

1 Background
All organisms respond to changing conditions in their environment by controlling the expression of their genes. Depending upon the particular circumstances, cells can efficiently regulate metabolic pathways by appropriately increasing or decreasing the concentration of specific enzymes. The concentration of these regulated enzymes in turn controls the flux through a given pathway. Escherichia coli, like all organisms, can meet its energy demands by altering enzyme concentrations to take full advantage of the fluctuating food supplies in their environment. When glucose is abundant, the bacterium utilizes it exclusively as its food source, even when other sugars are present in the surroundings. However, when the glucose supplies become exhausted, E. coli has the ability to take up and metabolize alternative sugars such as lactose. The ability of the bacteria to switch from one metabolite to another was described by Monod as diauxic growth [2]. The diauxic growth pattern, illustrated in Fig. 1, occurs when metabolites are used sequentially rather than simultaneously. Monod observed that bacteria prefer glucose as an energy source and only when the glucose supplies are depleted will the bacteria switch to an alternate carbon sources. Understanding at the molecular level how an organism sequentially utilizes metabolites has been a fundamental problem in biology that has attracted tremendous interest over the last fifty years.
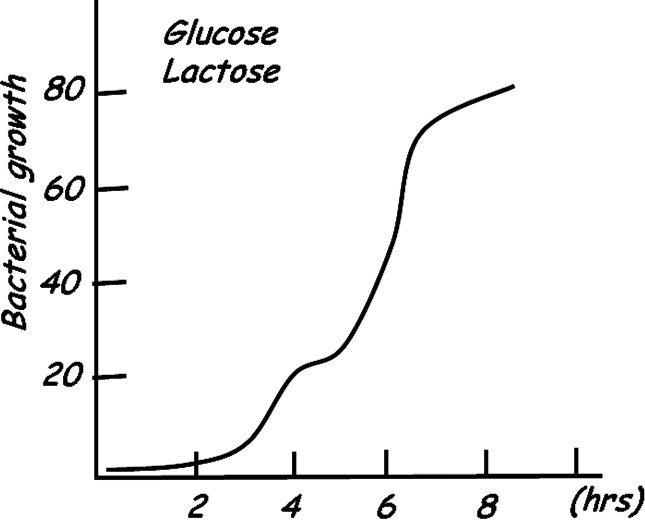
Diagram of diauxic growth of bacterial cultures adapted from Monod [2].
Jacob and Monod [3] conceptually outlined how bacterial cultures could switch their mode of growth from one state to the other so rapidly and completely. They described the operon as a group of structural genes that are coordinately regulated. The structural genes of an operon correspond to a group of proteins or enzymes that are responsible for a particular task or metabolic process. In the operon, the genes are regulated depending upon the metabolic needs of the cell. In order to regulate a gene, or a family of genes in a coordinated fashion, the operon requires a master switch. The switch of the operon is a repressor molecule, which itself is the product of a regulatory gene (R). The repressor associates with a regulatory element, called the operator (O), and controls the synthesis of the structural genes (A, B). A schematic representation of the operon is shown in Fig. 2. Binding of the repressor to the operator negatively regulates or blocks the expression of structural genes of the operon. If the repressor is to function as a switch, it must be inducible; the switch must be able to turn on or turn off in response to a given chemical signal. In this model the repressor not only binds to the operator it also binds an inducer (I), a metabolite that monitors the metabolic state. The inducer is a chemical signal that either directly or indirectly modulates the affinity of the repressor for operator. In the presence of the inducer the repressor dissociates from the operator, which relieves the negative regulation and allows the expression of the structural genes. The switch can also be controlled by positive regulation where a metabolite or co-repressor increases the binding affinity of the repressor for its operator. In both cases, the repressor controls the rate of a metabolic process by increasing or decreasing the concentrations of the structural proteins. Jacob and Monod proposed two possible models for gene regulation. The first model acts as described above while the repressor in Model II acts on messenger RNA rather than operator DNA. Model II, in fact was favored by Jacob and Monod; at the time it was not known that the repressor was a protein. Jacob and Monod based their general model on the system, which they had long studied, the lactose metabolism of Escherichia coli (E. coli).
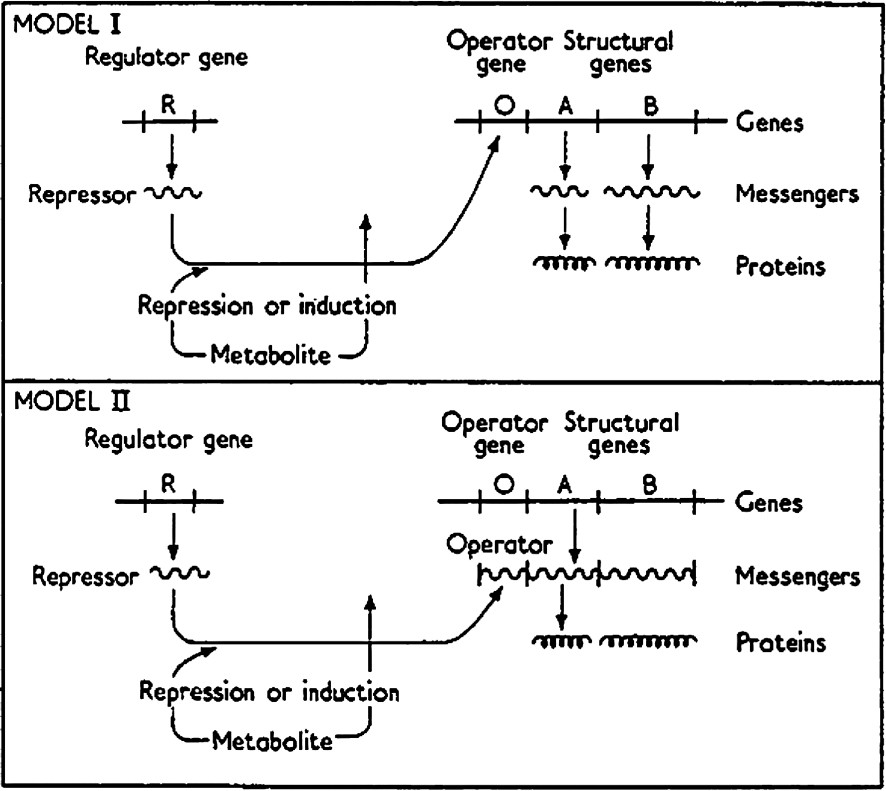
Diagram of the operon model [3]. Model I: Jacob and Monod predicted the existence of a ‘repressor’ molecule that would be produced from a regulator gene (R), would bind to an operator site (O), and hinder transcription of a set of structural genes (A, B) that follow the operator site. The repressor could also bind an inducer (I) that could lower or eliminate the affinity of the repressor for operator and allow transcription of the set of structural genes. Model II: Same as Model I except the repressor binds to messenger RNA (mRNA) rather than operator gene.
