

1 Introduction
Surveillance of Creutzfeldt-Jakob disease (CJD) was re-established in 1990 in the UK in order to identify any changes in the incidence or characteristics of human prion diseases which might be the consequence of human exposure to the bovine spongiform encephalopathy (BSE) agent. The subsequent identification of a novel human prion disease in the UK, variant Creutzfeldt-Jakob disease (vCJD) in 1996 was based on detailed neuropathological and clinical findings in a series of 10 patients 〚1〛. One hundred cases of vCJD have so far been identified in the UK, with 3 cases in France and one in Ireland. All cases of vCJD have occurred in individuals who were homozygous for methionine at codon 129 in the prion protein gene (PRNP). The evidence for a causal relationship between vCJD and bovine spongiform encephalopathy (BSE) has been considerably strengthened by the results of experimental strain typing and biochemical studies of the disease-associated form of the prion protein (PrPRES) in mice 〚2,3〛, and subsequent transmission studies of BSE and vCJD in bovine transgenic mice 〚4〛. Since it is likely that exposure to BSE has occurred in all prion protein genotypes in the population, the development of criteria to detect a BSE-related disease in individuals who are heterozygotes or valine homozygotes at codon 129 of the PRNP is important for surveillance purposes.
The World Health Organisation has indicated that neuropathological examination is mandatory for the confirmation of a diagnosis of vCJD 〚5〛. The neuropathological diagnostic criteria for vCJD are reviewed here, along with the results of biochemical analysis of PrPRES in the brain and recent studies to detect the abnormal form of the prion protein (PrPRES) outside the central nervous system.
2 Materials and methods
Since 1990, central neuropathological review has been performed in the CJD Surveillance Unit for all cases of CJD diagnosed in the UK. Tissues from autopsy cases were fixed in formalin for a minimum of three weeks prior to brain dissection. The brains were sampled to include the frontal, parietal, temporal and occipital cortex, the hippocampus, hypothalamus, thalamus, basal ganglia, midbrain, pons, medulla and spinal cord (when available). Other organs were examined histologically if appropriate permission was obtained and material was available.
All tissue blocks were immersed in 96 % formic acid for 1 h prior to routine processing. Sections were cut at 5 μm and stained by conventional neuropathological techniques and by immunocytochemistry for PrPRES using a range of monoclonal antibodies in a standardised validated technique 〚6〛. Microscopic sections were examined using a semi-automated image analysis system to quantitate spongiform change, PrP accumulation and astrocytosis as previously described 〚7〛.
2.1 Biochemical analysis
Frozen brain tissue was stored at –80 °C and investigated by Western blotting for the presence of PrPRES using a variation of the method of Collinge et al. 〚8〛. In summary, aliquots of the cleared 10 % brain homogenates were subjected to limited proteolysis by digestion with Proteinase K (BDH). Electrophoresis was performed on a 12 % T acrylamide SDS-PAGE mini-gel format (Bio-Rad Laboratories) and proteins transferred to Hybond ECL nitrocellulose or Hybond-P PVDF membranes (Amersham Parmacia Biotech). The anti-PrP monoclonal antibody 3F4 (Senetek) was used at a 1:10 000 dilution. Detection employed the ECL or ECL+ reagents and Hyperfilm ECL (Amersham Pharmacia Biotech). The molecular weight of PrPRES was estimated by reference to biotinylated ECL molecular weight markers (Amersham Pharmacia Biotech) visualised by the inclusion of streptavidin peroxidase at a dilution of 1:1 500 in the secondary incubation.
3 Results
3.1 Neuropathology of vCJD
The principal neuropathological features of vCJD are summarised in table I. In most cases, the brain weight after fixation was within the normal range in relation to the age of the patient, but in cases with a lengthy clinical history (> 18 months) there was evidence of cerebral cortical and cerebellar atrophy (particularly involving the vermis). No other significant macroscopic abnormalities were detected in the CNS.
Diagnostic neuropathological features of vCJD.
| Cerebral and cerebellar cortex: | Multiple florid plaques in H&E sections |
| Numerous small cluster plaques in PrP stained sections | |
| Amorphous pericellular and perivascular PrP accumulation | |
| Caudate nucleus and putamen: | Severe spongiform change |
| Perineuronal and axonal PrP accumulation | |
| Posterior thalamic nuclei and midbrain: | Marked astrocytosis and neuronal loss |
| Spinal cord and brainstem: | Reticular and perineuronal PrP accumulation in grey matter |
Florid plaques were identified on haematoxylin and eosin stains in all cases in the cerebral cortex (particularly the occipital cortex) and the cerebellar cortex. These comprised an eosinophilic central core with radiating fibrils in the periphery of the plaque, which was surrounded by a rim of spongiform change in an otherwise intact neuropil 〚1, 9〛 (figure 1). Florid plaques occurred in all layers of the cerebral cortex, often in a random focal distribution. In the cerebellum, florid plaques were most easily identified in the molecular layer, occasionally projecting into the subpial space, but were also present in aggregates in the granular layer without spongiform change. Amyloid plaques were often present close to blood vessels, but a true amyloid angiopathy was not identified.
