


 CC-BY 4.0
CC-BY 4.0
1. Introduction
Processes of generation, alteration, and recycling of the oceanic crust have a major role in the long-term C cycle on our planet since they control exchanges between mantle and surface C reservoirs [Bebout 1995, 2013; Staudigel 2007]. Specifically, alteration of the oceanic crust causes chemical and mineralogical transformations of the C-bearing matter that is ultimately recycled to the deep mantle in subduction zones. Isotopic compositions of volatile elements (C, H, O, etc.) record information about the mechanisms and conditions of these processes. Over the past decades, stable isotope systematics of these elements have been widely studied, providing better insight into physico-chemical processes associated with alteration of oceanic crust [e.g., Alt et al. 2003; Früh-Green et al. 2004; Delacour et al. 2008; Shilobreeva et al. 2011]. Total C in altered oceanic crust comprises inorganic (carbonate) and reduced (elemental and/or organic C) components, but the origin of these components is still not entirely clear. The reduced C component is significantly depleted in the heavy 13C isotope, with δ13C values ranging typically from −22 to −34‰. Some studies [e.g., Delacour et al. 2008; Alt et al. 2003; Lever et al. 2013] support a biogenic origin for reduced C, while others [Tingle et al. 1991; Shilobreeva et al. 2011] advocate an abiotic formation through reactions such as Fischer–Tropsch-type (FTT) synthesis. The inorganic C generally has δ13C values around 0‰ and is commonly considered as carbonate added to the rocks through seawater infiltration, but δ13C values ranging to negative values (to ∼−15‰) are taken to indicate incorporation of a component of organic carbon [Alt and Teagle 2003; Delacour et al. 2008; Coggon et al. 2004, 2010; Shilobreeva et al. 2011; Gillis and Coogan 2011; Lever et al. 2013; Coogan and Dosso 2015]. The isotopic composition of total C corresponds to the weighted sum of the 13C-depletedreduced C (biogenic or abiogenic) and the 13C-enriched carbonate C.
In the present work we analyze, both inorganic carbon (as carbonate) and reduced carbon in altered ocean crust from 170 Ma Ocean Drilling Program (ODP) Site 801C, the oldest section ever drilled that could be considered as a good representation of subducting lithosphere. Previous studies at this site analyzed only carbonate veins or bulk rock C contents [Alt and Teagle 1999, 2003]. We focus on the contents and isotope composition of carbon in the host altered basalts, complemented by a subset of the carbonate-veined portion of the basalts that provides a comparison with previous work. We developed an extended C extraction procedure for measuring the concentration and isotope composition of inorganic (carbonate) and reduced C fractions as well as of the total C. This is more comprehensive than that traditionally used where only the C isotope composition of total C, or total and reduced C are measured [e.g., Früh-Green et al. 2004; Delacour et al. 2008; Shilobreeva et al. 2011]. We first describe the contents and partitioning of reduced and inorganic C in altered oceanic crust. Then, based on C isotope compositions, we discuss the processes through which C is incorporated into altered oceanic crust. Finally, we consider the C budget in altered oceanic crust focusing on the significance of the reduced C component.
Map of the ODP Hole 801C location in the West Pacific (Izu and Mariana arcs). Modified after Alt and Teagle [2003]. Upper left corner displays the lithostratigraphy of oceanic crust at Hole 801C. Arrows mark the depths of the analyzed samples.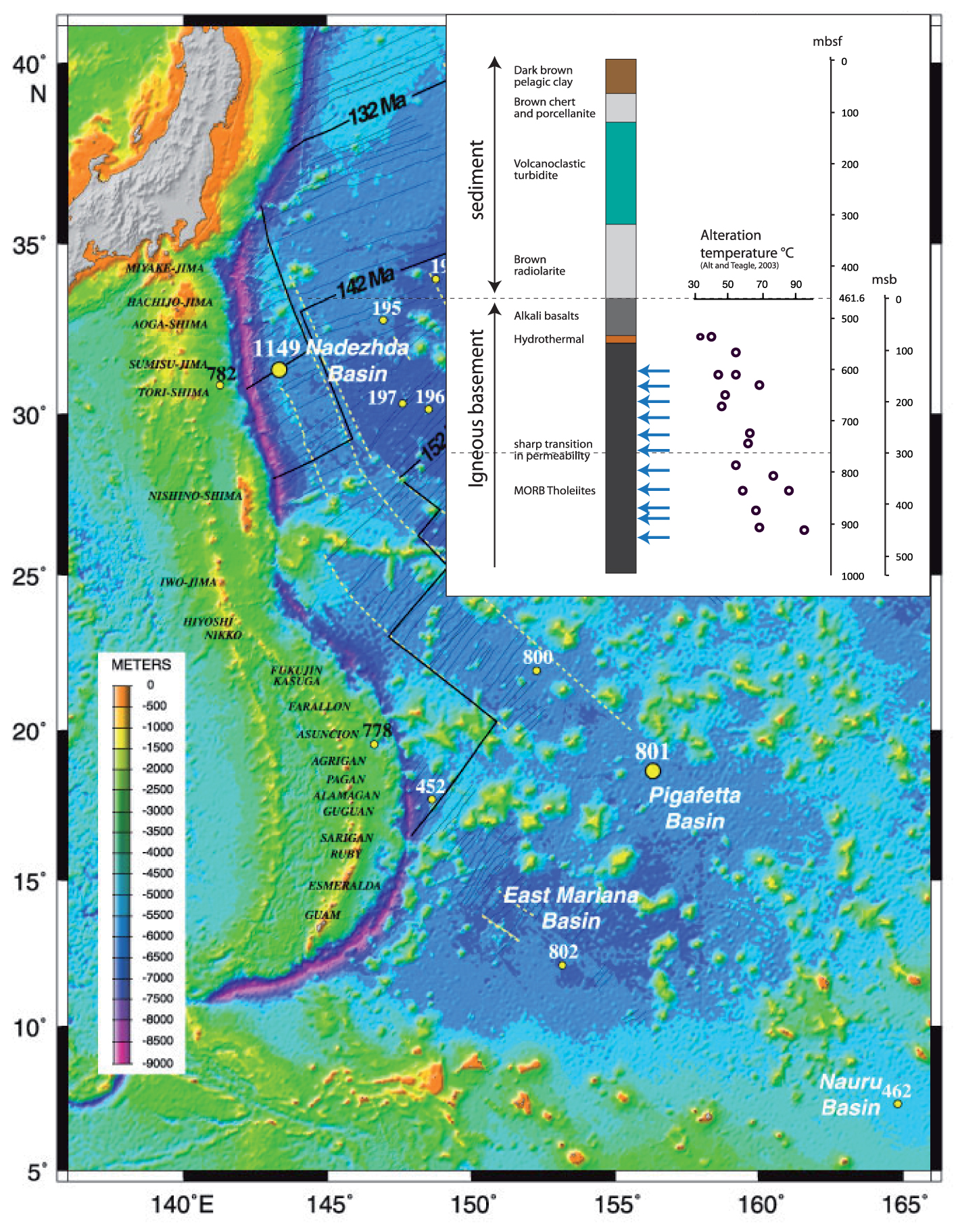
Sample descriptions
| Sample | % Alt | Olivine, Vesicles, Interstitial | Plagioclase | Veins | Alteration Halo |
|---|---|---|---|---|---|
| 14R1 108–112 | 5–10 | Replaced/filled by sap + cc | 2 mm sap + cc + chalc, 150 μm sap + py; 40 μm cc | ||
| 17R2 27–32 | 5–10 | Replaced/filled by sap + cc + minor py | Slightly alt to sap + cc | 2 cm cc with multiple generations | 5 mm green halo; celad + cc replace olivine, fill vesicles ; plag partly altered to cc |
| 20R2 62–65 | 10 | Replaced/filled by sap + cc | 2 mm sap + cc | 2 mm dark halo ; celad + Fe-ox replace olivine and fill vesicles | |
| 24R3 18–22 | 10 | Replaced/filled by sap + cc | 1.5 mm sap + cc + minor py | ||
| 28R1 2–6 | 10 | Replaced/filled by sap + cc | Several 100 μm–2 mm cc ± sap | ||
| 31R1 37–40 | 10 | Replaced/filled by sap | Slightly alt to sap | 5 mm celad + Fe-ox | 5 mm light halo; celad + Fe-ox + sap replace olivine and interstitial |
| 35R1 99–103 | 15 | Replaced/filled by sap + trace celad + Fe-ox + cc | Slightly alt to sap | Three 2–4 mm sap + cc + chalc | |
| 39R2 72–76 | 5–10 | Replaced/filled by sap | 1 cm cc + minor chalc + qz + sap | 4 mm dark halo; celad + Fe-ox + sap replace olivine and fill vesicles | |
| 43R2 121–124 | 15 | Three 0.5–1 mm celad + Fe-ox + sap | brown halo; celad + sap + trace Fe-ox replace olivine, interstitial, fill vesicles; plag slightly alt to sap | ||
| 45R2 100–103 | 5–10 | Replaced/filled by sap | Slightly alt to sap | 150 μm celad + Fe-ox + minor sap | |
| 49M1 34–36 | 5–10 | Replaced/filled by sap = minor py | 5 mm sap + cc + chalc | ||
Sap: saponite; cc: carbonate; chalc: calcedonite; qz: quartz; py: pyrite; celad: celadonite; Fe-ox: iron oxyhydroxide; plag: plagioclase; alt: altered.
