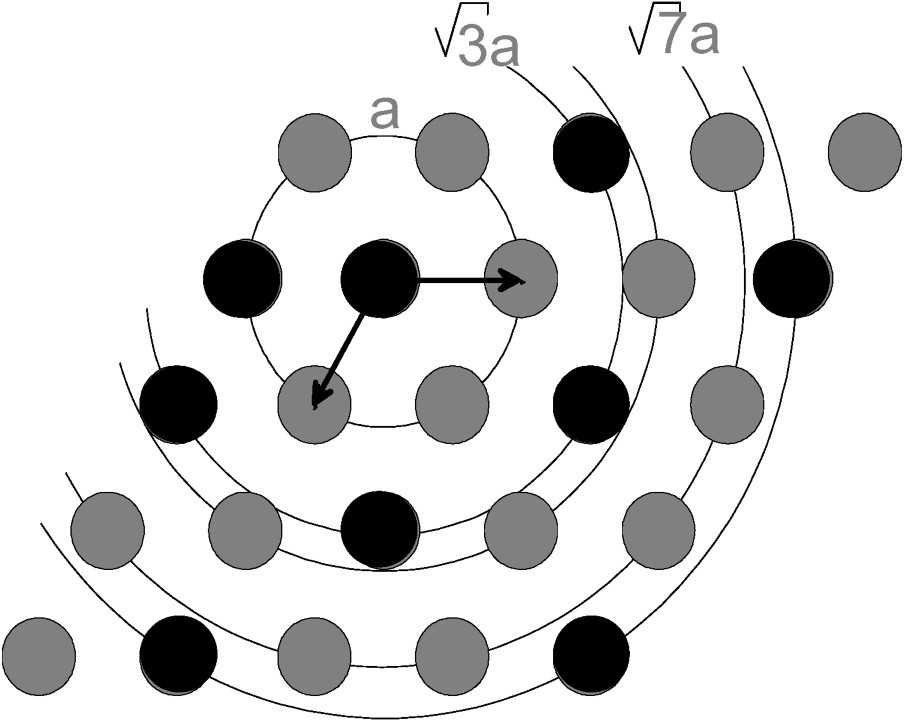
Upper structural limit of the distribution of divalent (grey spots) and trivalent metals (black spots) in the hydroxide layer where the presence of two neighbouring Fe(III) induces an oxolation reaction.
Limite structurale supérieure de la distribution des métaux divalents (ronds gris) et trivalents (ronds noirs) dans le feuillet d'hydroxyde où la présence de deux Fe(III) voisins entraîne une réaction d'oxolation.
Mg substitutes for Fe in the mineral. Evidence for this is that the radial distribution function obtained for the mineral is intermediate between those of GR1s and of synthetic pyroaurite, that is between Fe2+–Fe3+ and Mg2+–Fe3+ hydroxycarbonates. The peak positions are about the same, which proves that the mineral pertains to the group of GRs. However, the peak intensities observed for the mineral are different from the peak intensities of synthetic GRs and intermediate between those of GR1s and of synthetic pyroaurite. This is not surprising as many natural minerals are mixed Fe(II)–Mg(II) compounds (pyroxenes, micas, amphiboles, clay minerals, etc.).
The local ratio Mg/Fe is about ; this was estimated by fitting a linear combination of the contributions of Mg and Fe to the back-Fourier transform of PRDF curve: in the case of synthetic GR1s, no Mg is present and backscattering atoms are Fe atoms, whereas in synthetic pyroaurite, each Fe atom is surrounded by Mg atoms, so that Mg atoms are the only backscattering atoms. A much better fit is obtained when provision is made for both Mg and Fe contributions than with only Fe contribution.
The global mole ratio Mg/Fe cannot be directly obtained as the mineral is labile and small sized. However, additional data are given by microprobe analyses and by selective extraction techniques [15].
Microprobe analyses performed on 40 volumes from Fougères containing fougerite particles show that Mgtotal/Fetotal ratio ranges from 0.18 to 0.62. Each point is the average volume analysis of 2 μm3.
Selective extraction techniques using CB and CBD [45] give another argument. In the saprolite from Fougères, 48% Mg is extracted by CB, and all this Mg is extracted in 6 h, the global mole ratio (Mg/Fe)CB being close to 1. No additional Mg is extracted by CBD. This shows that Mg is present in the silicated phase (52%), but not in well crystallized Fe oxides.
In the reductic silty horizon in Fougères, 59% Mg is extracted by CB, Mg and Fe being simultaneously released with time.