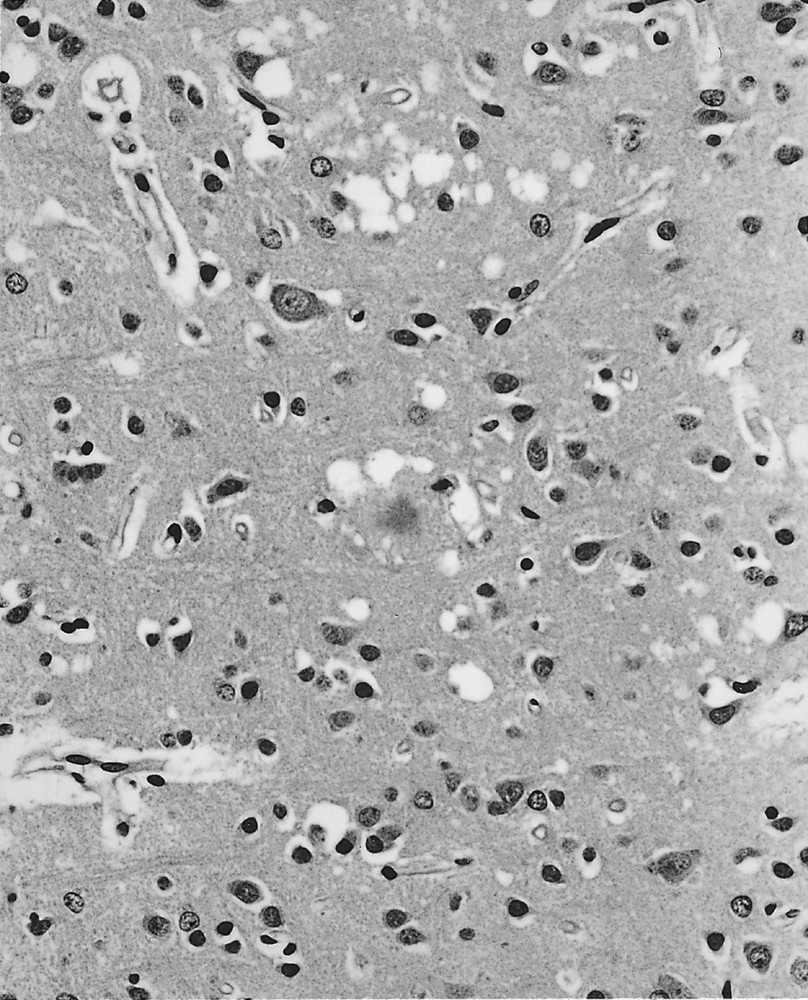
The frontal cortex in variant CJD contains a florid plaque (centre), with a dense core, pale-staining periphery and surrounded by spongiform change. Focal spongiform change, neuronal loss and gliosis are also evident. Haematoxylin and eosin, × 250.
Spongiform change within the cerebral cortex was usually most prominent in the occipital and inferior frontal regions; the hippocampus was relatively spared. Spongiform change was also a prominent feature in the cerebellar hemispheres and vermis. Extensive confluent spongiform change was present in the cerebral cortex, in comparison to cases of sporadic CJD. Extensive spongiform change was a constant feature in the caudate nucleus and putamen, and appeared severe in relation to the small numbers of amyloid plaques. Similar changes were present in the anterior thalamus, but focal spongiform change was seldom present in the posterior thalamic nuclei. In these regions, the neuropathology was dominated by severe neuronal loss (almost total loss in the pulvinar) and marked astrocytosis. The distribution of astrocytosis did not relate to the presence of amyloid plaques. Spongiform change was most evident in the paraventricular and supraoptic nuclei of the hypothalamus, which also contained occasional amyloid plaques. Spongiform change was also detected in the midbrain (particularly in the periaqueductal grey matter) and in the pontine nuclei, but was not present in either the medulla or spinal cord. In the midbrain, severe neuronal loss and astrocytosis occurred in the colliculi and the periaqueductal grey matter.