2. Geological setting and sampling
2.1. Geological setting and lithostratigraphy of Hole 801C
The ODP Site 801C is located in the ∼170 Ma-old oceanic crust of the western Pacific Ocean, at a water depth of 5674 m (Figure 1). The crust at this site formed at a fast-spreading ridge having an estimated full spreading rate of 160 km/Ma, and the lithostratigraphy has been described in detail elsewhere [Lancelot et al. 1990; Pringle 1992; Plank et al. 2000; Alt and Teagle 2003]. Hole 801C is penetrated through 461.6 m of sediments and 474 m of igneous basements. The basement section has been divided into eight major units: Unit I consists of 60.2 m of alkali basalt sills that intruded sediments at 157 Ma; Units II and V are 10–20 mm and 1-mm-thick hydrothermal silica–iron deposits at the top of the tholeiite section and at 150 msb (meters sub-basement), respectively; the remaining Units III, IV, VI, and VIII comprise of 400 m tholeiitic basalts including massive, sheet, and pillow flows, with a 10 m thick breccia Unit VII at ∼360 msb. The volcanic basement is similar to other oceanic sites, with the exception of the off-axis alkalic sills and the two hydrothermal silica–iron deposits and associated alteration that formed from low-temperature hydrothermal fluids at the spreading axis [Alt and Teagle 2003; Alt 2003]. Basement porosity averages 11 ± 4% from 0 to 300 msb but is lower (7 ± 1%) below 300 msb [Jarrard et al. 2003]. We refer to these zones as the upper volcanic section (UVS) and lower volcanic section (LVS).
2.2. Petrography of the samples
We studied 11 samples of tholeiitic basalt from 143 msb down to the bottom of the Hole at 467 msb (605–928 m below seafloor). The mineralogy and chemistry of basement alteration in Hole 801C has been documented by Alt and Teagle [2003], and descriptions of samples analyzed in this study are given in Table 1. In general, the basalt samples are slightly recrystallized (5–15%), with saponite ± carbonate ± minor pyrite replacing olivine and interstitial material and filling vesicles. Five samples also exhibit 2–7 mm wide alteration halos that are 5–10% altered into celadonite + iron oxyhydroxide ± saponite, and one sample (43R2 121–124) consists entirely of such alteration halos. All samples contain veins, which range from 40 μm to 2 cm wide, and can be divided into two types: (1) carbonate ± saponite ± chalcedony ± minor pyrite, and (2) celadonite + iron oxyhydroxide ± saponite.
Using the same approach as our previous work on Hole 1256D, we selected zones away from macroscopic veins and associated visible alteration halos in order to characterize alteration of the host basalt “background” alteration. We first removed visible secondary veins by sawing. Thus, we have two sample types: the background basalt samples and a subset of veins and associated alteration halos separated from the basalts. Samples were then crushed and sieved to a grain size fraction smaller than 150 μm in an agate mortar.
3. Analytical method
At IPGP, we used three different procedures to measure C concentration and isotopic composition: (1) online extraction of all volatiles (C, H) by step heating; (2) HCl attack of powders to remove carbonate with subsequent online extraction of volatiles for analyses of the reduced C component; and (3) H3PO4 attack for carbonate analysis. These three techniques are described below.
3.1. Step-heating extraction of total C and H
Step-heating experiments were used for the extraction of total C and H, as in previous work on Hole 1256D [Shilobreeva et al. 2011]. Approximately 100 mg of sample powder were loaded in a low C and H, blank fused quartz tube, which was connected to a vacuum line. After one-night degassing at room temperature, the samples were then heated at 100 °C under vacuum for 1 h to remove surface contaminants. Volatiles were then extracted during a one-step combustion by heating the samples at 1100 °C for 1 h, with an oxygen gas pressure of 4 mbar (set with hot CuO) because we have shown that most of the C release occurs during this step [Shilobreeva et al. 2011]. During this combustion, all C species were transformed into CO2 that was quantified using a Toepler pump, with a precision better than 5% using a procedure described by Javoy and Pineau [1991]. Carbon concentrations are thus given in wt% of CO2. Hydrogen, occurring as OH or H2O bound in rock minerals, was extracted as H2O and converted into H2 over hot uranium for subsequent quantification using a Toepler pump with a calibrated volume.
3.2. Step-heating extraction of reduced C
In order to determine the reduced C content in each sample, about 1 g of powder was loaded in a Falcon 50 mL tube for reaction with ultrapure 6N HCl acid for 12 h at room temperature [Thomazo et al. 2009]. This step removes carbonates from samples that contribute to the production of CO2 during the online combustion. After the HCl reaction, samples were rinsed several times (n ∼ 5 or 6) with ultrapure deionized CO2-free water till the solution reached a pH value close to 6. After removing the water, the solid residue was stored in a dry oven at 50 °C for 1 week. Finally, online combustion of approximately 100 mg of powder was performed as described in Section 3.1. Analytical blank, determined for this protocol, is 1.7 ± 0.2 μmol (i.e. 0.07 ± 0.01 wt% CO2) with δ13C ≈−26.0 ± 5.3‰. C contents and isotope compositions were corrected in order to remove this blank contribution. The corrections are smaller than 30% for C content and uncertainties are better than ±2‰ for δ13C value.
Depth profiles of total C, inorganic C, and reduced C concentrations in the altered basalts from Hole 801C, as well as inorganic C from veins. At 300 msb (meters sub-basement) lies the permeability transition that separates the upper volcanic section (UVS) and the lower volcanic section (LVS). Alteration temperatures reported along the depth profile are from Alt and Teagle [2003]. They are the temperature where seawater circulates the most but hydrothermal fluids, which certainly also contribute to alteration at higher temperatures.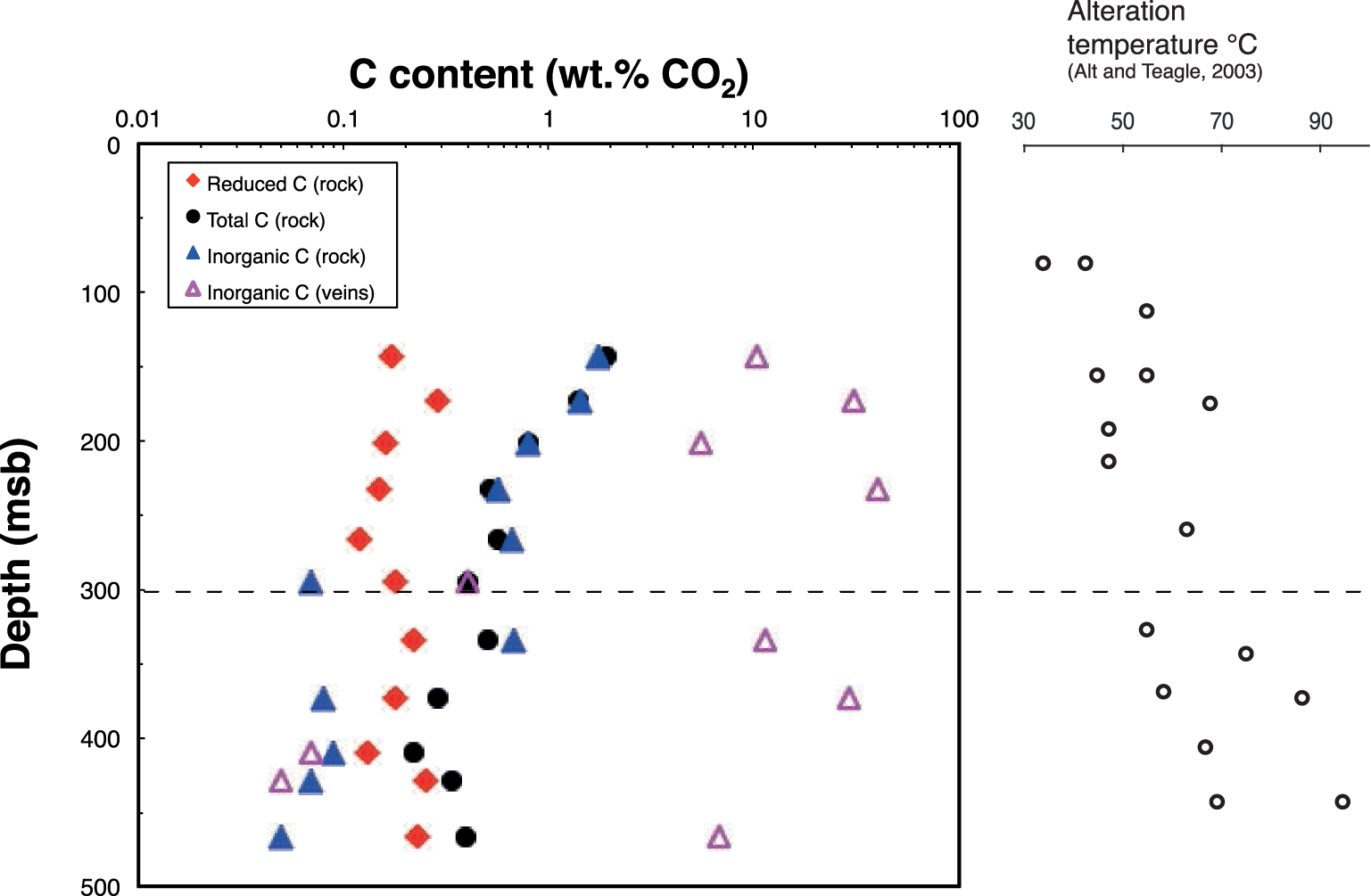