When the primary carbon source in the growth medium is lactose, the repressor is induced thus allowing transcription of three structural genes that coordinate lactose utilization. The structural genes regulated by lac repressor are the lacZ, lacY, and lacA, which code for three proteins involved in lactose metabolism, β-galactosidase, lac permease, and a transacetylase, respectively. β-galactosidase acts to cleave lactose into galactose and glucose, the first step in lactose metabolism, lac permease is a transmembrane protein, which helps lactose get into the cell, and transacetylase transfers an acetyl group from coenzyme A (CoA) to the hydroxyl group of the galactosides. Although the transacetylase is not essential for lactose metabolism, it is physiologically important for maintaining the viability of the cell. When the metabolite, lactose, is the primary carbon source in the growth medium, the repressor is induced thus allowing transcription of three structural genes that are necessary for lactose utilization. If lactose is to be used as a carbon source for energy production then the transcription of the three structural genes needs to be up regulated in order to increase the flux through the pathway. Lactose simultaneously induces the production of β-galactosidase, permease, as well as acetylase, from just a few molecules per cell to several thousand molecules per cell. The idea that metabolism can be regulated at the genetic level was of course an important conceptual breakthrough, as was the notion that regulatory genes could sense the environment and respond accordingly.
A year after Jacob and Monod received their Nobel Prize for their contributions to gene regulation, Muller-Hill and Gilbert isolated the repressor [4]. This was a difficult task; the concentration of the repressor in the cell is vanishingly low and constitutes less than 0.002% of the protein in a bacterial cell. In order to isolate the molecule, Muller-Hill and Gilbert genetically altered the bacterial system to increase the relative amount of repressor made in the cell by several orders of magnitude [5]. They screened for a mutation that would increase the constitutive expression of the repressor. The mutation altered the promoter region of the lacI gene. Once the concentration of the repressor was sufficiently elevated, the protein could be monitored and isolated using standard techniques. Preliminary studies suggested that each monomer was composed of 347 residues and the repressor associates as a tetramer [6]. However, with the sequencing of the gene, it was observed that a dozen amino acids had been missed [7]. The lac repressor is a protein of 360 amino acids that associates into a homotetramer with a 154 520 Dalton molecular mass. Further analysis revealed that the repressor has a modular structure and when the repressor is cleaved, by a limited protease digestion, it is cut into distinct fragments [8]. The tetrameric repressor dissociates into four NH2-terminal fragments (∼60 residues) that bind specifically to operator DNA and a COOH-terminal tetrameric ‘core’ that binds inducers. Connecting the amino- and carboxy-terminal domain is a portion of the structure, which is referred to as the hinge region because it is extremely susceptible to proteolytic cleavage and probably lacks structure. The last 30 amino acids of the core are partially responsible for the oligomeric state of the repressor and essential for tetramerization.
The operator of the lac operon, originally identified from cis-acting constitutive mutants, is located between the end of the lacI gene and the beginning of the lacZ gene [1]. Mutations in the operator region either greatly reduce or eliminate the ability of the repressor to regulate transcription. The primary operator site for the lactose operon was isolated by Bourgeois and Riggs [9]. The actual size of the operator was established by digesting the DNA with pancreatic DNase in the presence of the repressor [10]. The operator fragment protected by the repressor from digestion was about 27 base pairs, and it encompassed all of the known operator constitutive mutations. Subsequently, Gilbert and Muller-Hill demonstrated that the repressor directly binds to the operator by radiolabeling the repressor and observing that it sediments with DNA containing the lac operator [10]. When the repressor was combined with DNA that contained operator constitutive mutations it would no longer co-sediment. Similarly, in the presence of inducer, the repressor and the DNA do not form a stable complex. More than ten years after the publication of the Jacob and Monod model, Gilbert and Maxam [10] sequenced the 27-base pair section of double stranded deoxyoligonucleotides (Fig. 3). The operator is pseudosymmetric, or possesses an approximate dyad axis, about a central G⋅C base pair [10]. The minimal operator required for specific binding was later shown to be only the 17 base pairs in the center of the Gilbert and Maxam 27-base pair sequence [11] (shown in bold type in Fig. 3A). Many of the operator mutations reduce the apparent symmetry of the wild type operator sequence. Moreover, mutations in the left half of the operator site appeared to be more deleterious to repressor binding than the mutations in the right half of the operator, suggesting that the left half site contributes more to the binding affinity than the right half site. Based upon these observations, Sadler and Betz concluded that the natural operator is ‘flawed’ with respect to DNA binding and demonstrated that a completely symmetric DNA fragment binds 10-fold more tightly to the repressor than does the natural sequence [12]. This tight-binding fragment contains a true inverted repeat and has a sequence that corresponds to the left half of the natural operator (Fig. 3D). As described below, in addition to the primary operator , the lactose operon of E. coli has two ancillary operators, and [13]. Fig. 3B illustrates the position of the 3 operators with respect to the other elements of the operon, including the promoter site where RNA polymerase binds to initiate transcription, and the catabolite activating protein (CAP) binding site. The repressor binds specifically to an operator and is centered 11 bp downstream from the start of transcription of the gene for β-galactosidase [14]. Fig. 3C shows the nucleotide sequences of the three operators, illustrating their similarities and differences.
