


 CC-BY 4.0
CC-BY 4.0
1. Introduction
Carbonatites are magmatic rocks with more than 50% of carbonates [Le Maitre 2002] and constitute the main rare earth element (REE) deposits on Earth [Verplanck et al. 2016]. With only 527 described occurrences [Woolley and Kjarsgaard 2008], carbonatites are rare on the terrestrial surface. Only one occurrence of active carbonatitic magmatism is known: the Ol Doinyo Lengai volcano, in Tanzania [Keller and Zaitsev 2012]. Although this volcano mainly produces highly peralkaline silicate lavas such as phonolites and nephelinites, it is also the only one to produce alkaline carbonate lavas called natrocarbonatites [Klaudius and Keller 2006].
The CaCO3–Na2CO3–K2CO3 system has been experimentally studied to understand natrocarbonatite genesis. Cooper et al. [1975] established a phase diagram in the system Na2CO3–CaCO3, showing the saturation of nyerereite Na2Ca(CO3)2 to divide the system into two subsystems, an alkali- and a calcium-dominated one, with their own eutectics. However, Jago and Gittins [1991] showed that F drastically decreases the temperature of calcite saturation, potentially unifying the two subsystems by suppressing the calcite–nyerereite eutectic.
As natural natrocarbonatites are halogen-rich [Keller and Zaitsev 2012], Weidendorfer et al. [2017] tested this hypothesis by saturating a natural natrocarbonatite with calcite, and proposed a phase diagram in which the calcite–nyerereite eutectic is shifted into a peritectic point at a lower temperature, effectively making a genetic link possible between calcio- and natrocarbonatites.
Extrusive carbonatite occurrences are often described as coexisting with alkaline silicate rocks in the field [Bell et al. 1999; Mitchell 2005; Woolley and Kjarsgaard 2008]. The production of carbonatites by immiscibility with an alkaline silicate melt is a well-studied and recognised process [Freestone and Hamilton 1980; Brooker 1998; Martin et al. 2013; Nabyl et al. 2020]. Among the experimental studies on the immiscibility processes between carbonate and silicate melts, some have tackled the REE behaviour between both melts [Veksler et al. 1998, 2012; Martin et al. 2013; Nabyl et al. 2020]. REE enrichments in the carbonate melt at immiscibility are highly variable. However, the results from Nabyl et al. [2020] demonstrate that carbonate melts can be extremely Ca- and REE-rich if the immiscible silicate melt is strongly differentiated. Such REE-rich carbonate melts could be related to carbonatitic REE deposits.
In natural carbonatites related to REE deposits, REE are mostly contained in different REE minerals, with bastnaesite-(Ce) (LREE)CO3F being the main mineral of interest [Verplanck et al. 2016]. Field literature still debates the magmatic or hydrothermal origin of this mineral [Giebel et al. 2017; Cheng et al. 2018; Guo and Liu 2019; Anenburg et al. 2020]. Experimental literature on the subject is scarce and inconclusive, with Jones and Wyllie [1986] failing to crystallise magmatic bastnaesite in a CaCO3–Ca(OH)2–La(OH)3 system. As such, we currently have little knowledge on the whereabouts of magmatic REE minerals in carbonatites.
To better understand REE mineral crystallisation in carbonatites, magmatic crystallisation processes need to be assessed. As Ca- and REE-rich carbonate melts can be produced by immiscibility [Nabyl et al. 2020], the crystallisation sequence of such melts inside the CaCO3–Na2CO3–K2CO3 system needs to be defined, as well as the REE behaviour along this sequence. Here, we present new crystallisation experiments on a REE-rich calciocarbonatitic composition, in order to define the phase assemblage and characterise the REE behaviour between REE minerals and carbonate melt at the magmatic stage. These crystallisation experiments have been completed with a bastnaesite saturation experiment to determine the magmatic saturation of bastnaesite.
2. Experimental approach
2.1. Starting mixture
Because of extensive alteration, natural carbonatite compositions are not representative of primary carbonatitic melts [Bell et al. 1999; Verplanck et al. 2016]. To determine a theoretical, REE-rich primary carbonatitic composition, we used a natural rock composition database [GEOROC database (http://georoc.mpch-mainz.gwdg.de/georoc/, accessed: 2019/01/25) and De Moor et al. 2013; Mattsson et al. 2013; Mana et al. 2015] to first determine a silicate melt composition likely to be in equilibrium with an immiscible REE-rich carbonate melt, based on the work of Nabyl et al. [2020]. This silicate melt, alkaline and differentiated, corresponds to a phonolitic composition. We then used an experimental immiscibility database [Freestone and Hamilton 1980; Hamilton et al. 1989; Jones et al. 1995; Kjarsgaard et al. 1995; Brooker 1998; Kjarsgaard 1998; Veksler et al. 2012; Martin et al. 2013; Massuyeau et al. 2015; Nabyl et al. 2020] and the model of Nabyl et al. [2020] to determine partition coefficients for major and trace elements.
The carbonatite composition has been calculated using these partition coefficients. This composition was then synthesised using carbonates and oxide powders (Table 1). Details on the calculation to infer a primary carbonatite composition and on the preparation of the starting mixture are given in the Supplementary material.
2.2. Experimental strategy
At each P–T condition, up to three samples have been prepared: (1) with the starting composition (“EXP-P” set, Table 2); (2) with the starting composition and additional water, at a ratio of 95:5 (“EXP-W5” set, Table 2); and (3) with the starting composition and additional pure graphite, at a ratio of 97:3 (“EXP-G3” set, Table 2). Because of powder hygroscopy, the starting mixture holds around 2 wt% H2O (CHNX analysis, Flash 2000 Thermo). The addition of 5 wt% of water makes EXP-W5 hold around 7 wt% H2O. The addition of graphite in EXP-G3 reduces the system and consumes water, making them near-anhydrous. Twenty crystallisation experiments have been conducted at a pressure of 1 kbar, and at temperatures between 900 and 600 °C with 50 °C steps (Table 2). Experiments lasted from three days to two weeks.
Bastnaesite is stable below 760 °C at 1 kbar [Hsu 1992]. To test magmatic bastnaesite saturation, one saturation experiment has been conducted at 600 °C using the starting mixture and additional crushed bastnaesite from the Mianing-Dechang REE belt, at a ratio of 80:20 (“EXP-600B” experiment, Table 2).
Starting mixture composition, a REE-rich calciocarbonatite
| Components | Starting mixture |
|---|---|
| SiO2 | 0.36 |
| TiO2 | 0.13 |
| Al2O3 | 0.05 |
| FeO | 5.20 |
| MnO | 0.50 |
| MgO | 1.58 |
| CaO | 32.75 |
| Na2O | 12.91 |
| K2O | 1.61 |
| P2O5 | 0.59 |
| SrO | 0.11 |
| BaO | 0.24 |
| Cl | 1.44 |
| F | 4.43 |
| CO2 | 34.56 |
| La | 10,610 |
| Ce | 12,760 |
| Pr | 1,765 |
| Nd | 5,753 |
| Sm | 751 |
| Eu | 108 |
| Gd | 543 |
| Tb | 65 |
| Dy | 368 |
| Y | 2,108 |
| Ho | 81 |
| Er | 152 |
| Yb | 127 |
| Lu | 16 |
| Nb | 248 |
| Total | 100.00 |
Upper part: major elements (wt%). Lower part: trace elements (ppm). Total counts the abundance of trace elements and is in wt%.