3.3. Extraction of the carbonate (inorganic) fraction
The concentration and isotope composition of carbonates (CaCO3 and (Ca,Mg,Fe)CO3) were measured using gas chromatography coupled to a He continuous flow isotope ratio mass spectrometer (GC-IRMS, AP2003). The GC column (stainless column 60_1/800_2 mm, packed with HayeSep Q 60/80 mesh, Chrompack) separates the CO2 gas from other residual gas components (e.g., N2, O2, NO2, Ar) [Assayag et al. 2006]. Depending on the sample, between 5 and 500 mg of powder were loaded into exetainer® vials. After flushing with ultrapure He gas to eliminate atmospheric gases, orthophosphoric acid (H3PO4) was introduced in the vial to transform carbonates into gaseous CO2. After 4 h of reaction at ambient temperature (≈22 ± 3 °C), only CaCO3 is expected to be transformed into CO2 [McCrea 1950]. The He–CO2 gas mixture was thus transferred into a mass spectrometer for analysis of the CaCO3 component (CO2 amount and δ13C). The vial was flushed again with pure helium gas and heated at 80 °C for 2 h to release CO2 from other carbonate species ((Ca,Mg,Fe)CO3) for analysis of C and O isotope composition [Lebeau et al. 2014]. Internal standards of pure calcite and dolomite were run with the samples for calibration of both concentration and isotopic composition. The isotopic ratio 13C/12C is reported using the conventional δ-notation versus PDB international standard, whereas the SMOW international standard is used for δ18O. The external reproducibility was ±0.1‰ for δ13C, ±0.5‰ for δ18O and ±10% (relative) for the carbonate fraction content.
3.4. C and H isotope analyses
The isotope compositions of CO2 and H2 collected from step-heating experiments were measured on a Thermofisher Delta +XP dual inlet mass spectrometer. For H2, the interference factor was measured before any hydrogen isotope measurement. The isotope compositions are reported versus PDB for δ13C and SMOW for δD. The external repeatability of H isotopic measurement is about ±2‰ (2σ). Repeated measurements of the international graphite standard USGS24 gave δ13C values of −15.96 ± 0.06‰ (2σ; n = 10). This value is indistinguishable from the recommended value [−15.99 ± 0.11‰ 2σ; Stichler 1995]. Thus, the accuracy of the present procedure on δ13C measurements is better than ±0.20‰ (2σ).
Concentrations and isotope compositions of C and water extracted by one step-heating technique from background altered rock samples from Hole 801C
| Rock | Depth | Total C | Reduced C∗ | Total water | |||||
|---|---|---|---|---|---|---|---|---|---|
| (mbsf) | (msb) | Concentration (wt% CO2) | δ13C (‰ VPDB) | Concentration (wt% CO2) | δ13C (‰ VPDB) | Concentration (wt%) | δD (‰ VSMOW) | ||
| The upper volcanic section (UVS) | 801C-14R | 605.1 | 143.5 | 1.92 | −1.6 | 0.17 | — | 0.42 | −41.5 |
| 801C-17R2 | 634.5 | 173.0 | 1.38 | −2.8 | 0.29 | −26.3 | 1.15 | −35.5 | |
| 801C-20R2 | 662.7 | 201.1 | 0.80 | −2.9 | 0.16 | −27.8 | 0.89 | −52.2 | |
| 801C-24R3a | 694.3 | 232.7 | 0.52 | −5.4 | 0.15 | −26.0 | 0.47 | −59.8 | |
| 801C-28R1 | 728.7 | 267.1 | 0.57 | −3.6 | 0.12 | −25.4 | 0.77 | −17.6 | |
| 801C-31R1 | 757.0 | 295.4 | 0.40∗∗∗ | −19.1∗∗∗ | 0.18∗∗∗ | −22.6∗∗∗ | 0.89∗∗∗ | −59.3∗∗∗ | |
| Average: | 1.04 ± 0.59∗∗ | −2.7 ± 1.1 | 0.18 ± 0.07 | −25.7 ± 1.7 | 0.74 ± 0.30 | −39.7 ± 14.0 | |||
| The lower volcanic section (LVS) | 801C-35R1a | 795.9 | 334.3 | 0.50∗∗∗ | −4.7∗∗∗ | 0.22∗∗∗ | — | 0.67∗∗∗ | −61.1∗∗∗ |
| 801C-39R2 | 834.4 | 372.8 | 0.29 | −18.7 | 0.18 | −25.7 | 0.98 | −49.7 | |
| 801C-43R2 | 871.8 | 410.2 | 0.22 | −22.3 | 0.13 | −26.5 | 0.95 | −81.7 | |
| 801C-45R2 | 890.8 | 429.2 | 0.34 | −23.5 | 0.25 | −27.8 | 0.86 | −83.3 | |
| 801C-49M1 | 928.6 | 467.0 | 0.39 | −22.3 | 0.23 | −27.5 | 1.00 | −64.7 | |
| Average: | 0.31 ± 0.07 | −21.8 ± 1.8 | 0.20 ± 0.05 | −27.0 ± 0.9 | 0.95 ± 0.06 | −69.3 ± 13.7 | |||
∗After HCl acid attack removing the carbonate fraction.
∗∗All uncertainties correspond to the 1 σ interval.
∗∗∗Not used for the estimation of the average value (see text for details).
4. Results
4.1. C and water content
Total, reduced, inorganic carbon (all expressed as CO2 content) and water contents in background altered samples are reported in Tables 2, 3, and 5. Total C in the UVS decreases with depth, whereas no trends are observed with depth in the LVS (Table 2 and Figure 2). Although small variations exist, the reduced C content ranging from 0.12 to 0.29 wt% CO2 is roughly constant throughout the basement. In contrast, if we except 2 samples located around 300 msb (31R1 and 35R1a), inorganic C content exhibits a drastic decrease close to the UVS/LVS boundary, from 0.66 down to ∼0.08 wt% CO2 (Tables 3, 5).
The relative proportions of inorganic and reduced C thus change at the UVS/LVS boundary, from inorganic/reduced C >1 in the UVS to <1 in the LVS. Samples 31R1 in the UVS and 35R1a in the LVS are not following this general trend probably because the transition is not so sharp, and some variation may occur around that depth. These two samples will thus not be considered for average estimations of the UVS/LVS in Tables 2, 3, and 5 since they are not representative of the sections as we have defined them.
The total water contents vary from 0.42 to 1.15 wt% H2O (Table 2) but do not show any significant correlation with depth nor difference between the UVS and LVS. The average water content calculated for this portion of the crust is 0.83 ± 0.24 wt% H2O.
4.2. C, O, and H isotope compositions
Depth profiles of δ13C values for the total, reduced and inorganic C in background altered basalts are shown in Figure 3, along with δ13C data for carbonate C in the veins. The altered rock depth profiles for δ13C values are generally similar to those for C content (Figure 2). In the UVS, δ13C values for total carbon in bulk basalt become more negative with depth (from −1.6 at the top to −3.6‰ at the UVS/LVS boundary) whereas those of inorganic C do not (Figure 3). Except for samples 31R1 in the UVS and 35R1a in the LVS, a strong and sharp negative shift in δ13C values is observed for total and inorganic C at the UVS/LVS boundary. In contrast, δ13C values of reduced C remain at about −26‰ throughout the UVS and LVS.