The kinetics obtained is: (, ). The intercept is Fe/Mg= 0.5981, which corresponds to a global Mg/Fe ratio = 1.67. We can thus conclude that Mg present in the CB-extractible compartment is associated with Fe, i.e. originates from fougerite. The values obtained for the global mole ratio by selective extraction techniques in Fougères are thus in the range 0.1–1.7. This range is compatible with the local mole ratio obtained by XAS.
The following structural formula can thus be proposed for fougerite:
4.2 The nature of the interlayered anion: an open question
The nature of the anion present in the mineral cannot be determined by XAS, because spectra of various GRs proved to be independent of the nature of the interlayer anion. Parameter a is accessible by XAS and determined as 0.31–0.32 nm [37]. This is compatible with the values obtained for pyroaurite as 0.3113 nm [3], for as 0.3160 nm [13,23] and for GR(Cl−) as 0.319 nm [35].
Parameter c is more sensitive to the nature of the interlayered anion, but it is not accessible for the natural mineral.
It has been recently observed that synthetic can be partially deprotonated, with FeOFe bonds [19,24] by oxidation. It was argued that this could explain Fe(III) mole ratios larger than 1/3, and that this could be the case for the natural mineral fougerite. However the compound obtained, * is not well characterized, and its a and c parameters are smaller than the same parameters for synthetic , due to the shrinkage of the lattice; specifically, a “shrinks from 0.317 to 0.3014 nm” [19]. Such a shrinkage is not observed in the natural mineral, whose a parameter is larger than 0.31 nm. Moreover, there is a discrepancy between the stability of and of other GRs [10], which implies that carbonate is probably mixed with hydrogenocarbonate in the interlayer. It has been demonstrated that either the stoichiometry or the Gibbs free energy of formation of must be revaluated. The recent study of by FTIR [23] was made at very large concentrations of (1 M) and only showed that this technique is not sensitive enough to detect concentrations of hydrogenocarbonate at the levels commonly found in acid brown soils, 10−4 M.
Anyway, it is likely that the nature of the interlayer anion changes according to the environment. So, we can expect fougerite can accommodate a variety of anions, either inorganic or organic, the same as synthetic GRs and many other layered double hydroxides (LDH) [11]. This is similar to the situation encountered with smectites, which can accommodate a variety of interlayered cations. From the chemical composition of soil waters where fougerite from Fougères occurs, activities of Cl− and anions are very small, respectively 10−3 and 10−10. We suggested [15] that the most probable anion is OH−, which can be obtained by a simple deprotonation of a water molecule in the interlayer when Fe(II) is partly oxidized, without the necessity for the pH in the environment to be very high. Check of fougerite/solution equilibriums, based upon the development of a ternary solid solution model with different anions, OH− [10], Cl−, and mixed [16] shows that the soil solution (i) is always undersaturated when Cl− is the interlayer anion; (ii) always oversaturated when OH− is the interlayer anion. Depending on the Mg mole fraction in the mineral, the solutions are either undersaturated or oversaturated when or mixed / are the interlayer anion. This can be different in other environments, depending on the ratios of concentrations of the anions and on the Mg content in the layer.
In situ, fougerite transforms into lepidocrocite by oxidation [1] excluding high level of carbonate, dissolved aluminium and silicon in surrounding waters, which are inhibitors of lepidocrocite formation [39]. Moreover, in the experimental study on the electrochemical synthesis of [23], the authors noted: “The surface of the iron exhibited a greenish-blue colour after the rinsing and drying procedure. No colour change apparently occurs in air indicating that the oxidation layer is rather stable”. This is contrary to the classical observation that the colour of gleysols readily changes from bluish-green to ochre when exposed to the air. It appears then that the results from those syntheses cannot be straightforwardly extrapolated to natural conditions and to the mineral formation.
It can thus be concluded that for fougerite, the question of the exchangeable character of anions is open. We could speak of CO3-fougerite, SO4-fougerite, OH-fougerite or Cl-fougerite as we speak of Ca-montmorillonite, Na-montmorillonite, or of fluoro-apatite, carbonate-apatite, etc. Selectivity constants have not been determined, though qualitative results exist. According to [11], “the following sequence of exchange for both mono- and divalent anions in the LDHs has been derived:
Though the selectivity constants are in favour of divalent anions, which are the same mutatis mutandis for smectites and divalent cations, the exclusive presence of carbonate cannot be established as a rule, and the presence of OH− or Cl− in the interlayer cannot be ruled out. Homologous compounds have been described with OH− in the interlayer, such as meixnerite, Mg6Al2(OH)18⋅4 H2O, and with Cl− in the interlayer, such as iowaite, Mg6Fe2(OH)16Cl2⋅4 H2O and woodallite Mg6Cr2(OH)16Cl2⋅4 H2O.