Immunocytochemistry for PrPRES showed strong staining of the florid plaques in all areas of the cerebral and cerebellar cortex. This technique also revealed numerous smaller plaques which were not visible on routinely stained sections, often arranged in irregular clusters within the neuropil (figure 2). These ‘cluster plaques’ were present in all cases, including several cases which had undergone brain biopsy. Furthermore, there was a widespread deposition of PrPRES in a pericellular (neuronal and astrocytic) distribution. These unusual deposits could also be visualised on Gallyas silver impregnation. Quantitative assessment confirmed the marked PrPRES accumulation in the occipital cortex and the cerebellar cortex , in comparison to other cortical regions, basal ganglia and thalamus 〚10〛. In the hippocampus, the cornu ammonis showed little PrPRES deposition, but there was a dense accumulation in the dentate fascia and the subiculum.
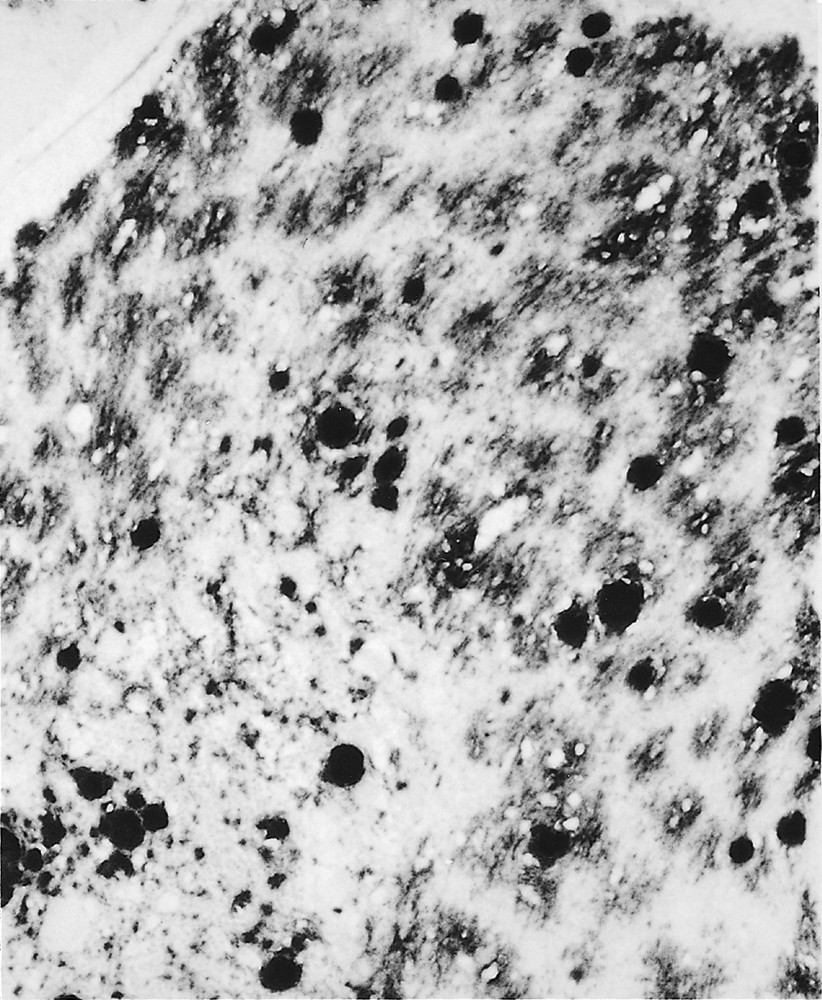
Immunocytochemistry for PrPRES in the cerebellum shows strong staining of large rounded plaques, with smaller clusters of plaques (centre) and abundant amorphous PrPRES deposits. KG9 anti-PrP monoclonal antibody, × 180.
PrPRES accumulation in the basal ganglia and thalamus occurred as in a linear and perineuronal pattern of accumulation, often with multiple small PrPRES plaques. A reticular pattern of immunoreactivity for PrPRES was also detected in the thalamus and hypothalamus, with occasional PrPRES plaques. Synaptic and neuronal PrPRES positivity was also present in the midbrain, pons and medulla and spinal cord (particularly in the substantia gelatinosa). PrPRES immunoreactivity was not detected in the dura mater or arachnoid mater.
3.2 Non-CNS tissues
Positive staining for PrPRES was confirmed (as previously reported) in follicular dendritic cells within germinal centres of the tonsil, spleen and lymph nodes 〚11, 12〛. Positive staining for PrPRES was also identified in cells that corresponded to follicular dendritic cells in lymphoid follicles in the wall of the appendix, and in Peyer’s patches in the ileum. PrPRES immunocytochemistry in the other main organs (including the heart, lung, muscle, liver, kidney, bladder, testes, and pelvic organs and skin) was all negative in vCJD. PrPRES immunocytochemistry was negative in all lymphoid tissues and other organs in sporadic CJD.