Depth profiles of carbon isotope compositions for total C, inorganic C (in rocks and veins), and reduced C in the altered basalts of Hole 801C. At 300 msb lies the permeability transition that separates the upper volcanic section (UVS) and the lower volcanic section (LVS). When error bars are not visible, it means that they are smaller than the size of the symbols. Alteration temperatures reported along the depth profile are from Alt and Teagle [2003].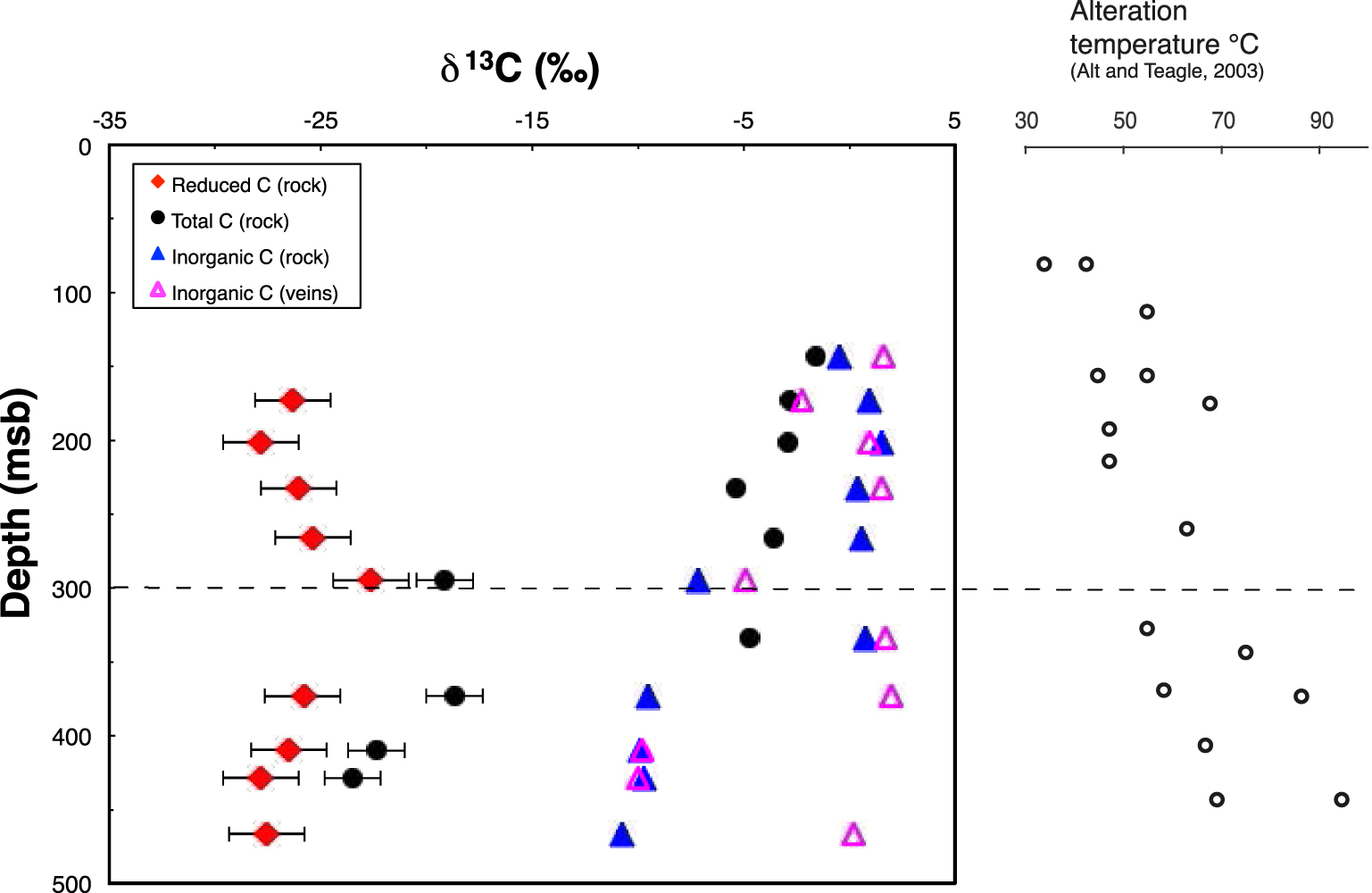
δ18O values of the inorganic C fraction dispersed in the altered basalt host rock and in veins, compared with vein data from Alt and Teagle [2003]. Alteration temperatures are from Alt and Teagle [2003].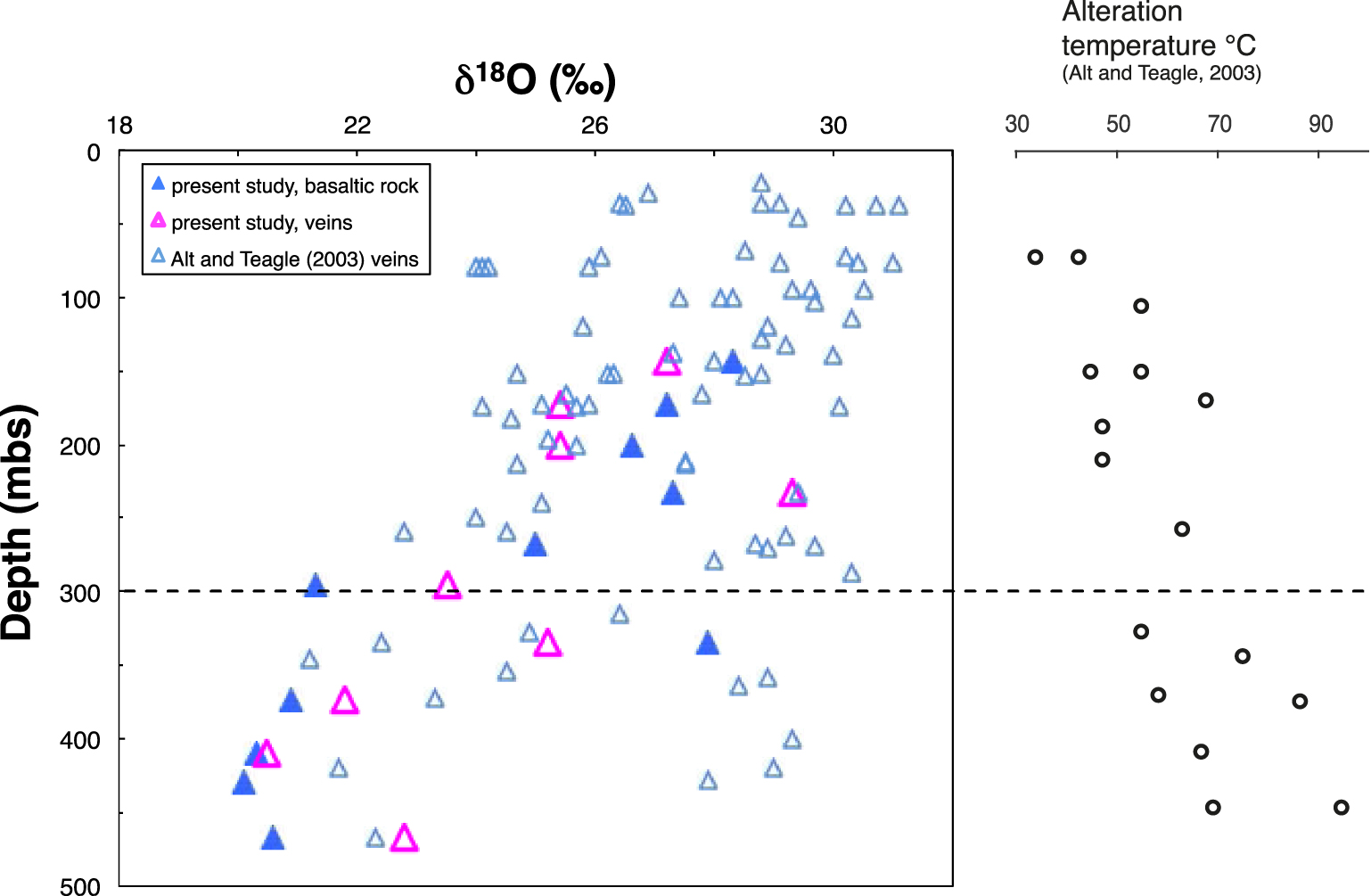
δ13C values of the carbonate fraction in veins vary from −10 to + 2‰ (Table 4), which is in the range for carbonates precipitated from seawater in the altered basaltic crust with the incorporation of oxidized carbon from intercalated sediments for the more negative values [e.g., −11.2‰ to 2.9‰ reported in carbonate veins from Hole 801C by Alt and Teagle 2003; Alt et al. 1996]. However, the very negative δ13C values of −9.8 and −10‰ are two silicate veins (801C-43R2, 801C-45R2) that have very low C concentrations, similar to inorganic C in the basalt samples from the same locations in the LVS (cf. Tables 2 and 3). A third silicate + carbonate vein (801C-31R1a) has an inorganic C content of 0.4 wt% CO2 and δ13C = −4.90‰, which are intermediate between those of LVS rock samples and vein carbonates.