Experiments were performed in a vertical internally heated pressure vessel (IHPV) at the Institut des Sciences de la Terre d’Orléans (ISTO). 20 to 30 mg of material were loaded into Au capsules with a diameter of 2.5–2.9 mm and a length of 10 to 15 mm. Sensor capsules were added to the experiments to measure the oxygen fugacity inside the IHPV. These sensor capsules were water-saturated and contained two Ni–Pd–NiO pellets separated by zirconium oxide powder. Ni and Pd molar proportions in the pellets were 0.5–0.5 and 0.15–0.85. We used a double-winding Kanthal furnace allowing near-isothermal conditions (gradient < 2–3 °C/cm) along a 3 cm long hot spot [Andújar et al. 2013]. The capsules were loaded in an alumina tube fixed at the top of the furnace (hot spot) by a Pt wire. Two K-type thermocouples were placed on top and bottom of the alumina tube to monitor temperature, with an accuracy up to ± 5 °C [Andújar et al. 2013]. The capsules were pressurised with 998 bars of Ar and 2 bars of H2. Pressure monitoring was performed through a transducer calibrated against a Heise Bourdon gauge, accurate up to ±2 MPa [Andújar et al. 2013]. At the end of the experiments, a fast isobaric quench (100 °C/s) was ensured using a drop technique, by electrically fusing the Pt wire [Di Carlo 2006].
Experimental conditions and run products. Partial pressure of H2 is always 2 bars. Gold capsules were used
| Experiment | Set | H2O (wt%) | C (wt%) | Bast (wt%) | P (bar) | T (°C) | 𝛥NNO | Duration | Phases |
|---|---|---|---|---|---|---|---|---|---|
| EXP-900P | EXP-P | 0 | 0 | 0 | 1000 | 900 | − 0.29 ± 0.73 | 38 h and 45 min | Liq, Fl, Brt, OxFe, Dpt |
| EXP-900W5 | EXP-W5 | 5 | 0 | 0 | 1000 | 900 | − 0.29 ± 0.73 | 38 h and 45 min | Liq, Fl, Brt, OxFe, Dpt |
| EXP-850P | EXP-P | 0 | 0 | 0 | 1000 | 850 | n.a. | 73 h and 50 min | Liq, Fl, Brt, OxFe, Dpt |
| EXP-850W5 | EXP-W5 | 5 | 0 | 0 | 1000 | 850 | n.a. | 73 h and 50 min | Liq, Fl, Brt, OxFe, Dpt |
| EXP-850G3 | EXP-G3 | 0 | 3 | 0 | 1000 | 850 | n.a. | 73 h and 50 min | Liq, Fl, Brt, OxFe, Cal, Dpt |
| EXP-800P | EXP-P | 0 | 0 | 0 | 1000 | 800 | 0.71 ± 0.41 | 89 h and 30 min | Liq, Fl, Brt, OxFe, Cal, Dpt |
| EXP-800W5 | EXP-W5 | 5 | 0 | 0 | 1000 | 800 | 0.71 ± 0.41 | 89 h and 30 min | Liq, Fl, Brt, OxFe, Cal, Dpt, Hl |
| EXP-800G3 | EXP-G3 | 0 | 3 | 0 | 1000 | 800 | 0.71 ± 0.41 | 89 h and 30 min | Liq, Fl, Brt, OxFe, Cal, Dpt |
| EXP-750P | EXP-P | 0 | 0 | 0 | 1000 | 750 | 1.11 ± 0.51 | 135 h and 45 min | Liq, Fl, Brt, OxFe, Cal, Dpt |
| EXP-750W5 | EXP-W5 | 5 | 0 | 0 | 1000 | 750 | 1.11 ± 0.51 | 135 h and 45 min | Liq, Fl, Brt, OxFe, Cal, Dpt |
| EXP-750G3 | EXP-G3 | 0 | 3 | 0 | 1000 | 750 | 1.11 ± 0.51 | 135 h and 45 min | Liq, Fl, Brt, OxFe, Cal, Hl |
| EXP-700P | EXP-P | 0 | 0 | 0 | 1000 | 700 | 0.55 ± 0.33 | 163 h and 00 min | Liq, Fl, Brt, OxFe, Cal, Pyr |
| EXP-700W5 | EXP-W5 | 5 | 0 | 0 | 1000 | 700 | 0.55 ± 0.33 | 163 h and 00 min | Liq, Fl, Brt, OxFe, Cal, Hl |
| EXP-700G3 | EXP-G3 | 0 | 3 | 0 | 1000 | 700 | 0.55 ± 0.33 | 163 h and 00 min | Liq, Fl, Brt, OxFe, Cal, Pyr |
| EXP-650P | EXP-P | 0 | 0 | 0 | 1000 | 650 | 0.54 ± 0.27 | 396 h and 20 min | Liq, Fl, OxFe, Cal, Pyr, Phg, Ap |
| EXP-650W5 | EXP-W5 | 5 | 0 | 0 | 1000 | 650 | 0.54 ± 0.27 | 396 h and 20 min | Liq, Fl, Brt, OxFe, Cal, Pyr, Phg, Ap |
| EXP-650G3 | EXP-G3 | 0 | 3 | 0 | 1000 | 650 | 0.54 ± 0.27 | 396 h and 20 min | Liq, Fl, OxFe, Cal, Pyr, Phg, Ap |
| EXP-600P | EXP-P | 0 | 0 | 0 | 1000 | 600 | n.a. | 311 h and 20 min | Liq, Fl, OxFe, Cal, Pyr, Ap |
| EXP-600W5 | EXP-W5 | 5 | 0 | 0 | 1000 | 600 | n.a. | 311 h and 20 min | Liq, Fl, OxFe, Cal, Pyr, Ap |
| EXP-600G3 | EXP-G3 | 0 | 3 | 0 | 1000 | 600 | n.a. | 311 h and 20 min | Liq, Fl, OxFe, Cal, Pyr, Phg, Ap |
| EXP-600B | EXP-B | 0 | 0 | 20 | 1000 | 600 | n.a. | 311 h and 20 min | Liq, Fl, Brt, OxFe, Cal, Dpt, Frg, Bast |
H2O, C and Bast are respectively pure water, pure graphite and natural bastnaesite added to the starting mixture. 𝛥NNO values are representative of the oxygen fugacity inside the IHPV. The global 𝛥NNO of the experiments is equal to 0.53 ± 0.21 (n.a. = not analysed). Liq: carbonate melt. Cal: calcite. OxFe: iron oxide. Fl: vesicle (fluid indicator). Dpt: deposit-like vesicle filling. Hl: halite. Brt: britholite. Phg: phlogopite. Ap: fluorapatite. Pyr: pyrochlore. Bast: bastnaesite. Frg: fergusonite.