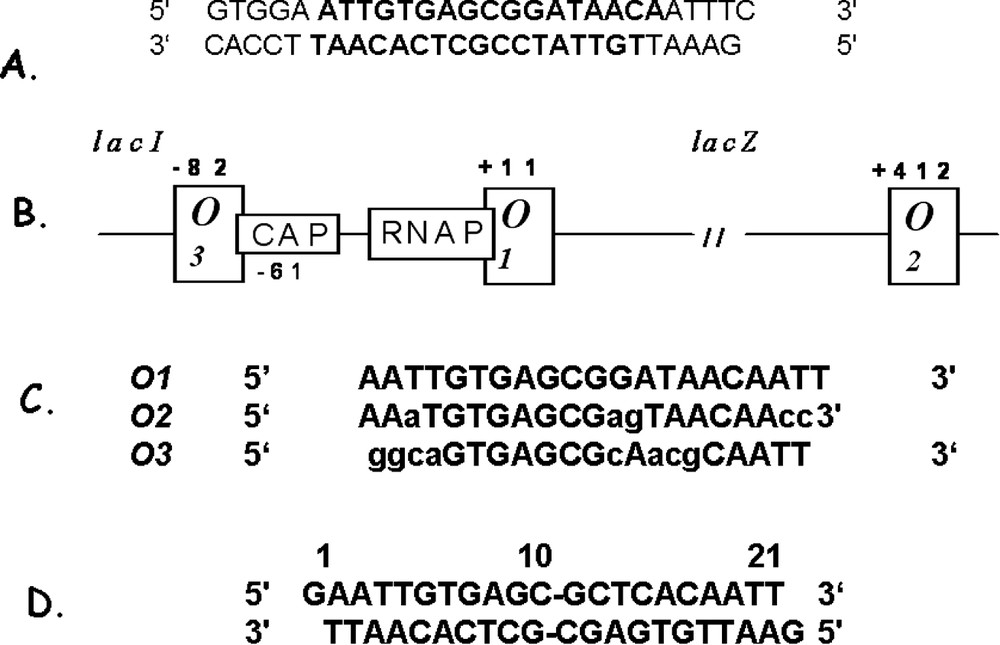
The operators of lactose operon system. (A) The deoxyoligonucleotide first identified (in bold, Bourgeois and Riggs [9]) and sequenced (Gilbert and Maxam [10]) as the operator (O) region of the lactose operon. (B) Diagrams the operon with the location of the three operators shown. (C) The sequences of the three operators are shown aligned with the center of each operator in bold type. Bases are listed with capital letters are identical to bases in the primary operator . (D) The sequence of DNA duplex which binds to repressor with higher affinity than wild-type operator [12]. This sequence lacks the central base and is a palindrome of the left half-site of .
Inducer and anti-inducer molecules alter the equilibrium between the induced and the repressed states. The inducer molecules reduce the affinity of the repressor for its operator while anti-inducers do the contrary. The natural inducer of the repressor molecule is allolactose, an analog of lactose created by a side reaction of β-galactosidase [15]. With the inducer bound, the repressor binds to the operator with a greatly reduced affinity, which allows the polymerase to bind its promoter and transcribe the genes necessary for lactose utilization. A gratuitous inducer, 1-isopropyl-β-d-thiogalactoside (IPTG), was discovered by Monod [3], and although it is not a substrate for β-galactosidase, it can be used to ‘turn on’ transcription of the lactose operon. The equilibrium between the induced and the repressed states is also affected by anti-inducer molecules. An anti-inducer binds to the repressor and performs the opposite function; it effectively ‘turns off’ or prevents transcription by increasing the stability of the repressor–operator complex. The most potent anti-inducer, orthonitrophenyl-β-d-fucoside (ONPF), is a galactoside and increases the affinity of the repressor for the operator [16]. Interestingly, the anti-inducer has no known regulatory function in E. coli and is not a naturally occurring metabolite. So how do these molecules alter the ability of the repressor to bind the operator and perform its biological function?
The repressor undergoes a conformational transition in response to bound ligands. Monod, Changeux, and Jacob described the structural changes that result when a ligand associates with the protein and alters its ability to perform a given function and coined the term allostery [17]. In theory, an inducer or a co-repressor changes the conformation of the repressor such that different conformations of the repressor have different binding affinities for the operator DNA. If the repressor is allosterically regulated, then it has the ability to react to its environment. In the absence of lactose, the repressor functions as a negative regulator. It binds to the operator and prevents transcription of the genes that code for lactose utilization. Only when the inducer, lactose, is present in the environment does the repressor adopt an altered conformation, which decreases its affinity for the operator. The inducer molecule, therefore, relieves repression of the operon by altering the repressor–operator equilibrium [18]. The rather low dissociation constant of lac repressor for operator DNA ( at a low salt concentration [19]) indicates the repressor binds to its operator DNA with very high affinity. Upon binding the gratuitous inducer IPTG, the affinity of lac repressor for DNA is lowered 1000 times ( [20]). The sigmoidal shape of the binding curve, shown in Fig. 4, over a small range in concentration of added inducer, illustrates the fact that the repressor is an allosteric protein. Binding of a small molecule alters the structure and its ability to perform its biological role. In the lac system, these small molecules or inducers act as couriers of external molecular signals. Inducer binding affects the DNA binding of the repressor by decreasing its affinity for the operator.
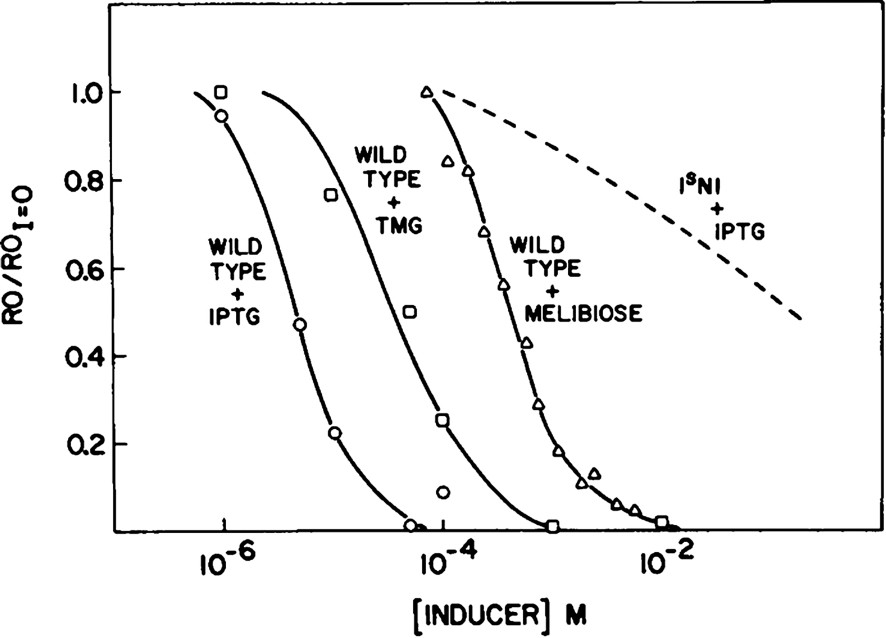
The allosteric nature of lac repressor (Barkley and Bourgeois, [18]). The fraction of repressor bound to operator is plotted as a function of inducer concentration.