Concentrations and isotope compositions of the inorganic (carbonate) fractions in altered background rock samples from Hole 801C
| Rock | Depth | Extraction at 25 °C | Extraction at 80 °C | Total inorganic fraction | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| (mbsf) | (msb) | Concentration (wt% CO2) | δ13C (‰ VPDB) | δ18O (‰ VSMOW) | Concentration (wt% CO2) | δ13C (‰ VPDB) | δ18O (‰ VSMOW) | Concentration∗∗ (wt% CO2) | δ13C∗∗∗∗ (‰ VPDB) | δ18O∗∗∗∗ (‰ VSMOW) | ||
| The upper volcanic section (UVS) | 801C-14R | 605.1 | 143.5 | 1.26 | 1.4 | 28.0 | 0.50 | −4.9 | 29.1 | 1.76 | −0.4 | 28.3 |
| 801C-17R2 | 634.5 | 173.0 | 0.55 | 1.1 | 26.0 | 0.88 | 0.8 | 27.9 | 1.43 | 1.0 | 27.2 | |
| 801C-20R2 | 662.7 | 201.1 | 0.35 | 1.5 | 27.4 | 0.44 | 1.4 | 26.0 | 0.79 | 1.5 | 26.6 | |
| 801C-24R3a | 694.3 | 232.7 | 0.18 | 0.3 | 26.4 | 0.38 | 0.5 | 27.8 | 0.56 | 0.4 | 27.3 | |
| 801C-28R1 | 728.7 | 267.1 | 0.24 | 0.4 | 24.8 | 0.42 | 0.7 | 25.1 | 0.66 | 0.6 | 25.0 | |
| 801C-31R1 | 757.0 | 295.4 | 0.02∗∗∗ | −7.9∗∗∗ | 22.0∗∗∗ | 0.05∗∗∗ | −6.8∗∗∗ | 21.03∗∗∗ | 0.07∗∗∗ | −7.1∗∗∗ | 21.3∗∗∗ | |
| Average: | 0.52 ±0.44∗ | 1.2 ±0.4 | 27.1±1.1 | 0.52 ±0.20 | -0.2 ±2.2 | 27.3 ±1.4 | 1.04 ±0.53 | 0.5 ±0.7 | 27.2 ±1.1 | |||
| The lower volcanic section (LVS) | 801C-35R1a | 795.9 | 334.3 | 0.34∗∗∗ | 1.0∗∗∗ | 26.4∗∗∗ | 0.33∗∗∗ | 0.6∗∗∗ | 29.4∗∗∗ | 0.67∗∗∗ | 0.8∗∗∗ | 27.9∗∗∗ |
| 801C-39R2 | 834.4 | 372.8 | 0.03 | −8.9 | 20.3 | 0.05 | −9.8 | 21.2 | 0.08 | −9.5 | 20.9 | |
| 801C-43R2 | 871.8 | 410.2 | 0.06 | −10 | 19.6 | 0.02 | −9.8 | 22.1 | 0.08 | −9.9 | 20.3 | |
| 801C-45R2 | 890.8 | 429.2 | 0.05 | −9.7 | 19.5 | 0.02 | −9.6 | 22.1 | 0.07 | −9.7 | 20.1 | |
| 801C-49M1 | 928.6 | 467.0 | 0.03 | −10.7 | 20.2 | 0.02 | −10.7 | 21.1 | 0.05 | −10.7 | 20.6 | |
| Average: | 0.04 ±0.02 | −9.8 ±0.7 | 19.8 ±0.3 | 0.03 ±0.02 | −9.9 ±0.3 | 21.5 ±0.4 | 0.07 ±0.02 | −9.8 ±0.5 | 20.5 ±0.3 | |||
∗All uncertainties correspond to the 1σ interval.
∗∗Calculated by summation of concentration of carbonate fractions extracted at 25 and 80 °C.
∗∗∗Not used for the estimation of the average value (see text for details).
∗∗∗∗Calculated from δ13C, δ18O values extracted at 25 and 80 °C averaged by the equation
Oxygen isotope compositions of carbonate fractions of the altered rock and veins are given in Tables 3 and 4. The transition seen in δ13C values at ∼300 msb is also visible in δ18O values measured in the altered rocks (Table 3; Figure 4). The weighted average δ18O value of the carbonate fraction in the UVS rocks (27.2‰, n = 5) is clearly higher than that for the LVS (20.5‰, n = 5). These values are within the range determined by Alt and Teagle [2003] for vein carbonates in the UVS, whereas those in the LVS are at the low end of this range (Figure 4). δ18O value of the vein carbonates, however, show similar variations than those reported by Alt and Teagle [2003], suggesting alteration temperatures increasing downward and always below 100 °C.
Comparison of δ13C versus C concentration data at Site 801C measured in this study and previous literature data. Three main domains can be defined: (1) carbonates precipitated from seawater DIC, (2) MOR basalt glasses and (3) reduced C.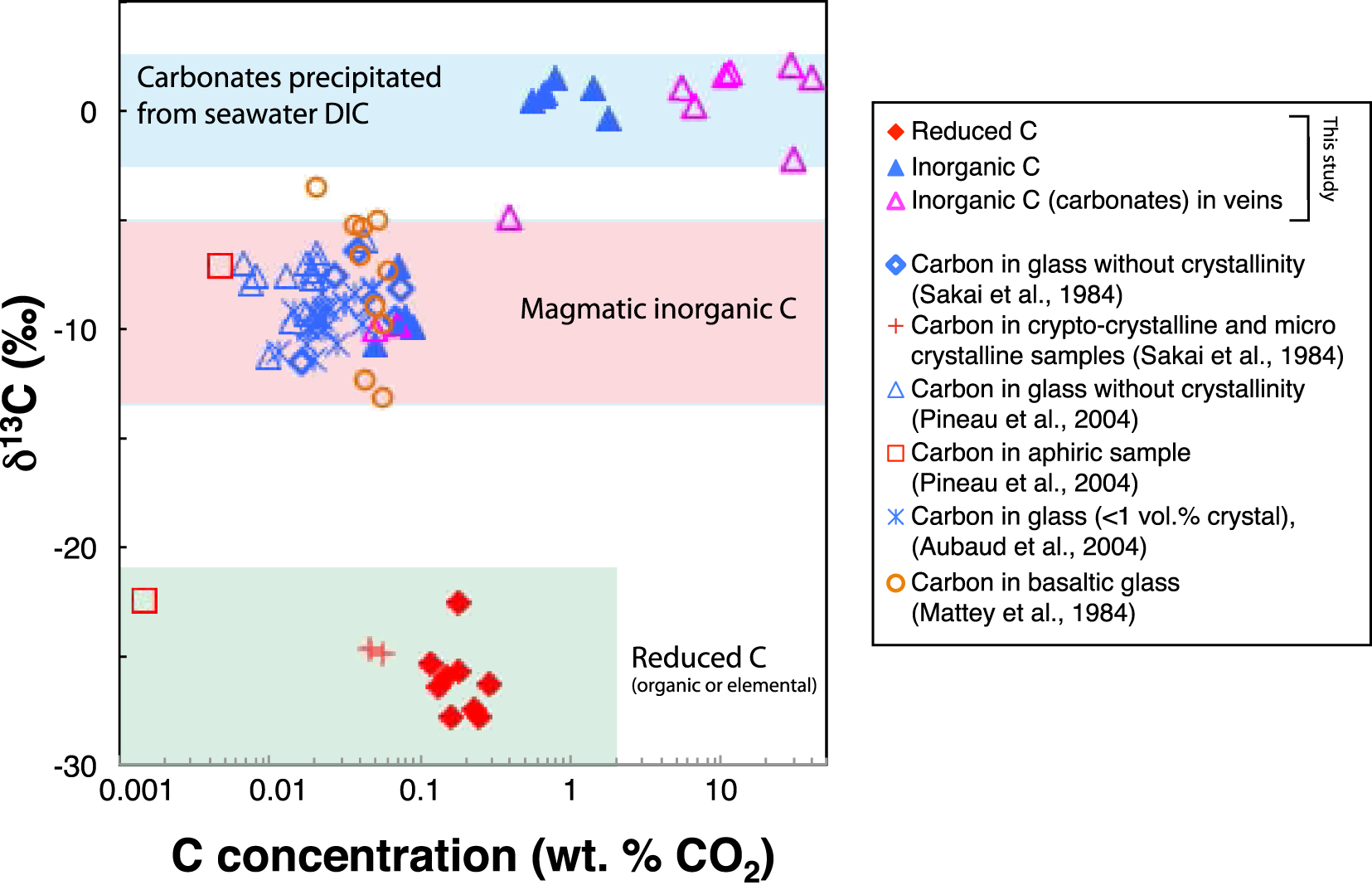
δD values in the UVS (mean = −39.7 ± 14‰,) are higher than those in the LVS (−69.3 ± 13.7‰; Table 2), but strong variability appears in both sections.