2.3. Analysis methods
Electronic images (back-scattered electrons: BSE) of the samples have been taken with a Merlin Compact ZEISS scanning electron microscope (SEM), equipped with a GEMINI I column (ISTO, Orléans). Energy dispersive spectroscopy (EDS) has been done with a Bruker QUANTAX XFlash® 6|30 to identify the composition of the different phases. Acquisition parameters were: working distance 10 mm; voltage 15 kV. A TESCAN SEM (FEG-Schottky cathode) has also been used (Bureau de Recherches Géologiques et Minères – BRGM, Orléans), with a working distance of 15 mm and a 15 kV voltage.
Major elements have been analysed with a CAMECA SXFive electron probe micro analyzer (EPMA) (ISTO, Orléans), using wavelength dispersive spectrometry (WDS), with acquisition parameters equal to 15 kV and 6 nA. Standards and counting times were: albite (Na: 10 s, Si: 10 s) – Al2O3 (Al: 10 s) – orthoclase (K: 10 s) – andradite (Ca: 10 s) – MnTiO3 (Mn: 10 s, Ti: 10 s) – Fe2O3 (Fe: 10 s) – vanadinite (Cl: 30 s) – topaz (F: 30 s) – dolomite (Mg: 10 s) – NdPO4 (P: 10 s). The carbonate melt does not quench into a homogeneous glass, so the melt quench products have been analysed with a 5 to 50 μm diameter defocused beam to account for this heterogeneity and to avoid element loss. Routine EPMA analyses were conducted on the sensor pellets to analyse the NiPd alloys. The 𝛥NNO was then calculated using Taylor et al. [1992] and Frost [1991] equations. 𝛥NNO values are available in Table 2. We specify here that these values are a measurement of the oxygen fugacity inside the IHPV, and may be different than the oxygen fugacity inside the capsules.
LA-ICP-MS (Laser Ablation Inductively Coupled Mass Spectrometry) has been used to quantify the trace elements composition of the experimental run products. Two devices have been used: an Agilent 7500 CS with a 193 nm laser (Laboratoire Magmas et Volcans – LMV, Clermont-Ferrand), and an Agilent 8900-QQQ with a RESOlution-SE 193 nm Ar-F laser (ISTO, Orléans). Analysed isotopes were 43Ca, 44Ca, 88Sr, 89Y, 93Nb, 137Ba, 139La, 140Ce, 141Pr, 146Nd, 147Sm, 153Eu, 157Gd, 159Tb, 163Dy, 165Ho, 166Er, 172Yb and 175Lu. The fluence was set to 2 J⋅cm−2; and the frequency was set to 2 Hz for the analysis of melts, and to 1 Hz for the analysis of crystals. Phases were analysed with a 5 to 50 μm diameter beam. The analysis sequence consisted of 20 to 30 s of background followed by 60 to 80 s of ablation, with dwell times of 50 ms per isotope. Raw data was processed using Glitter 4.4 [Van Achterberg et al. 2001]. NIST610 glass [Pearce et al. 1997] was used as an external standard. NIST612 glass [Pearce et al. 1997] and BCR-2G glass [Rocholl 1998] were used as quality controls. The CaO wt% content of phases, determined at EPMA, was used as the internal standard, for 43Ca contents. Repeat analyses on selected phases and standard analyses ensured that data collected with both devices were within uncertainty.
3. Results
3.1. Carbonatite crystallisation
Table 2 summarises the experiments performed during this study. Figure 1 shows the different phases observed in all the samples.

BSE images of samples. (A) melt and segregated calcite, iron oxides and britholite. (B) Euhedral britholites and iron oxides. (C) Vesicle filling hosting anhedral halite inside a vesicle. (D) Bastnaesite and fergusonite in the saturation experiment. Liq: carbonate melt. Cal: calcite. OxFe: iron oxide. Fl: vesicle (fluid indicator). Dpt: deposit-like vesicle filling. Hl: halite. Brt: britholite. Bast: bastnaesite. Frg: fergusonite.
The carbonate melt quenched into dendritic calcites intergrown with quenched Ca,Na,K-carbonates (Figure 1A). As the temperature decreases, quenched Ca,Na,K-carbonates become dominant and coarser. Calcite is the dominant crystallising phase in the system. Calcite crystals are round to sub-euhedral (Figure 1A), and more abundant with decreasing temperature.
Several minor phases are also observed. Ti-rich iron oxides form sub-euhedral crystals which are often grouped (Figure 1B). Britholite (REE, Ca)5((Si,P)O4)3(F,OH), a REE-phosphosilicate isomorphic with apatite, occurs only at high temperatures (700–900 °C). Britholite crystals are often grouped, forming small euhedral, hexagonal, columnar prisms (Figure 1B), around 5 to 15 μm in size. Phlogopite is observed at low temperatures, crystallising as euhedral hexagonal prisms, or anhedral elongated crystals. Small, euhedral fluorapatites are found as clusters at low temperatures. Pyrochlore (Ca,Na,REE)2Nb2O6(OH,F) [Verplanck et al. 2016] crystallises below 700 °C in small anhedral clusters.
A fluid phase is present as hinted by spherical vesicles in all experiments (Figure 1A,C). The vesicular texture is significantly more abundant in EXP-G3. A Na-rich carbonate lining may be observed on the vesicle walls, which will be referred to as vesicle filling (Figure 1C). This vesicle filling is significantly more present in EXP-W5 and is only observed at temperatures higher than 700 °C. Halite is occasionally seen both inside vesicles and fractures. Mostly anhedral (Figure 1C), it can also be found as euhedral cubes.
The bastnaesite saturation experiment features a different mineralogy than all of the crystallisation experiments set (Table 2). Large anhedral crystals of bastnaesite are common in the saturation experiment (Figure 1D), whereas bastnaesite is never found in crystallisation experiments. Fergusonite (REE)NbO4 is also found in the saturation experiment as euhedral crystals (Figure 1D). Britholite is found as large clusters of small anhedral crystals even at this low temperature (600 °C).
At high temperatures (⩾750 °C), we observe crystal-free zones in carbonate melts and cumulates of crystals (Figure 1A). Some small cumulates are observed along the melt–fluid vesicles interface.
Figure 2 presents a phase diagram in a T–H2O space for our crystallisation experiments, with H2O being the wt% H2O content of the system, for all type of samples as described in Section 2.2). We specify here that the hydration degree of the system (wt% H2O in the capsule) is not the same as the hydration degree of the carbonate melt (wt% H2O dissolved in the melt). The water content of the melt could not be quantified in this study, therefore the water effect discussed here is only qualitative.