The diauxic growth of bacterial cultures observed by Monod remained baffling until the discovery of cyclic AMP and the observation that this secondary messenger indirectly activates the lac operon [21]. Transcription of the operon is positively activated by a cyclic AMP-dependent catabolite gene regulator protein (CAP) (see Fig. 3B). In glucose starved cells, the level of cyclic AMP increases dramatically. The cyclic AMP binding to CAP increases its ability to bind to its activator site. CAP binding, in turn, activates transcription of the lac operon by increasing the affinity of RNA polymerase for its promoter [22]. When the bacteria are given both glucose and lactose, the cells will preferentially metabolize glucose. Even though the repressor is induced, the operon is not activated until the glucose is depleted. Only when the glucose levels in the cell are low and the cAMP levels are elevated will transcription of the enzymes necessary for lactose metabolism be activated. This combination of repression and activation accounts for the diauxic growth originally observed by Monod.
2 Mutational analysis of the repressor
In the absence of a structure, Miller and coworkers [23–25] embarked on a Herculean genetic analysis to learn more about the lac repressor. They created over four thousand single amino acid substitutions using suppressors of nonsense mutations. At each specific site in the repressor, nonsense suppressor tRNAs were used to replace every amino acid in the protein with different amino acids. The two most common types of mutations that occur in genes are missense and nonsense mutations. Missense mutations change a codon specific for one amino acid to a codon specific for another, while mutations that change a codon to a termination codon produce nonsense mutations. Both nonsense and missense mutations can be suppressed by mutant tRNAs. A genetic analysis of the repressor was performed by intentionally replacing every codon in the repressor gene with a nonsense mutation (usually the amber UAG mutation). Both natural and synthetic tRNAs were used to insert a range of amino acids at the UAG sites. A total of 14 suppressor tRNAs allowed 12 or 13 distinct amino acid substitutions to replace every amino acid in the repressor, from residue 2 to 329 [25]. Fig. 5 is a graph that summarizes the phenotypic behavior of the 4000 single amino acid substitutions in the lac repressor generated by Miller.
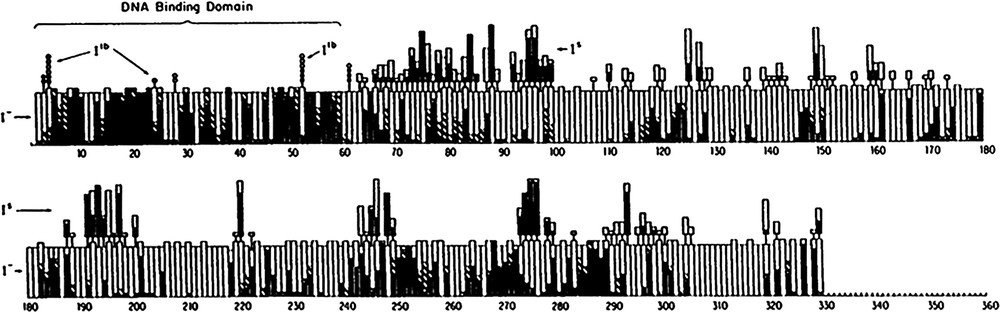
Summary of genetic analysis on E. coli lac repressor taken from Markiewicz et al. [25]. The results of over 4000 single amino acid substitutions are displayed. The height of the bar represents whether the number of amino acids substitutions was 12 or 13. When an amino acid substitution results in an phenotype, the corresponding section of the bar graph is filled in black. Substitutions which give intermediate results for phenotype are shown in stripes on the bar graph. mutations are marked in the main bars of the graph, while and mutants are listed above the main bars.
Repressor molecules that can no longer bind operator DNA have an phenotype. Mutant repressor molecules that fail to bind to the operator allow the cells to constitutively express β-galactosidase in vivo. These repressors display an altered phenotype; they produce blue colonies when grown on indicator plates that contain 5-bromo-4-chloro-3-indolyl-β-galactopyranoside (X-gal). Mutations that alter operator binding and change the phenotype can occur for a number of reasons. Changing the amino acids that are directly involved in DNA binding will create mutant repressor molecule that are incapable of recognizing the operator site. Similarly mutations that alter the kinetics or the thermodynamics of protein folding indirectly affect binding operator DNA. The mutations are not uniformly scattered throughout the sequence; there are large stretches of the sequence that are quite tolerant of substitutions and do not alter the phenotype and there are other regions that appear to be particularly important for repressor function. As seen in Fig. 5, there are particular stretches of sequence that are particularly susceptible to mutations and other regions that are more permissive. For example, the N-terminus, residues 1–60, is essentially intolerant of substitutions and the vast majority of the amino acid substitutions abolish the repressor's ability to bind DNA. In addition, mutations between residues 240 and 290 are significantly more likely to alter the repressor's ability to bind DNA than elsewhere in the protein. However, in the absence of the three dimensional structure it is difficult to deduce the functional significance of the vast majority of these substitutions.