4.3. C in the oceanic crust: inorganic and reduced C
Inorganic and reduced C are present throughout Site 801C basement. Reduced C contents and isotope compositions show little variations throughout the basement section (∼0.2 wt% CO2 and −22.6 to −27.8‰, Figures 2 and 3), but observed variations in inorganic C content (Figure 2) lead to differences in total C abundance and isotope composition. High concentrations of inorganic C in the UVS (up to 1.76 wt% CO2 at the top of the section) dominate total C there (1.92 wt% CO2), leading to relatively high δ13Ctotal C values of −1.6 to −5.4‰. In contrast, concentrations of inorganic C in the LVS are much lower (<0.1 wt% CO2), so total C in the LVS is dominated by reduced C, with δ13Ctotal C values of −18.7 to −23.5‰. It is interesting to note that the isotope composition of inorganic C also varies from the UVS to the LVS. The weighted average δ13C value of inorganic C in the UVS is + 0.5‰ and shifts to more negative values (−9.8‰) in the LVS. The oxygen isotope composition of inorganic C likewise changes from the UVS to the LVS: from a mean δ18O value of 27.2‰ to 20.5‰, respectively.
5. Discussion
5.1. Possible origins for reduced and inorganic carbon
5.1.1. Inorganic carbon
The δ13C values of inorganic C in the UVS fall within the range typical for carbonates precipitated from seawater (Figure 5). The δ18O values of inorganic C in the UVS basalts are also within the range of those for carbonate veins in Hole 801C basement, consistent with a seawater origin [Figure 4; Alt and Teagle 2003]. The decrease in inorganic C contents with depth in the UVS results from the decreasing accessibility of seawater due to the decreasing permeability with depth [Jarrard et al. 2003; Coogan and Gillis 2018]. This reflects a general downward decrease in the frequency of carbonate veins, and in the volume percent carbonate present in veins plus breccias in the drill core [Alt and Teagle 2003; Plank et al. 2000]. The δ18O values of the inorganic C fraction in the LVS are lower than in the UVS, for both background carbon and veins, suggesting an increase in fluid temperatures downward. Oxygen isotope data for the carbonate indicate formation temperatures ranging from 20 °C to 60 °C in the UVS and 30 °C–80 °C in the LVS calculated from mineral–water oxygen isotopic fractionation [Kim and O’Neil 1997] and assuming equilibrium with seawater (δ18O = 0‰), which is consistent with temperatures reported for silicate minerals by Alt and Teagle [2003].
Concentrations and isotope composition of carbonate fraction in vein samples from Hole 801C
| Rock | Depth | Extraction at 25 °C | Extraction at 80 °C | Total carbonate fraction | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| (mbsf) | (msb) | Concentration (wt% CO2) | δ13C (‰ VPDB) | δ18O (‰ VSMOW) | Concentration (wt% CO2) | δ13C (‰ VPDB) | δ18O (‰ VSMOW) | Concentration∗ (wt% CO2) | δ13C∗∗ (‰ VPDB) | δ18O∗∗ (‰ VSMOW) | |
| 801C-14R | 605.1 | 143.5 | 10.44 | 1.6 | 27.2 | −1.1 | 28.7 | 10.44 | 1.6 | 27.2 | |
| 801C-17R2 | 634.5 | 173.0 | 28.17 | −2.3 | 25.2 | 2.29 | −0.7 | 27.9 | 30.46 | −2.2 | 25.4 |
| 801C-20R2 | 662.7 | 201.1 | 5.07 | 1.0 | 25.0 | 0.42 | 0.8 | 29.3 | 5.49 | 1 | 25.4 |
| 801C-24R3(a) | 694.3 | 232.7 | 40.15 | 1.3 | 29.3 | 1.6 | 30.9 | 40.15 | 1.5 | 29.3 | |
| 801C-28R1 | 728.7 | 267.1 | |||||||||
| 801C-31R1 | 757.0 | 295.4 | 0.24 | −4.2 | 23.3 | 0.16 | −6 | 23.8 | 0.40 | −4.9 | 23.5 |
| 801C-35R1a | 795.9 | 334.3 | 10.49 | 1.7 | 25.2 | 0.85 | 1.5 | 28.7 | 11.35 | 1.7 | 25.2 |
| 801C-39R2 | 834.4 | 372.8 | 28.52 | 2 | 21.7 | 0.94 | 2 | 24.3 | 29.46 | 2 | 21.8 |
| 801C-43R2 | 871.8 | 410.2 | 0.04 | −10.1 | 19.6 | 0.03 | −9.4 | 21.9 | 0.07 | −9.8 | 20.5 |
| 801C-45R2 | 890.8 | 429.2 | 0.05 | −10.0 | 20.4 | −10.2 | 28.7 | 0.05 | −10.0 | ||
| 801C-49M1 | 928.6 | 467.0 | 6.67 | 0.2 | 22.8 | 27.9 | 6.67 | 0.2 | 22.8 | ||
∗Calculated by summation of concentration of carbonate fractions extracted at 25 and 80 °C.
∗∗Calculated from δ13C, δ18O values extracted at 25 and 80 °C averaged by the equation:
The inorganic C in the LVS, however, has low δ13C values of −10.7 to −9.5‰, which are not consistent with seawater DIC (Figures 3 and 5). Most carbonate veins in Hole 801C have δ13C values around 0‰, but negative values (down to −11.2‰) occur in the alkalic basalt section at the top of the basement, as well as locally in tholeiites of the UVZ [Alt and Teagle 2003]. The alkalic basalts are sills intruding into sediments, and interflow sediment is common in the UVS section [Plank et al. 2000], leading to the interpretation that the negative δ13C values for veins reflect (non-equilibrium) incorporation of oxidized sedimentary organic matter into fluids circulating in the basement [Alt and Teagle 2003]. Similar negative δ13C values of carbonate veins in basalts near the sediment–basement contact and in sills intrusive into overlying sediment on the Juan de Fuca ridge flank reflect the same process [Coggon et al. 2004]. While negative δ13C values of carbonate veins reflect solutions circulating in fractures, bulk rock inorganic C in the LVS rather reflects host basalt away from fluids in fractures in a non-permeable media. Moreover, no interflow sediment was observed in drill core from the LVS [Plank et al. 2000], and none of the carbonate veins analyzed from the LVS has a negative δ13C value [Alt and Teagle 2003] except for carbonate-poor samples 801C-43R2 and 801C-45R2 (Figure 3 and Table 4), suggesting that processes other than oxidation of organic carbon may account for negative δ13C values of inorganic C in the LVS basalts. As it is seen from Figure 5, both content and isotopic composition of the inorganic C fall in the ranges observed for C incorporated in non-crystalline basaltic glasses [Sakai et al. 1984; Mattey et al. 1984; Aubaud et al. 2004; Pineau et al. 2004]. In such glasses, C is present as ions in the highly CO2-outgassed melt after decompression and cooling of the basaltic magma, and is depleted in 13C relative to the mantle δ13C value of ∼−5‰ [Figure 5; Javoy et al. 1982; Pineau et al. 2004]. Thus, residual magmatic C, remaining from the formation of the basalts at the spreading axis, is the most likely source for inorganic C in the LVS basalts.
Data for H isotope compositions support this interpretation. δD values in the LVS range from −83.3 to −49.7‰ with a mean of −69.3‰ (Table 2). These values are typical for magmatic rock, in particular, for MOR basalts [Sheppard and Epstein 1970], indicating that the LVS basalts are not significantly affected by seawater interactions. In contrast, δD values in the UVS are significantly higher, −17.6 to −59.8‰, with an average value of −39.7‰ (Table 2). Such high δD values result from interactions of basalts with seawater (having a δD = 0‰), consistent with more intensive circulation of seawater in the UVS [Agrinier et al. 1995a,b].
The veins consist of variable proportions of carbonate plus silicate, so C contents are lower than for pure carbonate minerals. C and O isotope compositions of inorganic C in most of the veins are typical for vein carbonates as measured earlier by Alt and Teagle] (2003], Figures 4, 5), and consistent with carbonates forming in veins from circulating seawater. The two silicate vein samples in the LVS (801C-43R2, 801C-45R2) have low inorganic C concentrations and negative δ13C values like those for the basalts from the same sample (Tables 2, 3; Figure 2). It is possible that the inorganic C in these veins has a magmatic origin like in the host basalts. The third low C silicate–carbonate vein (801C-31R1) has higher inorganic C content and δ13C value (0.40 wt% CO2, δ13C = −4.90‰ and δ18O = 23.5‰) consistent with a mixture of magmatic C and seawater DIC.
