

1 Introduction
As the human genome has been mostly sequenced on genomic level [1,2], the identification, isolation and functional characterization of the genes and proteins remain the next challenges, especially in view of their role in disease processes. The lack of perfect gene prediction software and the need for physical clone resources of genes and gene products limits the use of the genomic sequence for direct use in functional genomics. The availability of full-length cDNAs has proven to be pivotal for the process of correct gene identification, and cDNAs also constitute the ideal physical clone resources for functional genomics' approaches. The German cDNA Consortium which was established in 1997 [3–5], also has been first to develop and apply systematic high-throughput strategies for the functional characterization of encoded proteins [6].
While the cDNAs analyzed by the German cDNA Consortium have already been successfully employed in single-gene approaches [7–9], their use in high-throughput applications is especially appealing as standardized set ups could be installed where a large number of cDNAs could be analyzed in parallel to generate a huge amount of functional information. We combine this information with existing knowledge (e.g., chromosomal mapping position, disease information) to allow for a comprehensive analysis of individual genes and gene products and their possible involvement in disease processes.
In our initial set-up we exploited the human cDNAs that are generated and sequenced by the German cDNA Consortium [3] by determining the sub-cellular localization of the encoded proteins [6,10,11], but recently we have extended our research to include a variety of functional genomics and proteomics approaches. Our research aims at the comprehensive functional characterization of the majority of the proteins and their splice variants, which have been identified by the German cDNA Consortium.
2 Materials and methods
2.1 cDNA resources and sequencing
cDNA libraries are constructed and systematically sequenced at the DKFZ within the German cDNA Consortium as described previously [3]. In brief, we use mostly the Smart system (Clontech), and size select prior to cloning. The size selection procedure allows for the cloning of long-insert sub-libraries. All libraries are arrayed in 384-well microtiter plates and distributed to the seven sequencing centers in the consortium. 5′ end-sequencing is done with all clones of every plate, ESTs are analyzed for novelty and likelihood to contain the complete open reading frame (ORF) of a gene. Positive clones are released for full-length sequencing. Annotation of finished sequences is done at MIPS [12], sequences are entered into the EMBL sequence database [13]. All clones are made available through the Resource Center (http://www.rzpd.de) via clone@rzpd.de. Annotations are made public also through the web-site of the consortium (http://mips.gsf.de/projects/cdna).
2.2 ORF identification and sub-cloning
The cDNA sequences are systematically screened for the presence of ORFs with the HUSAR-program ‘Frames’ (http://genome.dkfz-heidelberg.de/ ) and the graphical analysis tool ‘ORFfinder’ from NCBI (http://www.ncbi.nlm.nih.gov/gorf/gorf.html). ORFs are manually annotated and analyzed for their probability to be complete. To allow for their functional characterization, we use the Gateway cloning system (Invitrogen) [6,14], which is based on cloning by site-specific recombination. PCR primers for the amplification of ORFs are selected automatically using the PRIDE program [15]. Amplification is carried out systematically by two-step PCR using the Expand High Fidelity PCR System (Roche) to optimize the success rate also of long fragments, the fidelity and the yield of PCR products. Primers for first-step PCR are designed to contain both an ORF-specific and a Gateway compatible segment, which is extended in the second-step PCR to generate the complete recombination sites. The cloned ORFs are sequence verified. For functional analysis, the ORFs are subcloned into Gateway compatible expression vectors. The resulting expression clones allow for protein expression in either bacterial, baculoviral or eukaryotic systems in a native form or as fusion proteins. The ORFs are cloned without a native stop-codon in order to allow the generation of both N- and C-terminal protein fusions.
2.3 Sub-cellular localization
We use the green fluorescent protein (GFP) [16] to pursue systematic experiments in order to unravel the sub-cellular localization of the proteins that are encoded by full-length cDNAs [6]. We produce N-terminal (CFP) and C-terminal (YFP) fusions with derivatives of the GFP since the orientation of the fluorescent tag relative to the ORF of interest in many cases influences the localization of the fusion protein [11]. Plasmids are extracted from bacteria in 96-well format using a Millipore system that has been adapted to a Packard Biosciences Multiprobe liquid handling robot. Vero cells (ATTC CCL-81) are cultured in MEM medium supplemented with 10% fetal calf serum in 35 mm glass-bottom dishes. Transfection and image acquisition is done as described previously [6].
2.4 Functional assays
The GFP-fusion constructs are also employed in functional assays. Two types of assays have been established yet that are compatible with the 96-well format. In a protein transport assay the VSV-G protein is used as a marker for transport to the plasma membrane [17]. Proteins that have an effect on the transport of the VSV-G protein to the plasma membrane are candidate regulators of protein secretion. The proliferation assay relies on the detection of BrdU which is incorporated into the cellular DNA during S-phase of mitosis [18]. Enhanced BrdU incorporation is indicative of a proliferating effect of the analyzed proteins, decreased BrdU incorporation is expected to occur when proteins reduce the proliferation rate of cells.