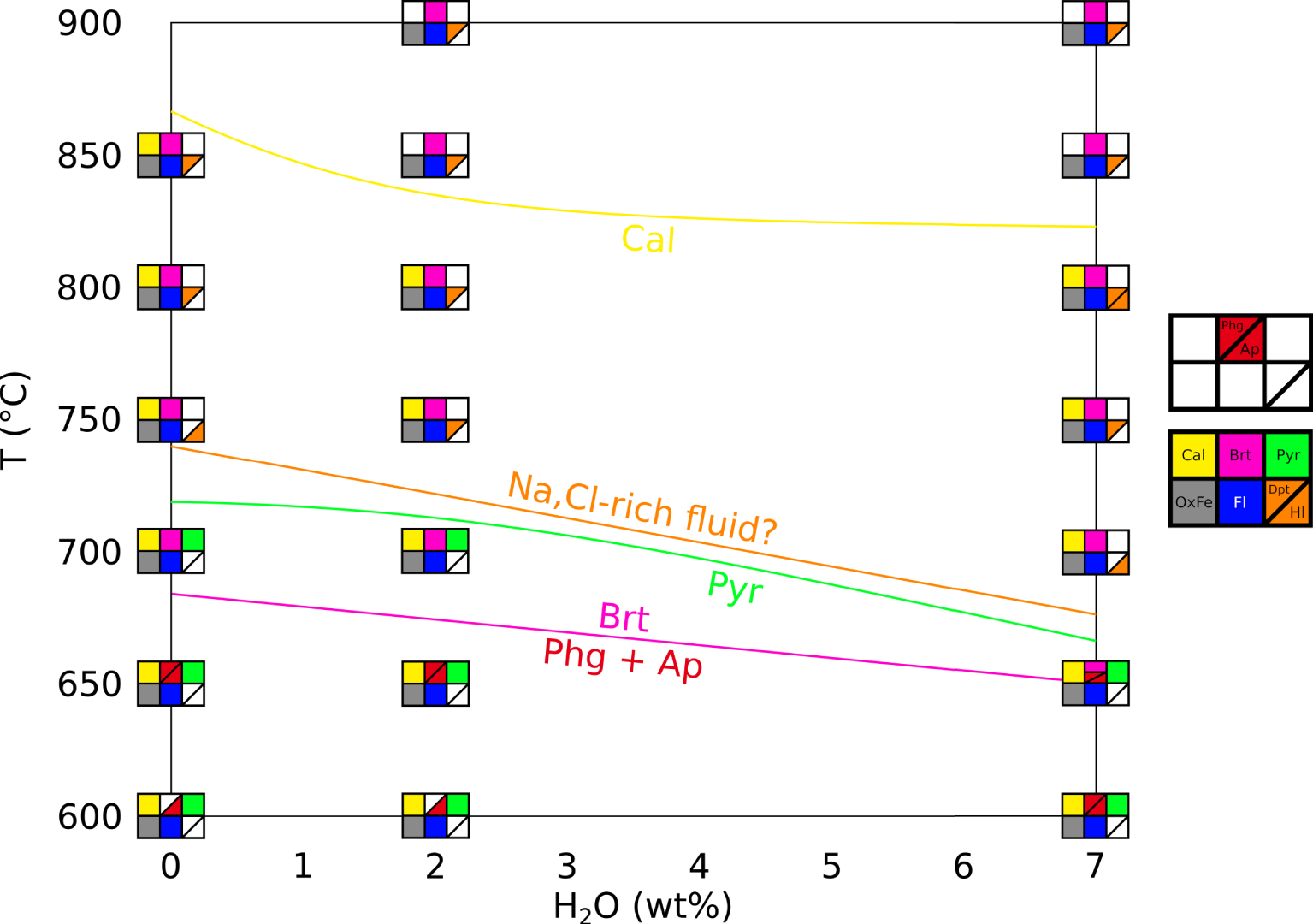
Phase diagram of the REE-rich carbonatite, showing temperature and water effects. Calcite (yellow) starts crystallising at lower temperatures in presence of water. Fluid phase (blue) and iron oxides (grey) are always observed. Britholite (pink) crystallises at high temperature and is replaced by phlogopite (rare, presumed in EXP-600G3 and EXP-600P) and fluorapatite at lower temperature (red). Pyrochlore (green) is a rare phase at low temperature. We may draw a stability domain for a Na,Cl-rich fluid (orange) at high temperature and high water content. Cal: calcite. OxFe: iron oxide. Fl: vesicle (fluid indicator). Dpt: deposit-like vesicle filling. Hl: halite. Brt: britholite. Phg: phlogopite. Ap: fluorapatite. Pyr: pyrochlore. Masquer
Phase diagram of the REE-rich carbonatite, showing temperature and water effects. Calcite (yellow) starts crystallising at lower temperatures in presence of water. Fluid phase (blue) and iron oxides (grey) are always observed. Britholite (pink) crystallises at high temperature and is ... Lire la suite
Our results suggest that britholite is stable at high temperatures, concentrating REE, Ca, Si, P and F, whereas phlogopite and REE-rich fluorapatite are stable at low temperatures, the former concentrating Si and the latter concentrating REE, Ca, P and F. Pyrochlore crystallises at low temperatures, concentrating Nb and REE. By using the Na-rich vesicle filling and the halite in fluid vesicles as markers for a Na,Cl-rich fluid, we may define a stability domain for this fluid at high temperatures and for hydrous conditions (Figure 2).
3.2. Carbonate melt and calcite composition
The composition of carbonate melts and calcite across all experiments is detailed in the Supplementary material. Carbonate melt from samples synthesised at 600 °C in crystallisation experiments have not been analysed, as melt areas were too small.
Below 850 °C, the CaO content of the carbonate melt decreases with temperature as calcite crystallises, from 36.60 ± 1.12 wt% at 900 °C to 20.92 ± 1.43 at 650 °C for EXP-P. The CaO content is ∼5 wt% lower in EXP-G3 than in EXP-W5 (Figure 3A). Calcite crystallisation is the main cause for CaO depletion and chemical evolution in the melt. Therefore, we can use the CaO content of the melt as a simple “differentiation index”: with CaO depletion in the melt, the differentiation increases. This qualitative index allows to compare all types of samples into one trend.