3.3 Biochemistry
In agreement with Parchi et al. 〚13, 14〛 we identified two different mobility variants in sporadic CJD: the first having a non-glycosylated PrPRES of ∼21 kDa and the other of ∼19 kDa. The PrPRES isoform pattern from patients with vCJD is indistinguishable in terms of mobility from the 19 kDa isotype in sporadic CJD, but it has a predominance of di-glycosylated PrPRES 〚13〛. This finding has been observed in all cases of vCJD studied in the Surveillance Unit, but in none of the cases of sporadic CJD or other human prion diseases. Comparison of these findings with other cases of CJD diagnosed in the Surveillance Unit since 1990 showed no major overlap in terms of neuropathological or biochemical features.
4 Discussion
Neuropathological and biochemical studies on vCJD allow a clear distinction from other forms of CJD; the relative uniformity of vCJD is in marked contrast to sporadic CJD (the main differential pathological diagnosis). This uniformity is also consistent with the effects of a single agent strain (BSE) in individuals with a uniform PRNP genotype. In the limited data available on kuru (which was probably also transmitted by the oral route), codon 129 PRNP genotype did not appear to exert a major influence on the neuropathological or clinical features in one series 〚15〛, although in larger study (without detailed pathological review) it was suggested that kuru plaques were more evident (but not exclusively present) in individuals who were valine homozygotes or heterozygotes at codon 129 〚16〛. There is only very limited data on PrP subtypes in kuru, and no information exists on the immunocytochemical localisation of PrP outside the CNS.
The diagnostic utility of the florid plaque as a neuropathological hallmark of vCJD is confirmed; these are present in a widespread distribution in the cerebral and cerebellar cortex in all cases. Florid plaques visible on routine microscopy have not been reported in cases of sporadic CJD, and there seems to be no major overlap of the neuropathology in any of the subtypes of sporadic CJD identified so far with the morphological diagnostic criteria proposed here for vCJD (table I). Florid plaques are not a unique to vCJD, and were first described in transmissions of Icelandic scrapie to mice 〚17〛. They also occur in chronic wasting disease 〚18〛. Intracerebral transmission of BSE to macaques produced a spectrum of neuropathology which was remarkably similar to vCJD, including the presence of florid plaques 〚19〛. Recently, small numbers of florid plaques have been reported in occasional cases of iatrogenic CJD in dura mater graft recipients in Japan. None of these cases, however, exhibited any of the other neuropathological features of vCJD 〚20〛. The small cluster plaques in the cerebral and cerebellar cortex appear to be more specific for vCJD and have not been reported in any other from of human prion disease. The rounded amyloid plaques occurring in kuru and in sporadic CJD can be distinguished easily from florid plaques by their restricted anatomical distribution, their compact plaque morphology and the absence of a rim of spongiform change in the surrounding neuropil 〚21〛.
The distribution of neuronal loss and gliosis in the thalamus in vCJD is distinct from fatal familial insomnia 〚22〛 and sporadic fatal insomnia 〚23〛. This may be of interest in relation to the sensory abnormalities in patients with vCJD, which may represent thalamic pain, particularly since there is no histological evidence of a peripheral neuropathy. Further studies are required to assess more precisely the distribution of the pathology in the posterior thalamic nuclei in vCJD, and to relate these changes to the clinical abnormalities occurring in this disorder.
Western blotting analysis of PrPRES in the brain is important in discriminating between CJD and other dementias. Our studies indicate that the PrPRES glycoform ratio can also help distinguish between vCJD and sporadic CJD as reported previously 〚8, 14〛; however, a glycoform pattern similar to that of vCJD is known to characterise fatal familial insomnia 〚24, 25〛 and GSS 〚26〛. Experimental transmission studies using transgenic mice have suggested that this characteristic glycoform ratio will be preserved following BSE infection in humans with valine homozygous or heterozygous PRNP genotypes 〚3〛.
In conclusion, these findings indicate the need for an integrated approach to the diagnosis of vCJD, using a combination of neuropathology, PRNP sequencing and PrPRES analysis 〚27〛. On this basis, there is no evidence to suggest that BSE infection has yet been identified in patients with valine homozygous or heterozygous PRNP genotypes. Continuing surveillance with detailed laboratory investigation of all suspect CJD cases is required in all countries with reported cases of BSE.
Acknowledgements
I thank all the neuropathologists in the United Kingdom, and my colleagues Prof R.G. Will and Dr M.W. Head. I thank Mrs L. McCardle and her staff for technical assistance. The CJD Surveillance Unit is funded by the Department of Health and the Scottish Executive Department of Health.