Pipetting of assays is carried out with help of Packard Multiprobe pipetting robots. Image acquisition is done using a 96-well automated fluorescent microscope. The microscope, which is a prototype developed at the EMBL (http://www.embl-heidelberg.de/ExternalInfo/pepperko/index.html), performs image acquisition and analysis automatically. Statistical evaluation of raw data (e.g., fluorescence intensities in the GFP, BrdU and Dapi channels of the proliferation assay) is done with R-statistical software (Version 1.6.0; Gentleman and Ihaka, University of Auckland).
2.5 Data integration and analysis
We have implemented a Microsoft SQL-server database which serves as a tracking database in the cloning process as well as for the processing of cell-based assays. Raw data and annotation is imported via a Web interface which also allows for example the uploading of microscopic images. The database is made publicly available on-line in a queriable format.
Common identifiers are the clone names, links to external resources are implemented, e.g., the clone request at the RZPD (http://www.rzpd.de), the sequence entry in the EMBL database [13], GoldenPath [19], and ENSembl [20] genome browsers, GeneCards [21] and Omim (http://www.ncbi.nlm.nih.gov/omim/ ). Blastp analysis in the Trembl database [22] is carried out on a weekly basis to provide up-to-date similarity information on the encoded proteins. The localization of fusion proteins within living cells is displayed with microscopic images. The integration of the results from the cell-based assays and from the proteomics approaches is in progress.
3 Results and discussion
3.1 cDNA resources
The German cDNA Consortium (Fig. 1) combines the capacities and competence of several sequencing centers, one of Germany's leading bioinformatics institutes and potent functional genomics laboratories. Biologists, chemists, physicists and informaticians synergistically produce gene sequence and functional information. The cDNA clones originate from 15 human libraries that are enriched in long and full-length cDNAs. ESTs from over 170 000 individual cDNA clones have been generated. The sequences of over 7500 cDNAs have been completely determined, resulting in 24 Mb of high-quality sequence. Our clones, which are distributed by the Resource Center, RZPD, are widely used in the community for single gene [7–9] and large scale approaches [23], as well as in the projects we have established in the consortium.
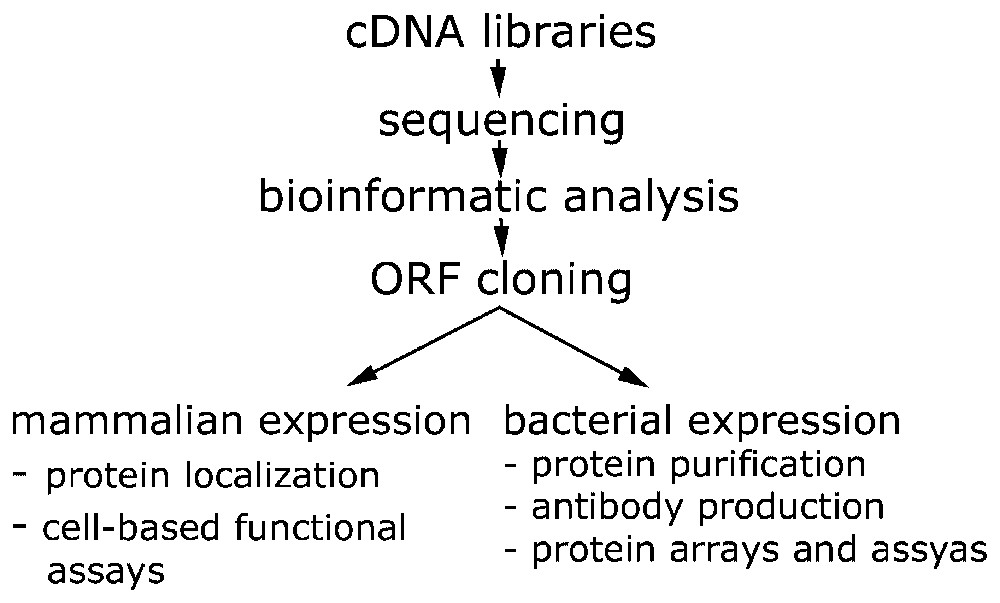
Cloning and data production pipeline within the German cDNA Consortium. cDNA libraries are generated at the DKFZ, sequencing is performed at: AGOWA GmbH (Berlin), DKFZ (Heidelberg), EMBL (Heidelberg), GBF (Braunschweig). Heinrich Heine University (Düsseldorf), Medigenomix GmbH (Munich) and Qiagen AG (Hilden). Bioinformatic analysis and annotation is done at MIPS-GSF (Munich). Functional genomics and proteomics approaches are carried out at the DKFZ and the EMBL.
3.2 ORF identification and cloning
The protein coding regions of over 1100 individual cDNAs have been subcloned into entry vectors and sequence verified thus far. ORFs are cloned lacking the native stop-codon to enable the generation of both N- and C-terminal protein fusions. The ORFs are further cloned to produce CFP and YFP fusion constructs (expression clones) and used in the sub-cellular localization and functional assays. We have modified a number of other expression vectors to be Gateway-compatible to increase the versatility of the entry clones. These vectors allow for bacterial expression with fusions to a variety of tags.