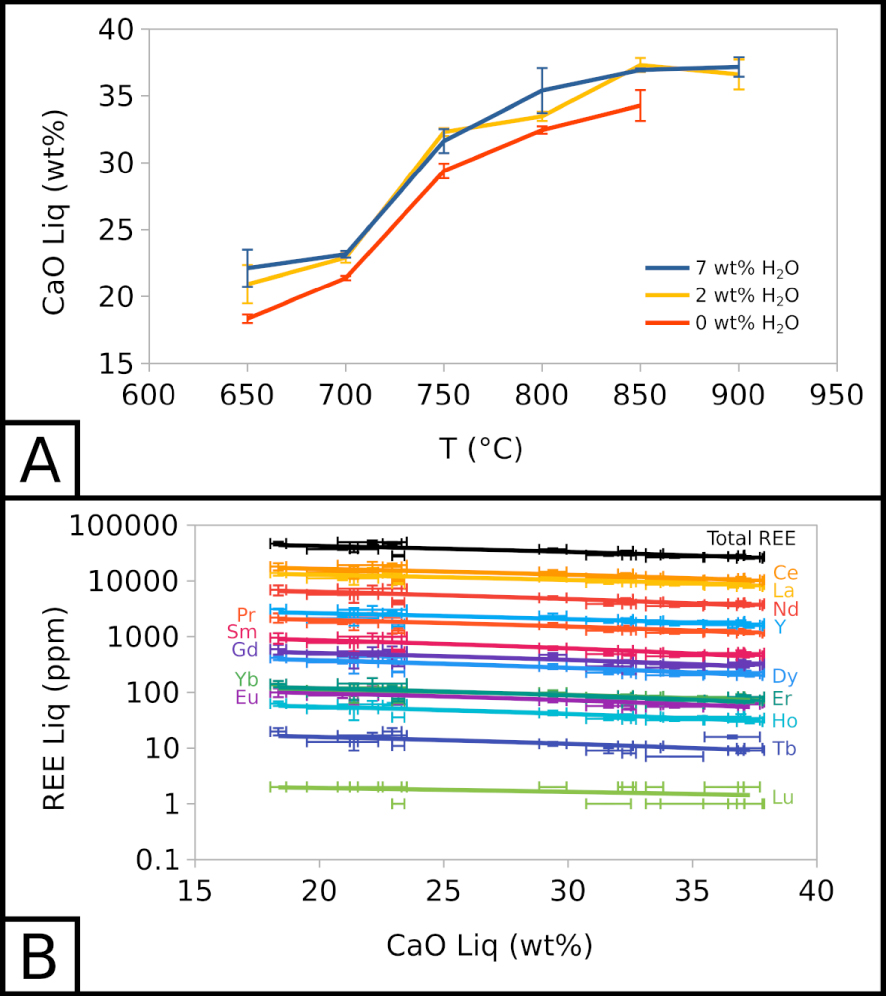
Compositional evolution of carbonate melt. (A) CaO in the carbonate melt as a function of temperature and water content. (B) REE in the carbonate melt as a function of CaO in the carbonate melt. 7 wt% H2O corresponds to EXP-W5, 2 wt% H2O to EXP-P, and 0 wt% H2O to EXP-G3.
Along this index, with a CaO decrease from ∼37.5 to ∼17.5 wt% in the melt, alkali (Na2O + K2O) increases from ∼13.5 to ∼27.9 wt% and halogens (Cl + F) from ∼6.1 to ∼9.1 wt%. The REE total also increases slightly in the melt, from ∼26,400 to ∼45,000 ppm (Figure 3B). REE enrichment trends are almost parallel in this semi-log space: all REE show a similar behaviour, and REE proportions stay globally the same. Water does not seem to influence the REE content of the melt.
REE content in calcite does not change with temperature, but evolves with the presence of water in the system. Ce content is similar in EXP-P and EXP-G3 (∼3,000 ppm), and is lower in EXP-W5 (∼2,000 ppm). Only EXP-850G3 yields higher Ce content (∼4,000 ppm).
3.3. Partition coefficients
Partition coefficients for carbonate melt and calcite (DLiq∕Cal) are detailed in the Supplementary material. REE are slightly incompatible with calcite compared to the carbonate melt: for all REE, the partition coefficients are comprised between 1 and 11. Light REE (from La to Sm) have slightly higher partition coefficients than the other REE. From Eu to Yb, partition coefficients display nearly flat patterns, with the exception of a small positive anomaly for Y (Figure 4). Lu concentrations are too low in carbonate melt (1 to 2 ppm, Figure 3B) and calcite (below detection limit to 1 ppm) to determine reliable partition coefficients.
All REE partition coefficients increase as the temperature decreases (Figure 4A), showing that REE are less favourably incorporated in calcite as the temperature decreases: DLiq∕Cal values increase by 3 to 5 with a 200 °C decrease. REE partition coefficients also seem to increase with the water content of the system (Figure 4B). At 800 °C, EXP-800G3 and EXP-800P show almost identical partition coefficients, whereas EXP-800W5 shows slightly higher values.
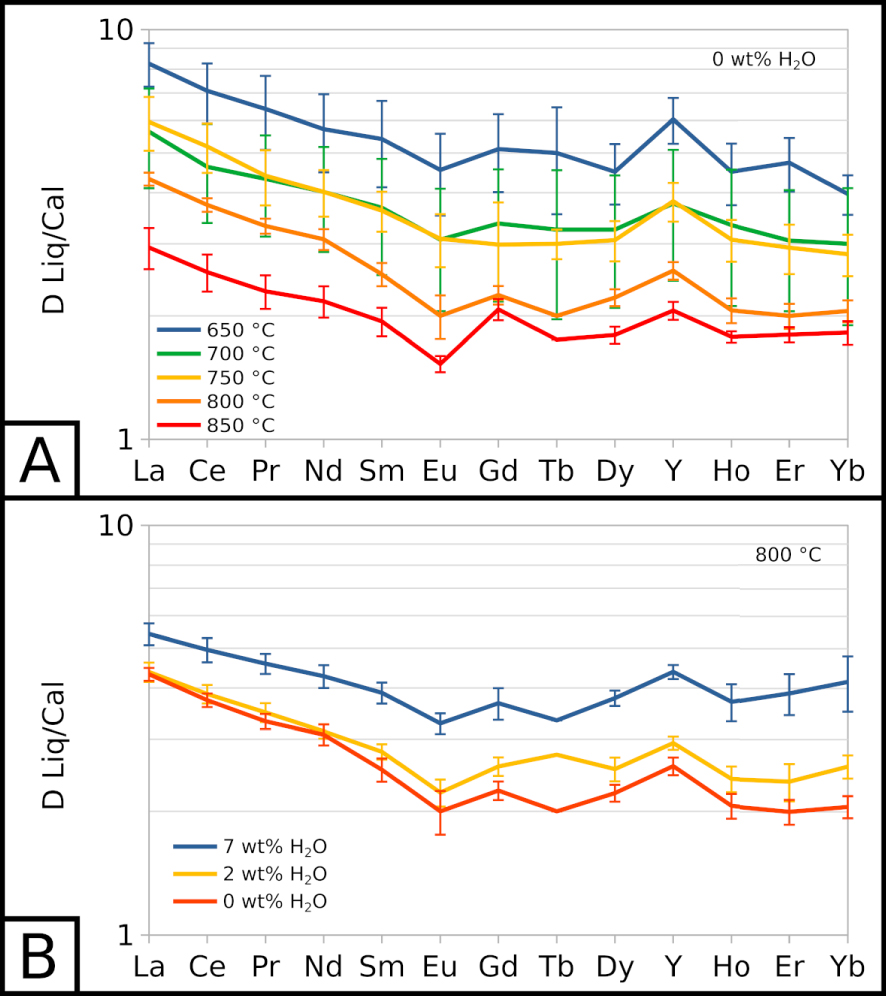
Partition coefficients for REE between carbonate melt and calcite (
3.4. Composition of the vesicle filling
As suggested by textural evidences, the vesicle filling is likely to originate from the fluid phase, thus providing an indirect evidence of the fluid composition (Figure 1C). Vesicle filling composition is detailed in the Supplementary material.
The mean alkali content of the vesicle filling is constant with temperature, but increases with the water content in the sample, evolving from 21.30 ± 1.56 wt% (EXP-W5) to 23.51 ± 1.70 wt% (EXP-P) to 27.18 ± 0.21 wt% (EXP-G3). However, EXP-850W5 (Table 2) is significantly different from other experiments, which may be due to off-target analysis.