Amino acid substitutions can also produce repressor molecules that do not respond to inducer molecules; they are referred to as super repressors and have an phenotype. Super repressors bind to the operator like the wild-type but do not respond to inducer. On indicator plates that contain IPTG and X-gal, the wild-type repressor produce blue colonies as a result of normal induction, but cells that contain mutant repressor molecules produce white colonies. The altered repressors do not release the operator and consequently repress the production of β-galactosidase. These mutations are scattered through the linear sequence of the repressor, although there appears to be some regions of the molecule that are more sensitive to substitutions than others. The phenotype will be observed if the mutant repressors have either lost their ability to bind the inducer or they are incapable of transmitting the allosteric signal. In the presence of inducer, these mutant repressors remain bound to the operator and therefore prevent polymerase from transcribing β-galactosidase. In the absence of structural data it is not possible to determine which mutations alter inducer binding from those that alter the signaling. Mutant repressors that no longer bind to the inducer molecule, or cannot propagate the signal from the inducer binding site to the operator binding motif have the same phenotype. As described below, the mutational data in conjunction with the structure provides important insight as to how the repressor functions and the molecular basis of allostery.
3 The three-dimensional structure of repressor
The three-dimensional structure of the lac repressor provides important clues at the atomic level as to how the repressor performs its biological role. In the early seventies several hundred milligrams of the repressor were purified and used for crystallization studies in many laboratories around the world [26]. Yet the three-dimensional architecture of the repressor remained elusive until the structures of proteolytic fragments of the NH2-terminal DNA binding domain [27] and the COOH-terminal tetrameric core bound to inducer [28] were determined. These structures provided the first insight into how the repressor functioned. The structure of the DNA binding domain showed that the repressor contained the helix-turn-helix (HTH) motif that was observed in other proteins that bind specifically to DNA [29]. The structure of the repressor's core demonstrated that the inducer binding domains resembled the periplasmic binding proteins and that molecules associate to form stable dimers. The dimers then further associate to form the functional tetramers that are held together by a four helix bundle. Shortly there after, the three-dimensional structures of the intact lac repressor, the lac repressor bound to the gratuitous inducer IPTG, and the lac repressor bound to symmetric operator DNA were elucidated [30]. Together these structures provided insight into how the repressor may function, as well as the three-dimensional framework for interpreting a plethora of biochemical and genetic information. Most importantly, when the biochemical and genetic data were viewed in the context of the structure, a detailed molecular model could be constructed to provide a physical basis for the allosteric response and a more detailed understanding of the genetic switch.
The repressor folds into four discrete functional units. The intact repressor monomer, illustrated in Fig. 6, consists of an NH2-terminal domain (shown in red), a hinge region (shown in yellow), a sugar binding domain (shown in blue), and a COOH-terminal helix (shown in purple). The NH2-terminal domain or ‘headpiece’ of the lac repressor contains a helix-turn-helix motif that is responsible for interacting with the operator. The headpiece is a small, compact globular domain with a rich hydrophobic core that is created by three α-helices. The first two helices form the classical HTH motif (residues 6 to 25). A linker (residues 46 to 62) connects the DNA-binding domain to the core of the repressor. This segment of the polypeptide chain, referred to as a ‘hinge’, which was thought to be devoid of secondary structure, is ordered in the presence of DNA and forms an α-helix that makes specific interactions with the lac operator DNA and also orients the headpiece. A coil-to-helix transition of the hinge occurs in the presence of DNA, when the repressor associates with the operator. In the absence of operator DNA, the hinge helices are disordered, giving the headpiece a broad range of structural freedom. The core of the repressor or sugar binding domain is composed of two subdomains (colored light and dark blue) that are topologically similar. Each subdomain contains a six stranded parallel β-sheet that is sandwiched between four α-helices. The two subdomains are structurally very similar despite the fact that there does not appear to be homology at the level of the amino acid sequence. The subdomains can be overlaid and although the superimposed is not exact (the rms errors in the alpha carbon positions is 1–2 Å), it is visually obvious that there is structural homology. The topology and the fold of these domains are not unique and, as illustrated in Fig. 7, the domain architecture is remarkably similar to flavodoxin. However, the subdomains are not completely continuous with respect to the primary sequence. For example, the N-terminal subdomain is created from residues 62 to 163. The polypeptide chain then forms the C-terminal subdomain, residues 164 to 292, before returning to the N-terminal subdomain to form an alpha helix (helix 13) and a beta strand (strand K). As illustrated in Fig. 8A, with respect to flavodoxin, helix 13 in the NH2-subdomain appears to have been swapped or exchanged with helix 8 of the C-terminal subdomain. The swapping of helices could be related to the evolution of the repressor. In Fig. 8B the intact monomer of lac repressor is redrawn to emphasize the position of helix 13 in the NH2-subdomain and helix 8 of the COOH-terminal subdomain. Perhaps in the past, single subdomains existed as common monomeric structures or folds, like flavodoxin, until a new function was needed which required the presence of multiple domains. The structure of the repressor illustrates that the molecule is modular and the domains are functionally self-contained.
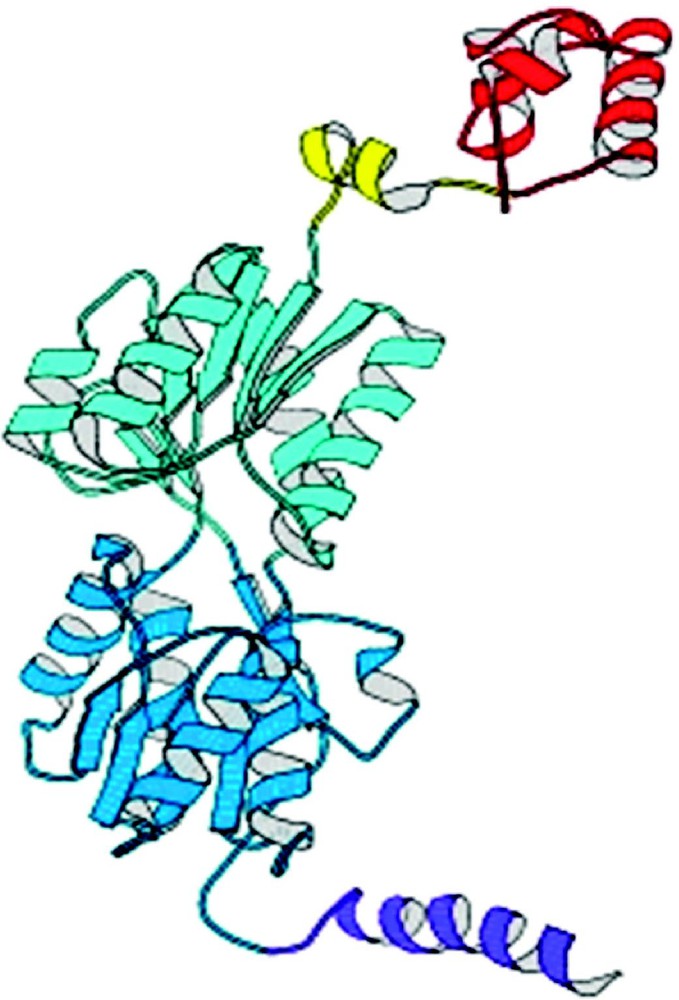
A view of the lac repressor monomer from the complex with DNA from Lewis et al. [30]. Four functional domains occur in repressor monomer: starting from the N-terminus (1) the DNA binding domain or headpiece (residues 1–45) is colored red; (2) the hinge region (residues 46–62) is yellow; (3) the ligand binding domain or core (residues 63–329) which has distinct N- and C-terminal sub-domains are colored in two shades of blue; and (4) the tetramerization helix (residues 340–357) is purple.