5 Conclusion
Fougerite is today recognized as a new iron oxide, which occurs in waterlogged soils. It belongs to the green rust group with particular properties quite different from the other iron oxides. Both laboratory experiments in abiotic or biotic conditions and field evidences suggest that fougerite may be an important precursor of many ferric oxides in many environments. Fig. 9 summarizes from literature the pathways between Fe2+ in solution and ferric oxides implying green rusts. The challenge to the future is now: (i) to reevaluate the stoichiometry or the Gibbs free energy of formation of and consequently the soil-solution equilibriums tests; (ii) to determine the exact nature of the interlayer anion in fougerite; (iii) to study the interactions between fougerite and other foreign ions and molecules present in the milieu and their consequences on the biogeochemistry of iron cycle in natural environment.
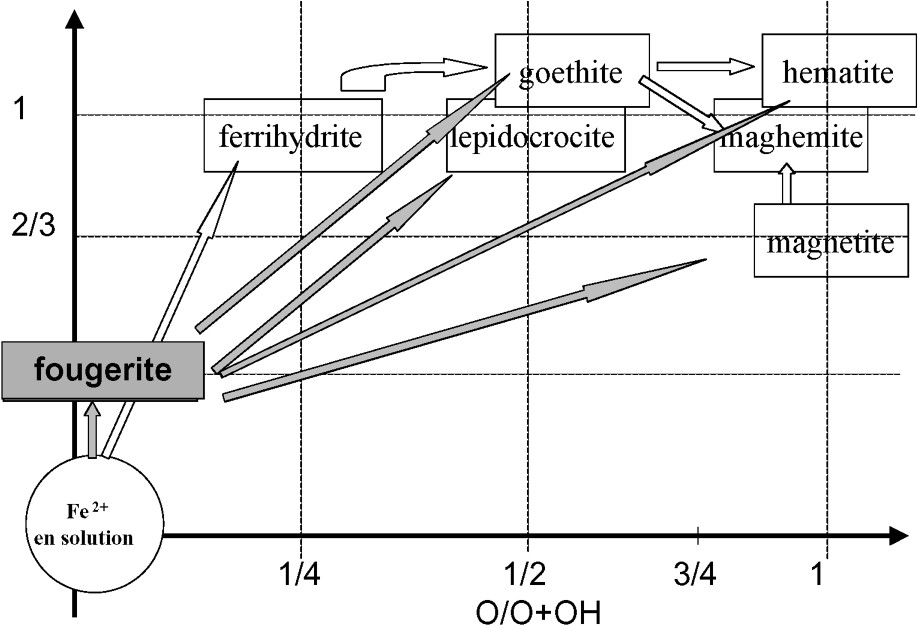
Place of the fougerite in the pathways of formation of iron oxides and oxihydroxides from Fe(II) in solution.
Place de la fougérite dans les voies de formation des oxydes et oxyhydroxydes de fer à partir du Fe(II) de la solution.
Acknowledgements
The results summarized here have benefited from the cooperation during many years of a large staff of colleagues and PhD students: M. Abdelmoula, Y. Bénard, B. Bernahrdt, D. Borschneck, G. Bourrié, E. Burke, L. Carteaux, P. Curmi, J.-J. Ehrhardt, F. Féder, F. Garnier, J.-M.R. Génin, J.-C. Germanique, A.J. Herbillon, B. Humbert, A. Jaffrezic, G. Klingelhöfer, V. Maître, M. Mayor, S. Moustier, P. Refait, A. Régeard, F. Rouault, A. Soulier. It was supported financially by INRA, CNRS, the European Union (FEDER), the French ‘Ministère de la Recherche’, the Brittany and Provence–Alpes–Côte d'Azur regions, the ‘Syndicat mixte de l'Arbois’, the ‘Communauté urbaine du pays d'Aix’, the PROSE–PNSE and PRIR programs. The help of Drs P. Chassin, J. Mamy, Profs A. Mariotti, Drs G. Pédro, P. Stengel, and P. Thivend is gratefully acknowledged. The help of an anonymous referee in improving the manuscript is acknowledged too.