Cl content in the vesicle filling seems unaffected by the water content in the system but is highly affected by temperature. Across all experiments, at 900 °C, Cl represents ∼2 wt% of the vesicle filling. This value decreases to ∼0.5–1 wt% at 850 and 800 °C, and increases to ∼3–3.5 wt% at 750 °C (Figure 5A). The vesicle filling is also F-rich, with concentrations between ∼8 to ∼14 wt% across all experiments, without a clear influence of temperature or water content in the system. As Cl and F, respectively, barely exceed 1 wt% and 8 wt% in crystallisation experiments, the vesicle filling is especially halogen-rich in comparison with the melt.
In all crystallisation experiments, we detected ∼10,000 to ∼20,000 ppm of Ce in the vesicle filling, without a clear influence of temperature and water content in the system (Figure 5B). This range of values is similar to the range of Ce concentrations in the carbonate melt.
Therefore, the vesicle filling hints at the existence of an alkali-, halogen-, REE-rich fluid phase. The effects of water and temperature on the composition of such a vesicle filling may reflect their influence on the fluid composition.
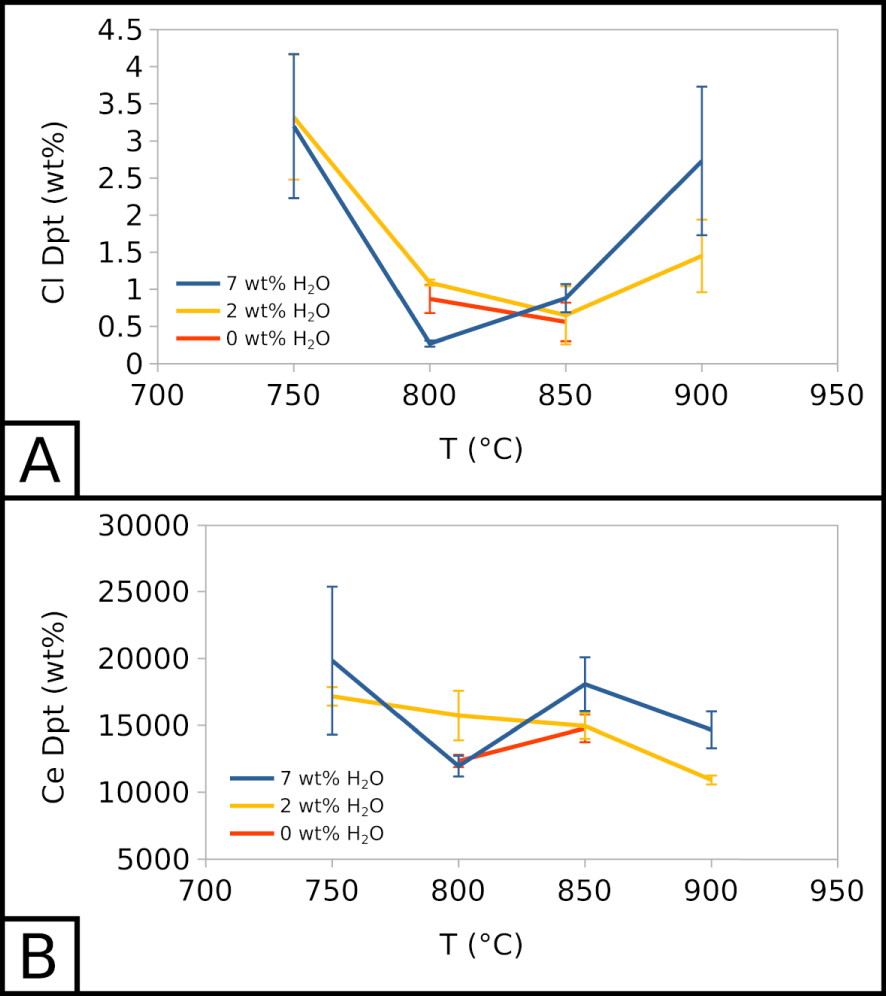
Compositional evolution of vesicle fillings. (A) Cl in the vesicle filling as a function of temperature and water content. (B) Ce in the vesicle filling as a function of temperature and water content. 7 wt% H2O corresponds to EXP-W5, 2 wt% H2O to EXP-P, and 0 wt% H2O to EXP-G3.
3.5. Bastnaesite saturation experiment
As no bastnaesite has been found in our crystallisation experiments, bastnaesite saturation has been forced in a dedicated saturation experiment at 600 °C with 20 wt% of bastnaesite added to the starting mixture (Figure 1D, Table 2). This EXP-600B experiment is also saturated with numerous small and clustered britholite crystals, fergusonite, calcite, fluid vesicles, vesicle fillings and iron oxides. Carbonate melt zones are larger in comparison with crystallisation experiments at the same temperature. The carbonate melt is extremely REE-rich, with the REE total representing 20.21 ± 1.87 wt% of the melt.
4. Discussion
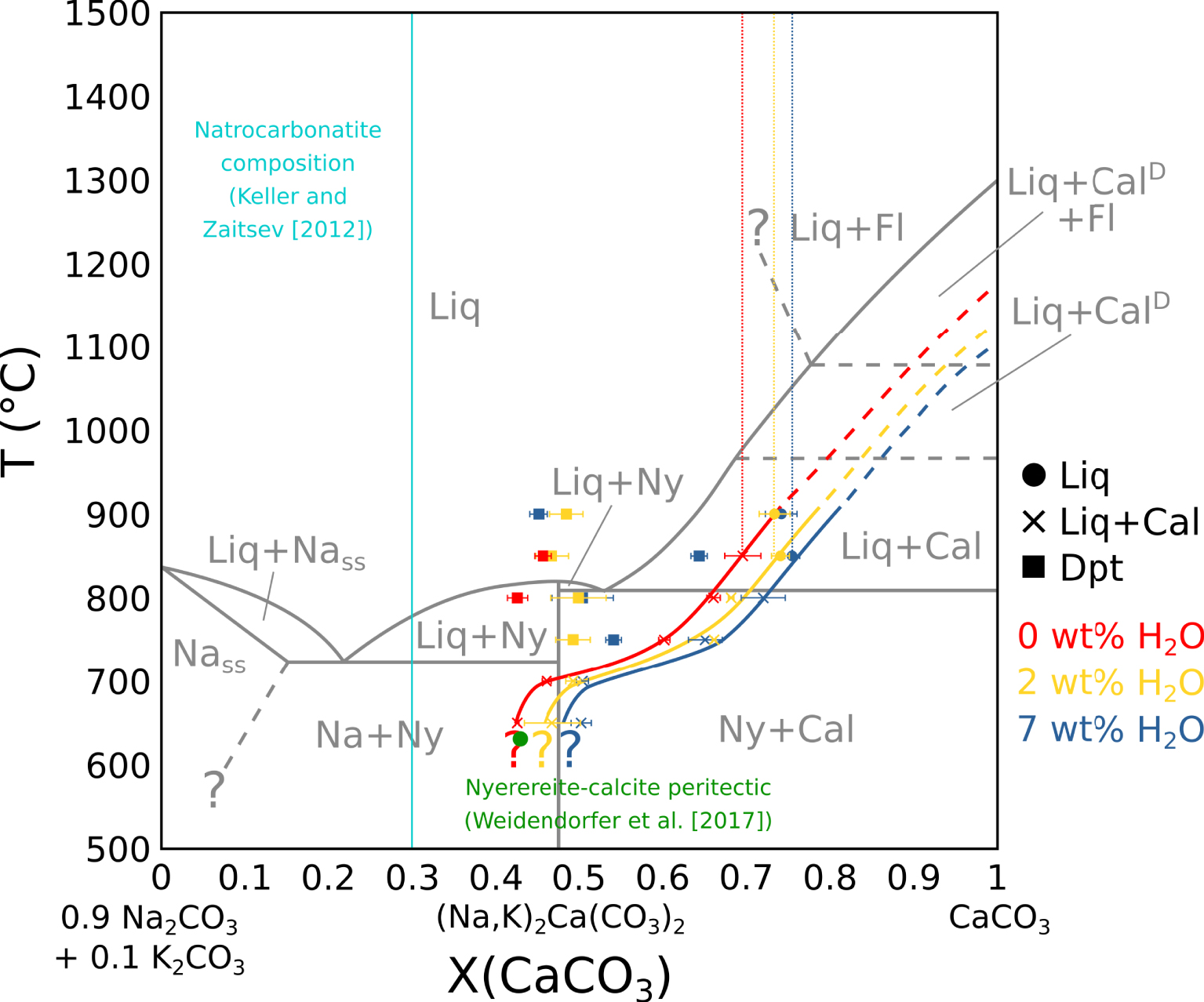
Pseudobinary phase diagram in the (0.9 Na2CO3 + 0.1 K2CO3)–CaCO3 system. In grey: Cooper et al. [1975] phase diagram, in a simple system. In red: carbonate melt and carbonate vesicle fillings composition in near-anhydrous EXP-G3. In yellow: carbonate melt and carbonate vesicle filling composition in 2 wt% H2O EXP-P. In blue: carbonate melt and carbonate vesicle fillings composition in 7 wt% H2O EXP-W5. Vertical dotted lines show the carbonate melt composition before calcite saturation. Plain lines show the carbonate melt evolution with temperature at calcite saturation. The curves are extended at high temperatures (dotted). The green dot indicates the calcite–nyerereite peritectic from Weidendorfer et al. [2017]. The sky blue line show the Ol Doinyo Lengai natrocarbonatite composition from Keller and Zaitsev [2012]. Liq: carbonate melt. Cal: calcite. CalD: disordered calcite. Fl: fluid. Dpt: deposit-like vesicle filling. Ny: nyerereite. Na: alkaline carbonate. Nass: alkaline carbonate solid solution. Na and Nass are equivalent to gregoryite in nature. Masquer
Pseudobinary phase diagram in the (0.9 Na2CO3 + 0.1 K2CO3)–CaCO3 system. In grey: Cooper et al. [1975] phase diagram, in a simple system. In red: carbonate melt and carbonate vesicle fillings composition in near-anhydrous EXP-G3. In yellow: carbonate melt and ... Lire la suite
4.1. The genetic link between calciocarbonatites and natrocarbonatites
Residual melt compositions have been compared to the literature in the CaCO3–Na2CO3–K2CO3 system. In all experiments, the (Na2CO3)∕(Na2CO3 + K2CO3) ratio in the melt is constant with a ∼0.9 value. Therefore, the pseudobinary system CaCO3–(0.9 Na2CO3 + 0.1 K2CO3) is used in this study for data projection. As the vesicle filling also yield a ∼0.9 (Na2CO3)∕(Na2CO3 + K2CO3) ratio, its composition can also be projected on this pseudobinary.