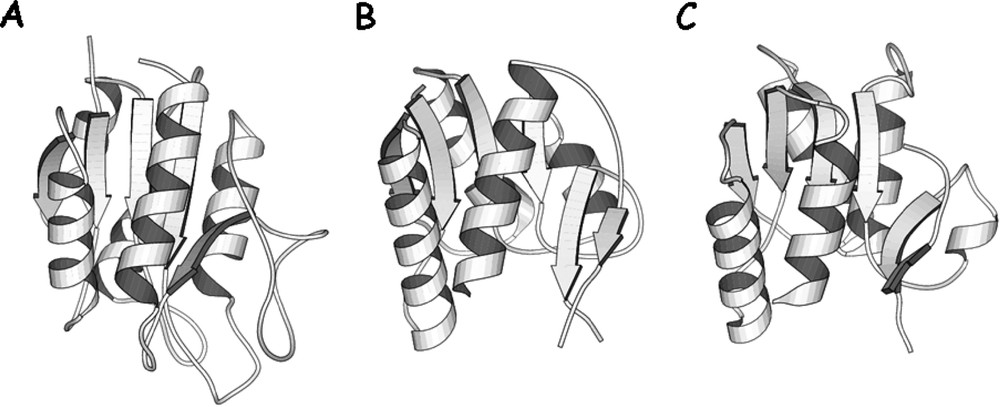
Comparison of domain folds. (A) The structure of oxidized flavodoxin from Anabaena (PDB entry – 1flv) (B) and (C) the NH2-terminal and COOH-terminal subdomains of the core of lac repressor. The subdomains are in approximately the same orientation to illustrate the similarity of the fold.
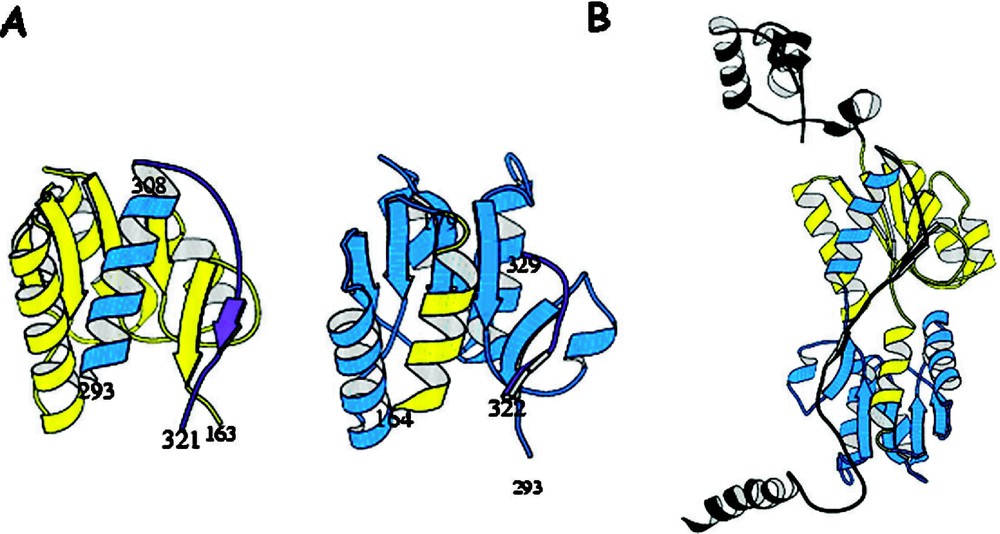
Comparison of the subdomains of the core of lac repressor redrawn to emphasize the discontinuously folded sections. (A) The N-terminal and C-terminal subdomains of the core. Colored yellow are the residues in the primary sequence at the N-terminus and the blue are residues from the C-terminus of the sequence. The figure illustrates that a helix from the C-terminus is used to form part of the N-terminal subdomain and similarly a helix from the N-terminus of the sequence is used to form the C-terminal subdomain. (B) is the structure of the repressor monomer illustrating the covalent domain swapping in lac repressor between subdomains of the core.
The quaternary structure of the lac repressor is an unusual tetramer. The repressor forms stable dimers that are tightly held together through an extensive interface. The interface between two monomers of a dimer is quite extensive, and buries ∼2200 Å2 of surface area. There are five principal clusters of amino acids create this dimer interface: residues 70 to 100, 221 to 226, 250 to 260, and 275 to 290. The buried surface area at the monomer–monomer interface is nearly equally distributed between the interface of the N-terminal and C-terminal subdomains. With the exception of residues 250 to 260, point mutations within these clusters result in monomeric forms of lac repressor. The dimers then associate to form the tetramers that are created by the self association of the C-terminal α-helices (residues 340 to 357). Each helix contains two leucine heptad repeats, which are responsible for the association and the formation of a four helix bundle. The repressor does not maintain the point group symmetry of other oligomeric proteins of known structures and is essentially a dimer of dimers that appears to be roughly V-shaped (Fig. 9). By contrast, virtually all homotetramers of known structure have three mutually perpendicular two-fold axes. Apart from the helical bundle, the contacts that stabilize the tetramer are quite tenuous and there are very few interactions between the oligomerization domain and the core domain to maintain this specific quaternary structure. The arrangement of strong dimer contacts and weak tetramer interactions suggests that the observed tetrameric structure of the repressor is essentially a tethered dimer. There are no obvious reasons why the two dimers associate with this particular geometry and one might expect that the pair of tethered dimers could adopt a variety of conformations. Comparing the quaternary structures of repressor from a variety of crystal forms suggests that the orientation of the two dimers is not fixed precisely, and repressor dimers are likely to adopt a number of alternate conformations.