3.3 Sub-cellular localization
The context of a protein determines the activity (e.g., availability of substrates), interaction and function of a protein. The milieu of other proteins, lipids and nucleic acids regulates the potential of the protein to interact with these other molecules. Therefore, the determination of the natural habitat of any protein is a first step towards the understanding of its function [6]. Our results of overexpressing novel human proteins as fusions with the GFP have drastically demonstrated that the position of the GFP relative to the ORF has dramatic effects on the resulting localization [11]. For example, signal peptides of secreted proteins are frequently located at the N-terminus of proteins, which are masked by the GFP fusion when it is located N-terminal to the protein under investigation. However, other proteins carry their targeting signals at the C-terminal end resulting in a mis-localization of fusion proteins that have the orientation ORF-YFP. Consequently we always analyze both, N- and C-terminal fusion proteins in the determination of a protein's sub-cellular localization.
3.4 Functional assays
While knowledge of a potential domain often helps to predict a possible activity and/or localization of a protein, in many cases database searches still do not reveal any similarities to known proteins or domains. However, even the presence of a domain does not immediately provide information on the specific activity or activities and substrate(s) of a protein, but merely guides (or misguides) the investigator towards a possible direction. Instead of only relying on bioinformatic predictions, we aim at the systematic functional characterization of proteins. We initially group the proteins into functional categories by applying cell-based screens. These screens focus on disease relevant cellular processes like proliferation, apoptosis and signal transduction. In order to allow for the parallel analysis of a large number of proteins, these assays need to be simple and robust. Because of the heterogeneity of our set of proteins, the number of assays will be steadily increasing in order to obtain functional data on a growing fraction of the total proteins. The data produced and the results of these assays are published via the Web site that is described below. Once candidates have been identified in these screens, they are deeply characterized to elucidate their cellular roles and possible disease relation (Fig. 2).

Information from a suite of functional genomics and proteomics approaches and bioinformatic analyses are integrated in the LIFEdb database (http://www.dkfz.de/LIFEdb). The work is supported within the German Genome Project (DHGP) and the National Genome Network (NGFN) by the Bundesministerium für Forschung und Technologie (BMBF).
3.5 Data integration and analysis
We have implemented LIFEdb, a Microsoft SQL database, to serve as tracking database for the experimental processes, and which also integrates and disseminates the information that is produced in the diverse projects. Data on protein structure and similarity, protein localization, protein–protein interaction, and functional information from cell assays and protein expression are made publicly available (http://www.dkfz.de/LIFEdb). Expression profiling data [24,25] is integrated to link the information of differential expression of genes in cancer tissues with the functional information that is generated. Over- or under-expression of genes also serves as additional criterion for the selection of primary candidates to be put into the assays. The database serves as tool for the integrated evaluation of the diverse data that are acquired in the experiments. By linking internally generated with external data, the public user of the database is enabled to view functional information in a more global context.
4 Conclusions
We systematically generate and exploit cDNA resources in the German cDNA Consortium. Large-scale genomics in form of the production and analysis of full-length cDNA resources are connected to down-to-earth biology, as these cDNAs are applied in cell biology analysis that we adapt to cope with the large number of clones and proteins.
The overall aim is to identify novel human cDNAs and proteins and to functionally characterize these proteins in as much detail as possible using large-scale approaches and then to concentrate on promising candidates for in depth research. We focus on disease-relevant processes that are mostly connected to cancer in order to connect basic with applied research.
Acknowledgements
The German cDNA Consortium is: Stefan Wiemann, Regina Albert, Petra Moosmayer, Ingo Schupp, Detlev Bannasch, Alexander Mehrle, Ulrike Korf, Ruth Wellenreuther, Annemarie Poustka (DKFZ, Heidelberg); Wilhem Ansorge, Sabine Glassl (EMBL, Heidelberg); Helmut Blöcker, Michael Böcher (GBF, Braunschweig); Karl Köhrer, Andreas Beyer (University of Düsseldorf); Jürgen Lauber, Andrè Bahr (Qiagen, Hilden); Werner Mewes, Clara Amid (MIPS-GSF, Munich); Birgit Ottenwälder, Brigitte Obermaier (Medigenomix, Munich); Dagmar Heubner, Rolf Wambutt (AGOWA, Berlin); Rainer Pepperkok, Jeremy Simpson, Urban Liebel, Veronika Neubrand, Vytaute Starkuviene (EMBL, Heidelberg). Expression profiling data are generated by Holger Sültmann and Wolfgang Huber (DKFZ, Heidelberg). We thank Simone Schleeger, Christian Schmidt, Regina Zahn, Kerstin Hettler, Anny Duda, Sara Burmester, Ute Ernst, Nina Claudino, and Daniela Heiss for excellent technical assistance.
The work is supported by the BMBF with grant 01GR0101 in the National Genome Research Network (NGFN), and with grants 01KW9987 (German cDNA Consortium), 01KW0012 (DKFZ) and 01KW0013 (EMBL) within the German Genome Project (DHGP). Sequencing was supported in part by the EU grant BMH4-CT97–2284 (DKFZ and EMBL).