By projecting melt compositions into this pseudobinary, we can define the calcite saturation curves for each experimental set. Along these curves, calcite crystallisation effectively drives the carbonatite composition from the calcic pole towards the alkaline pole (Figure 6), as observed by Cooper et al. [1975]. The natrocarbonatite composition [sky blue line, Keller and Zaitsev 2012], however, is never reached in our experiments. As there is still residual melt at 600 °C, the solidus was not reached in our experiments.
Compared to the Cooper et al. [1975] data (grey lines, Figure 6), the calcite saturation curve is lowered by ∼125 °C for near-anhydrous EXP-G3 (red line) and ∼200 °C for 7 wt% H2O EXP-W5 (blue line). Therefore, according to Wyllie and Tuttle [1960], water lowers the curve, but another process is needed to explain the lowering in near-anhydrous EXP-G3. In their study, by adding 3.75 wt% F to the system, Jago and Gittins [1991] observed a ∼100 °C lowering of this curve compared to Cooper et al. [1975]. Because our starting mixture is halogen-rich (4.43 wt% F + 1.44 wt% Cl) (Table 1), halogens could have also strongly impacted the calcite saturation curve in our experiments.
The nyerereite saturation thermal maximum described by Cooper et al. [1975] in a simple CaCO3–Na2CO3–K2CO3 system is not observed in our system. Jago and Gittins [1991] suggest that in a halogen-rich complex system, this thermal maximum does not exist due to the lowering of the calcite saturation curve. Indeed, Weidendorfer et al. [2019] also show the absence of this thermal maximum when saturating a halogen-rich natrocarbonatitic composition with calcite. They also show the transformation of the nyerereite–calcite eutectic into a peritectic (green dot, Figure 6), allowing a genetic link between calciocarbonatitic and natrocarbonatitic melts. However, in our study, even though we reach the position of this peritectic around 650 °C (Figure 6), the peritectic itself seems not to be crossed, as nyerereite is still not observed at 600 °C (Table 2).
Furthermore, according to Keller and Zaitsev [2012], natrocarbonatitic melts are Fe-, Mg- and REE-poor (respectively 0.45 ± 0.19, 0.44 ± 0.07 and 0.14 ± 0.02 wt%). Our results show that calciocarbonatitic melts are likely to be enriched in these elements during differentiation towards alkali-rich compositions: with a CaO decrease from ∼37.5 to ∼17.5 wt% in the melt, the Fe, Mg and REE contents respectively increase from ∼0.7 to ∼2.4 wt%, ∼1.7 to ∼7.5 wt%, and ∼26,400 to ∼45,000 ppm (Figure 3B). Therefore, given the issues highlighted in this section, we argue that calciocarbonatitic melt differentiation cannot be responsible, at least alone, for natrocarbonatitic genesis.
The vesicle filling is significantly more alkaline than the carbonate melt. Its composition is constant with temperature and water content in the system between a ∼0.45 to ∼0.55 (CaCO3)∕(CaCO3 + Na2CO3 + K2CO3) ratio, with the exception of the vesicle filling from EXP-850W5 at a ∼0.64 ratio. This may be due to off-target analysis. At temperatures below the point where the calcite saturation curve crosses the range of vesicle filling compositions, the vesicle filling disappears (Figure 6). Guzmics et al. [2019] showed the existence of alkali-rich, carbonate condensates inside fluid vesicles at equilibrium with a calciocarbonatitic melt and a silicic melt. They suggested the remixing of a calciocarbonatitic melt with an alkali-rich fluid as a process to explain natrocarbonatitic melt formation. The vesicle filling in our experiments could be similar to such products. Given its position in the Cooper et al. [1975] and Weidendorfer et al. [2019] phase diagrams (Figure 6), it could be parental to natrocarbonatitic melts.