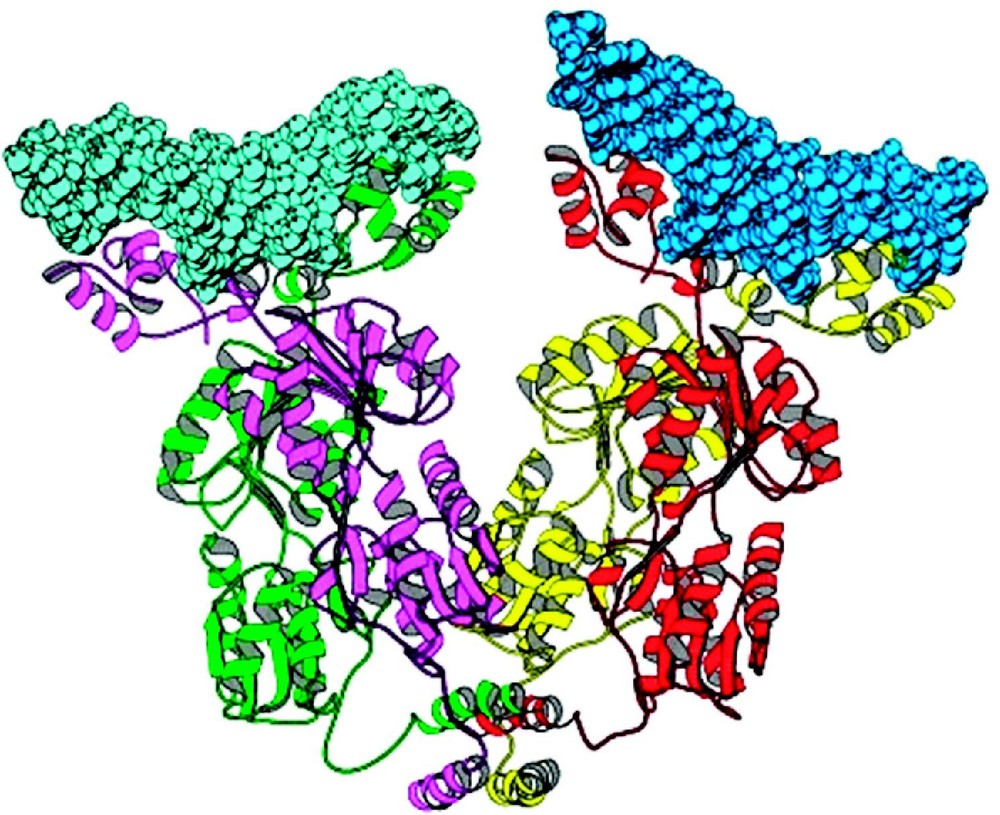
A ribbon diagram of the quaternary structure of the lac repressor complexed to DNA. Lac repressor is a tetrameric structure where each monomer is drawn in color. Each dimeric repressor binds to a 21 base pair duplex deoxyoligonucleotides (in space filling representation). The observed lac tetramer appears to be a tethered dimer of dimers. Adapted from Lewis et al. [30].
With every amino acid substitution there is some probability that the phenotype will be altered. As seen in Fig. 10A, the amino acid substitutions that result in an phenotype are scattered throughout the linear sequence of the protein. There are, however, a surprisingly large number of positions that tolerate substitutions quite well and close to 80% of the substitutions had little or no effect on the functioning of the repressor. When the mutations are mapped onto the structure it becomes obvious that those amino acid substitutions that alter the phenotype are not randomly dispersed throughout the protein but appear to cluster. There are 42 amino acid positions that are intolerant of substitutions and over half of these residues are located within DNA binding domain and the hinge helix.
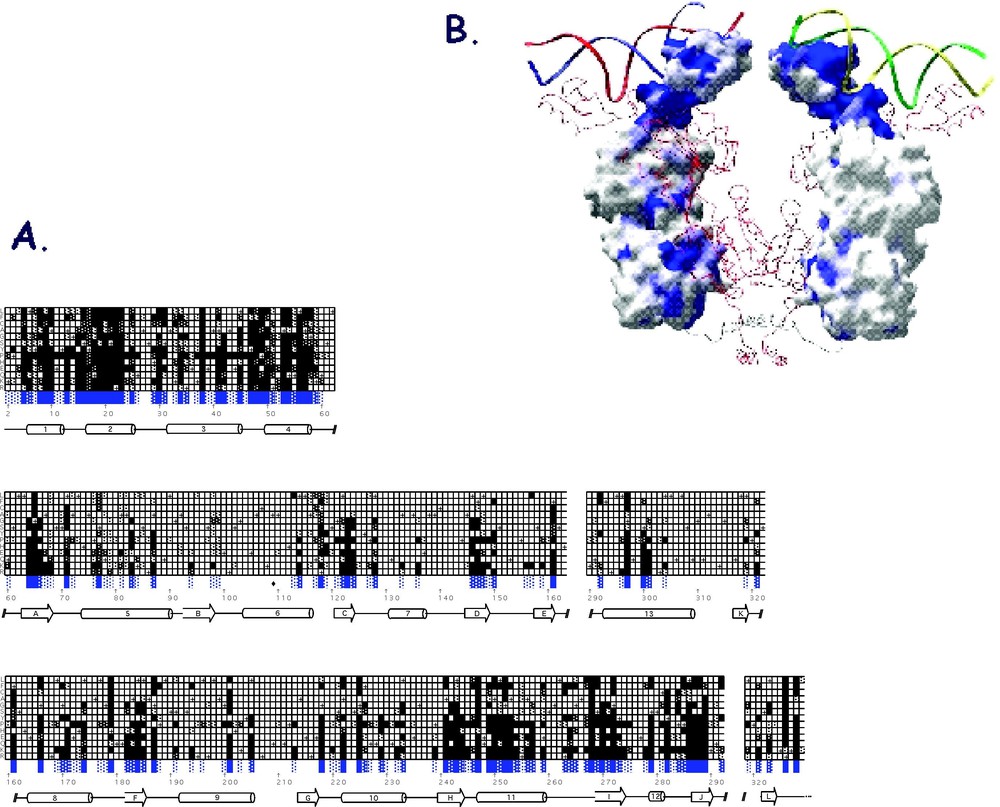
(A) A checkerboard that illustrates the genetic analysis data for I-mutants. The I-mutants are represented as three bar graphs corresponding to the DNA binding domain, the N-terminal subdomain and the C-terminal subdomain. The secondary structure is represented by cylinders and arrows. The vertical axis shows the single letter code for each of the 13 amino acids substituted and each square is shaded depending upon the observed phenotype. Black are ; dark gray are ; light gray indicates ; and white are positions that are tolerant of substitutions. The wild-type residues are shown with a +. B. The mutations are mapped onto the surface of the repressor. The location of mutants are shown in blue. (The figure was made by Helen Pace using GRASP [77].)