5.1.2. Reduced carbon
The reduced carbon component has a relatively constant concentration (average value: 0.19 ± 0.06 wt% CO2) and isotopic composition (average value: −26.4 ± 1.3‰) throughout the UVS and LVS (Table 2; Figures 2, 3). Possible sources of reduced C are: (1) seawater dissolved organic C (DOC), (2) organic C produced by microbial activity, and (3) abiotically formed carbon components from magmatic C. If we propose seawater dissolved organic C as a possible origin of this reduced C and assuming quantitative fixation of DOC from seawater; it would take a seawater/rock mass ratio greater than 860 in order to incorporate the observed amount of C in the rocks [0.19 wt% CO2 i.e., more than 43,000 μmol of C per kg of rock; seawater DOC ≈ 50 μM C per kg; Ogawa and Tanoue 2003]. Such uniformly high water/rock ratios are not consistent with indications of variable seawater fluxes through different zones of the basement [Alt and Teagle 2003]. In particular, Sample 43R2 121–124 in the LVZ consists of a brown alteration halo containing Fe oxyhydroxides (Table 1) that indicate higher water/rock ratios than other samples, yet the reduced C content and isotope composition of this sample are similar to those of the other basalts (Table 2).
Another possibility for the origin of reduced C is microbial C. Basalts from Hole 801C lost igneous sulfur through oxidation by seawater, but also contain secondary sulfide minerals that have δ34S values ranging down to −45‰ [Rouxel et al. 2008]. These data plus multiple sulfur isotope analyses (32S, 33S, 34S) all indicate that microbial reduction of seawater sulfate contributed sulfur to the basement at Site 801 [Rouxel et al. 2008]. Given the evidence for microbial activity throughout the basement, it is likely that a component of the reduced C in the rocks is organic carbon of microbial origin. Similar data for other basement sites (Holes 1256D in the Eastern Pacific and 1301B on the Juan de Fuca ridge) indicate formation of low-δ34S secondary sulfides (to −70‰) during microbial reduction of seawater sulfate in the basement [Alt and Shanks 2011; Lever et al. 2013], suggesting that microbially derived organic C could be present in the basalts. The DNA and RNA extracted from veins in basalt basement on Juan de Fuca as well as negative δ13C values for reduced C (−22 to −34‰) in the rocks and for DOC in basement fluids indicate the activity of bacteria in the basement [Lever et al. 2013; McCarthy et al. 2011]. Thus, the reduced C in altered seafloor basalts from Hole 801C and elsewhere may contain a component of organic C of microbial origin.
Summary of the C concentration data in investigated Hole 801C samples
| Rock | Depth | Total C (wt% CO2) | Reduced C (wt% CO2) | Inorganic C (wt% CO2) | Reduced C + Inorganic C (wt% CO2) | Reduced C fraction∗∗ | Inorganic C fraction∗∗ | ||
|---|---|---|---|---|---|---|---|---|---|
| (mbsf) | (msb) | ||||||||
| The upper volcanic section (UVS) | 801C-14R | 605.1 | 143.5 | 1.92 | 0.17 | 1.76 | 1.93 | 0.09 | 0.91 |
| 801C-17R2 | 634.6 | 173.0 | 1.38 | 0.29 | 1.43 | 1.72 | 0.17 | 0.83 | |
| 801C-20R2 | 662.7 | 201.1 | 0.80 | 0.16 | 0.79 | 0.95 | 0.17 | 0.83 | |
| 801C-24R3a | 694.3 | 232.7 | 0.52 | 0.15 | 0.56 | 0.71 | 0.21 | 0.79 | |
| 801C-28R1 | 728.7 | 267.1 | 0.57 | 0.12 | 0.66 | 0.78 | 0.15 | 0.85 | |
| 801C-31R1∗∗∗∗∗ | 757.0 | 295.4 | 0.40∗∗∗ | 0.18∗∗∗ | 0.07∗∗∗ | 0.25∗∗∗ | 0.72∗∗∗ | 0.28∗∗∗ | |
| Average: | 1.04 ± 0.59∗ | 0.18 ± 0.07∗ | 1.04 ± 0.53∗ | 1.22 ± 0.57∗ | 0.16 ± 0.05∗ | 0.84 ± 0.05∗ | |||
| The lower volcanic section (LVS) | 801C-35R1a∗∗∗∗∗ | 795.9 | 334.3 | 0.50∗∗∗ | 0.22∗∗∗ | 0.67∗∗∗ | 0.89∗∗∗ | 0.25∗∗∗ | 0.75∗∗∗ |
| 801C-39R2 | 834.4 | 372.8 | 0.29 | 0.18 | 0.08 | 0.26 | 0.69 | 0.31 | |
| 801C-43R2 | 871.8 | 410.2 | 0.22 | 0.13 | 0.09 | 0.22 | 0.59 | 0.41 | |
| 801C-45R2 | 890.8 | 429.2 | 0.34 | 0.25 | 0.07 | 0.32 | 0.78 | 0.22 | |
| 801C-49M1 | 928.6 | 467.0 | 0.39 | 0.23 | 0.05 | 0.28 | 0.82 | 0.18 | |
| Average: | 0.31 ± 0.07∗ | 0.20 ± 0.05∗ | 0.07 ± 0.02∗ | 0.27 ± 0.04∗ | 0.72 ± 0.10∗ | 0.28 ± 0.10∗ | |||
∗All uncertainties correspond to the 1σ interval.
∗∗Calculated as the ratio of reduced or oxidized C to the sum reduced + inorganic C.
∗∗∗Not used for the estimation of the average value (see text for details).
As a third possibility, the isotope composition of the reduced C is also consistent with an abiotic origin. In basalts from ODP/IODP Hole 1256D, Shilobreeva et al. [2011] observed reduced C with similar concentration and isotope composition as that from Hole 801C. Taking into account the depth of the samples (up to 1200 m), they proposed that this C could be formed abiotically at the ridge axis by reduction of degassed magmatic CO2 under hydrothermal conditions. Several mechanisms can be invoked to explain this abiotic CO2 reduction, such as FTT processes [Shock 1990; Shock and Schulte 1998; McCollom and Seewald 2006, 2007] or Boudouard disproportionation reaction (BDR), [Mathez and Delaney 1981; Mathez 1984, 1987; Pineau and Mathez 1990; Mathez and Mogk 1998] during hydrothermal alteration of Fe2+-rich minerals in the temperature range of 100 °C–300 °C. Even if experimental works have been able to reproduce abiotic organic compounds among gaseous or liquids products only, none of these works have focused on the production of condensed and solid organic compounds. In serpentinized peridotites, for instance, numerous lines of evidence for the abiotic formation of condensed carbonaceous matter (CCM) have been reported recently [Sforna et al. 2018; Menez et al. 2018]. Such CCM production may also occur during low-temperature (T < 150 °C) alteration of Fe-rich phyllosilicates (saponite), suggesting that it can be widespread even in mafic lithologies. Moreover, the CCM formation was also predicted by thermodynamic calculations [Milesi et al. 2016].
5.2. Carbon incorporation during aging of the crust
We thus propose a sequence of processes that affect the contents and isotope compositions of carbon in oceanic basalts. Abiotic (FTT and BDR) processes can leave their imprint on reduced and inorganic C throughout the rocks of the volcanic section at the ridge crest, resulting in the presence of reduced C having negative δ13C values (−22 to −28‰), and of small amounts (0.07 ±0.02 wt% CO2, Table 5) of inorganic C having δ13C values of −9 to −11‰. This could happen during cooling and crystallization of the basalts and/or through the passage of cooled hydrothermal fluids “leaking” upward from the underlying sheeted dikes at the spreading axis, such as those that produce the cool diffuse hydrothermal venting at the seafloor along ridge crests [e.g., Alt 1995]. Later, as cool seawater solutions percolate through the porous and permeable volcanic section and alter the basement at low temperatures, microbial activity results in the addition of microbial sulfide to the rocks, which can also contribute to organic C having δ13C values of −22 to −34‰ to the basalts throughout the lavas. Relatively late seawater solutions also deposit seawater DIC in carbonate veins in the basalt basement and within altered basalts, mainly in the uppermost portion of the volcanic section where high porosity and permeability allow greater fluid circulation [Alt and Teagle 1999, 2003; Alt 1995; Jarrard et al. 2003; Coogan and Gillis 2018].