4.2. REE incompatibility and melt enrichment
REE (especially LREE) are shown to be increasingly incompatible in calcite with decreasing temperature (
4.3. Exsolution of a Na- and Cl-rich, REE-bearing fluid
There are several hints in favour of the exsolution of a Na-, Cl-rich fluid phase in our experiments. Given the composition of the vesicle filling, such fluids are also probably CO
The presence of halite plus alkali- and halogen-rich vesicle fillings in fluid vesicles is qualitative evidence for the presence of Na and Cl in the fluid phase. The (CaCO3)∕(CaCO3 + Na2CO3 + K2CO3) ratio in the carbonate melt is slightly shifted from ∼0.7 (EXP-G3) to ∼0.73 (EXP-P) to ∼0.75 (EXP-W5) (dotted red, yellow and blue vertical lines, Figure 6), i.e., from near-anhydrous to highly hydrated experiments. As no high-temperature minerals allow alkali in their structure, this X(CaCO3) shift in hydrated experiments suggests that the formation of a water-dominated fluid withdrew some Na from the melt. When compared with the starting mixture, the carbonate melt is depleted in Cl, especially at high temperature. Since no mineral phase allows Cl in its structure, it is likely that Cl has been partially withdrawn from the melt by the fluid phase.
Williams-Jones et al. [2012] studied the solubility of REE in a halogen-bearing aqueous fluid. They showed that REE mainly exist in such fluids as REE–Cl complexes, making a Na-, Cl-rich fluid a good candidate for mobilising REE. According to them, if a high-temperature (∼400 °C), acidic fluid cools down (∼200 °C) and is being neutralised in pH, the REE solubility drops by several log units. Therefore, we may argue that the fluid phase can play a major role in the formation of carbonatitic REE ore deposits. Indeed, halogen-rich fluids will probably also be REE-rich, as the carbonate melt is increasingly REE-rich with calcite crystallisation. During fluid transport, cooling and neutralisation at the contact with surrounding rocks can then hamper REE mobilisation and trigger REE-bearing mineral crystallisation.
4.4. Bastnaesite: magmatic or hydrothermal crystallisation?
Magmatic crystallisation of bastnaesite in nature probably does not occur. Indeed, according to our results, for magmatic bastnaesite saturation, extremely high values of REE concentrations (∼20 wt% total REE) are needed at 600 °C. Such a value is unrealistic in nature: one of the most REE-rich carbonatitic ore deposits in the world, Mountain Pass, locally shows only up to 14 wt% REO (rare earth oxides) concentrations [Castor 2008]. Moreover, no bastnaesite has been found in crystallisation experiments, despite the melt being REE-rich (up to ∼5 wt% total REE). Fluid-related processes are probably needed to achieve bastnaesite crystallisation.
The following sequence can be proposed:
- (1) REE are mainly concentrated in a calciocarbonatitic melt during immiscibility with an alkaline silicate melt;
- (2) Differentiation slightly concentrates REE in the residual melt by calcite crystallisation;
- (3) REE are also concentrated in a hydrous, Na-, Cl-bearing fluid phase along the differentiation;
- (4) During the post-magmatic stage, the hydrothermal fluid cools down and is neutralised in pH, whereby the REE solubility drops [Williams-Jones et al. 2012];
- (5) REE minerals like bastnaesite can then crystallise from the fluid phase.
Halogens and water enhance this process by lowering the calcite crystallisation curve, allowing REE enrichment in the melt and the fluid at lower temperature, and increased REE mobilisation by the fluid.
4.5. REE-bearing minerals at magmatic conditions
We stress the existence of accessory REE-bearing magmatic minerals in our experiments. In particular, magmatic britholite (a REE, P-bearing silicate isomorphic with apatite) crystallises at high temperature (700–900 °C), even in Si-poor carbonate melts. Britholite crystals concentrate into calcite–britholite–iron oxides cumulates in melt-rich, high-temperature experiments (⩾750 °C). To date, britholite has only been described as a low-temperature, hydrothermal mineral in the literature [Verplanck et al. 2016; Giebel et al. 2017]. At low temperature (600–650 °C), REE-bearing apatite is stable instead of britholite. Pyrochlore, a Nb,REE-bearing oxide, crystallises in small amounts at low temperature (600–700 °C).
5. Conclusion
This study provides new insights about carbonatite genesis and REE behaviour inside a complex, natural-like CaCO3–Na2CO3–K2CO3 system. Experiments on Ca, REE-rich carbonatite differentiation show that REE-bearing calcite is the main crystallising phase, forming at low temperature in the presence of halogens and water. Its crystallisation controls the residual melt composition, which will evolve towards a more alkaline composition with decreasing temperature. However, neither the natrocarbonatitic composition [Keller and Zaitsev 2012] nor the nyerereite–calcite peritectic described by Weidendorfer et al. [2017] were reached in our experiments. Thus, a process other than carbonatite differentiation alone is needed to explain natrocarbonatite genesis. The answer may reside in the co-differentiation of a carbonate melt in presence of an immiscible silicate melt, or in fluid–melt interactions resulting in the formation of alkali-rich carbonate products, that could be parental or contribute to natrocarbonatites, as described in Guzmics et al. [2019].
REE are slightly incompatible in calcite with respect to the carbonate melt, especially at low temperature and with water in the system. This results in a minor REE enrichment in the carbonate melt with calcite crystallisation. However, such enrichment, even in a REE-rich system, does not permit the crystallisation of magmatic bastnaesite, as bastnaesite is never observed in our crystallisation experiments. Indeed, REE are highly soluble in carbonate melts, with around 20 wt% REE in the melt at bastnaesite saturation in our saturation experiment at 600 °C. Such concentrations are unlikely to be reached in nature.
Several hints for a Na-, Cl- and REE-rich fluid phase are found in our experiments. As bastnaesite crystallisation is unlikely to be magmatic, it is likely to be due to hydrothermal fluids at a post-magmatic stage, as Cl-rich fluids are reasonable candidates for REE mobilisation [Williams-Jones et al. 2012]. Thus, this experimental study highlights that hydrothermal fluids are probably the main actor for REE mineral crystallisation in carbonatitic REE ore deposits. Further investigations should focus on fluid composition and REE partitioning between carbonatites and fluids.
Conflicts of interest
The authors declare no competing financial interest.
Dedication
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
Acknowledgements
This work was supported by the LabEx VOLTAIRE project (ANR-10-LABX-100-01) and the EquipEx PLANEX (ANR-11-EQPX-0036). We thank colleagues for their advice and remarks throughout this work. We thank technicians and engineers at the ISTO, BRGM and LMV for their work and their help during the preparation and the analyses of the samples.