As the accumulated effect of these processes, reduced C contents and isotope compositions show little variation throughout the Hole 801C basement section ( ∼0.2 wt% CO2 and −22 to −28‰, respectively), but variations in inorganic C contents lead to differences in total C abundance and isotope composition. High concentrations of inorganic C in the bulk rocks of the UVS (up to 1.76 wt% CO2 at the top of the section) result from greater flux of seawater through this portion of the crust. This leads to inorganic C dominating total C in the UVS rocks and to relatively high δ13Ctotal C values of −1 to −5‰. In contrast, concentrations of inorganic C in the LVS basalts are much lower ( <0.1 wt% CO2), so total C in the LVS is dominated by reduced C with δ13Ctotal C values of −19 to −23‰. The contents and isotope compositions of C in the Hole 801C basement thus reflect a relatively uniform component of reduced C, derived from abiotic and biotic reactions, with variable addition of seawater DIC to the rocks, with seawater derived DIC also deposited in carbonate veins (Figure 6).
Bulk δ13C versus C concentration in altered basaltic rocks from Holes 801C and 1256D. Data from the UVS (for depths <300 msb) at Holes 801C show an apparent trend reflecting a correlation between the isotopic composition and the total C content. Data from Holes 1256D do not exhibit any correlation, and are similar to altered basalt samples from the LVS (depths >300 msb) at Holes 801C. We suggest a similar origin for C in the altered basalts at Hole 1256D and in the LVS at Hole 801C.
5.3. C budget at mid-ocean ridges
Except for the upper part of the basaltic basement, where both seawater DIC and biologically derived C are abundant, magmatic CO2 is a source for C in the background altered basaltic crust at mid-ocean ridges (MORs). This C is stored in the altered basaltic ocean crust as (i) inorganic C retained in the CO2-outgassed basaltic melt and as (ii) reduced C formed during abiotic reduction reactions or through microbial activity. Integrating the present results with data from Site 1256D obtained in a previous study [Shilobreeva et al. 2011], we can estimate the magmatic C budget at MOR. At Site 801, the reduced C concentration in the volcanic section is 0.19 wt% CO2 (average value between the UVS and LVS). We should add to this value the inorganic fraction of magmatic C (0.07 wt% CO2 in the LVS; Table 3), giving a total concentration of 0.26 wt% CO2. As we discussed above, some fraction of the reduced C is derived from biological activity. To get a rough estimate of each fraction (biological versus abiotic), we can use the C concentrations measured in Hole 1256D. Hole 1256D passed through all lithological units of the basaltic ocean crust, except gabbro, where it penetrated only the uppermost tens of meters of the gabbro section. The estimated temperatures in the crust today reach more than 120 °C at a depth greater than 1000 mbsf (meters below seafloor), indicating that microbial activity is not expected to be sustained at deeper levels [>600 msb: i.e., the transition zone, sheeted dikes, and gabbros, Shilobreeva et al. 2011]. Even if thermogenic carbon from the shallower part can be delivered downward via hydrothermal circulation, because of low permeability, we propose that most of the C measured at depths greater than 600 msb is magmatic carbon. The average value calculated for depths between 600 msb down to 1200 msb [Table 3 in Shilobreeva et al. 2011] is 0.126 wt% CO2, which is half of the value calculated at Site 801. We can compare this value to the depth-weighted average CO2 content estimated by Alt and Teagle [1999] from a compilation of different sites. From Table 1, a depth-weighted average CO2 content of 0.05 wt% CO2 is calculated for the deeper levels ( >600 msb) of the crust, which again can be assumed to be magmatic carbon at these depths. These two values probably represent the upper (0.126 wt% CO2) and lower (0.05 wt% CO2) limit for magmatic carbon stored in the altered crust. A way to explain this difference is that Site 801 was formed in the Jurassic age when atmosphere and ocean were enriched in CO2 and when CO2 degassing in association with magmatism at MOR was more active than the present [Gillis and Coogan 2011]. These values are lower than the mean total C content in the altered basaltic crust [cf. Alt and Teagle 1999; Shilobreeva et al. 2011]. The difference between the total and magmatic C contents results from C precipitated seawater DIC and DOC that is stored not only in the background basaltic rock but also in veins and alteration halos along veins. For this reason, C in the altered oceanic crust is dominated by magmatic C in the young basaltic crust where it may exceed 80% of the total carbon (e.g., Holes 504B, 1256D), whereas for an old basaltic ocean crust containing greater amounts of carbonate veins (e.g., Site 801) the magmatic C fraction may be less than 40%.
Considering a crustal production rate of 6.0 ± 0.8 × 1016 g⋅y−1 [Mottl 2003], the total flux of magmatic C stored in altered basaltic ocean crust ranges between 1.5 × 1012–2.0 × 1012 molC⋅y−1 (using Cmag = 0.126 wt% CO2). This magmatic flux stored in the crust can be compared with the flux of C degassed from the mantle during the formation of oceanic crust at mid-ocean ridges. The latter ranges from ∼1.0 × 1012 to 2 × 1013 molC⋅y−1 according to various estimations [e.g., Javoy et al. 1982, 1983; Des Marais 1985; Javoy and Pineau 1991; Marty and Tolstikhin 1998; Cartigny et al. 2001, 2008; Chavrit et al. 2014; Le Voyer et al. 2019]. Using the reevaluated value of Cartigny et al. [2008] or the most recent value by Le Voyer et al. [2019], 2.0 × 1012 molC⋅y−1 or 1.32 ± 0.8 × 1012 molC⋅y−1 respectively, the flux of magmatic C stored in the altered oceanic crust is similar to the C flux degassed from the mantle. Moreover, and except for the UVS, our data suggest that the dominant form of magmatic carbon that is retained in the crust is reduced C (70%, Table 5). This observation raises the question of its behavior during subduction and recycling in the mantle. The reactivity of carbonates during subduction has been intensively studied, yielding either to a large recycling into the deep mantle, or more recently, to a large transfer back to the surface via mantle wedge [Kelemen and Manning 2015]. Understanding the reactions of reduced forms of C at HP–HT in subduction zones conditions will be necessary in order to refine future global C cycle models.
6. Conclusions
In the UVS at Hole 801C (at depths <300 msb), the total C content and its isotope composition are controlled by inorganic (carbonate) C. δ13C values of the inorganic C range between −0.4 to + 1.5‰, which suggests an origin from precipitated seawater DIC. Infiltration of seawater DIC controls the total C content and its isotopic composition resulting in a positive correlation between these two quantities; in turn, the correlation may serve as an indicator of the seawater DIC infiltration into altered basaltic oceanic crust. The inorganic C content decreases drastically for depths >300 msb across the UVS/LVS boundary.
In the LVS at Hole 801C (depths >300 msb), the total C is largely (∼70%) dominated by reduced C. No variation of concentration and isotopic composition of the total C with depth, nor correlation between these quantities are observed in the LVS. In this section, the inorganic C content (from 0.05 to 0.09 wt% CO2) and δ13C values (from −10.7‰ to −9.5‰) fall in ranges characteristic of C dissolved in MOR basaltic glasses. Similar to C in MOR basaltic glasses, the inorganic C in the LVS at Hole 801C has a magmatic origin and represents residual 13C-depleted C, initially dissolved in the abundantly outgassed basaltic melt as ions. Origins of the inorganic C in the UVS and the LVS are thus different. The reduced C does not show significant variations in its content (from 0.12 to 0.29 wt% CO2) and isotopic composition (from −22.6 to −27.8‰) all along the section. Considering the δ34S values of sulfide minerals ranging down to −45‰ [Rouxel et al. 2008], it is likely that a portion of the reduced C in the rocks is organic carbon of microbial origin. Comparison with Hole 1256D also suggests that some reduced C may have formed by abiotic reduction of magmatic CO2 close to the ridge axis.
We have estimated the flux of magmatic C stored in altered basaltic crust using the C content measured in this study (at Sites 801 and 1256) or previously estimated by Alt and Teagle [1999]. Our estimate ranges between 1.5 × 1012–2.0 × 1012 molC⋅y−1 (using Cmag = 0.126 wt% CO2), which is the same order of magnitude than that of outgassed CO2 using the most recent value of Cartigny et al.] (2008], 2.0 × 1012 molC⋅y−1) and Le Voyer et al.] (2019], 1.32 ± 0.8 × 1012 molC⋅y−1). Further measurements are needed to refine this flux in order to constrain the fraction of magmatic CO2 that does not escape the oceanic lithosphere but remains stored during alteration. Finally, we suggest that reduced forms of C in the oceanic lithosphere also have to be considered in order to refine future global C cycle models, in particular, their recycling or not during subduction.
Acknowledgments
The authors thank Carine Chaduteau for her help with isotopic measurements. We are also grateful to Jean-Jacques Bourrand, Guillaume Landais, and Nelly Assayag for their technical assistance during Svetlana stays in IPGP. Helpful discussions with Vincent Busigny and Sergey Silantiev were greatly appreciated. S. Shilobreeva thanks IPGP for supporting her stays at IPGP. J. Alt’s contribution was supported by NSF-OCE-1334758. This study was also partly supported by IdEX Université de Paris ANR-18-IDEX-0001. We greatly thank Matthieu Galvez and an anonymous reviewer for their constructive comments. This is an IPGP contribution No. 4217.