The headpiece and the hinge region of the repressor are the most sensitive regions of the repressor with respect to DNA binding. The majority of the mutations in the headpiece alter the protein's ability to recognize the operator. Mutations in over 90% of the residues in the headpiece and the hinge helix alter the phenotype of the repressor. Some of the substitutions that alter the repressor phenotype are on the surface of the repressor and are directly involved in recognizing the DNA, while other substitutions affect buried amino acids that are responsible for maintaining the structural integrity of this domain. Since this domain is absolutely essential for making specific contacts with the operator, it is not surprising that most of the amino acid substitutions in the headpiece alter the repressor phenotype and destroy its ability to bind DNA.
Mutations in the core of the repressor can also result in a defective repressor molecule. Most of these mutations affect residues that are buried and are responsible for maintaining the integrity of the folded state. Within the core of the repressor there are 115 amino acids that are completely buried that are sensitive to substitutions. Since the two sub-domains that create the core of the repressor are approximately equal in size, it might be anticipated that mutations in these two domains would display a similar number of mutations. However, the N-terminal sub-domain is less affected by mutations than is the C-terminal sub-domain. Approximately 40% of the residues in the N-terminal sub-domain show some sensitivity to substitutions while 55% of the residues in the C-terminal sub-domain show the same level of sensitivity. However, the structure illustrates that in order to maintain a functional repressor dimer, the C-terminal sub-domain of the core must be correctly folded and a distortion of the internal structure would prevent the formation of repressor dimers. Only a small number of mutated surface residues within the core displays an phenotype (Fig. 10B). The molecular surface of the repressor monomer is created by 92 amino acids. Amino acid substitutions at only 14 positions alter the ability of the repressor to bind DNA and appear to have an phenotype. Of the 14 amino acid positions, 12 are at the interface between the C-terminal subdomains. Although the interactions between the N-terminal subdomains are important for allosteric signaling (described below), the C-terminal domains appear to be most important for creating a stable oligomer. Altering specific key surface residues at the subunit interface prevents the formation of the functional dimer and thereby inactivates the repressor. Analogous to the buried residues within the monomer, these surface residues, buried within the dimer interface, are essential to the overall oligomeric structure. The tetrameric repressor is established by the association of the C-terminal helices (residues 322–360). However mutations in the helix have little or no effect on the repressor phenotype and therefore the repressors' ability to bind to operator. While it is essential for the repressor to form stable dimers, the tetrameric interface is far less important. Mutant repressors that are devoid of the C-terminal helix and cannot form a tetramer are essentially indistinguishable from the native molecule, and the dimeric repressors perform as well as tetramers in genetic screens.
Some suppressor substitutions are more detrimental than others. Substituting proline and lysine alters the phenotype more frequently than other suppressor mutations. Changing each amino acid within the core to proline alters the repressor in a fashion that is distinct from all of the other suppressors and is the most lethal amino acid substitution. Proline substitutions at 257 positions in the repressor core produce 122 defective repressor molecules. Lysine and arginine substitutions are not tolerated well and alter the phenotype about 35% of the time. In contrast, replacing amino acids in the repressor with alanine or cysteine are well tolerated and these substitutions result in a change in phenotype less frequently than any other substitution.
4 Interactions between repressor and operator
The first structures of the lac repressor bound to DNA were determined in solution using the headpiece domain with a half operator site. The structure of this complex identified the key residues that were responsible for the specific recognition of the operator and observed that the DNA adopts the canonical B-form [31]. The conformation of the DNA, however, appeared quite different in the crystalline state. The first crystal structures of the repressor bound to DNA was determined using an ‘ideal-operator’ sequence, which has perfect palindromic symmetry and binds to the repressor with 10-fold higher affinity than the wild-type operator. The structure of the repressor bound to this operator sequence confirmed that the HTH motif fits snugly in the major groove and is consistent with the previous solution studies as well as a variety of biochemical studies [10,32]. The interactions of the headpiece with the symmetric operator bury over 3300 Å2 of solvent-accessible surface area, suggesting that their molecular surfaces are highly complementary. Fig. 11 illustrates the binding of the headpiece to the operator. Unexpectedly, binding of the repressor to this 21-base pair symmetric operator alters the conformation of the DNA. The operator fragment bends away from the protein with an approximate radius of curvature of 60 Å. As a consequence, the DNA is somewhat distorted from the canonical B-form. In the center of the operator, there is a bend or kink of that opens the minor groove. The width of the grove increases to over 11 Å and there is a significant reduction in the depth of the groove to less than 1 Å. The central portion of the operator has a helical rise and a twist angle of 6.1 Å and 22°, respectively. The average helical parameters that describe the conformation of the DNA in the environment of the HTH are consistent with the canonical B-form. Further solution studies, confirmed those observed in the crystalline state. When the solution measurements were made using the full operator and a headpiece domain that contained the hinge region (residues 1 to 60), the bending of the DNA was observed [33]. The solution studies and the crystallographic studies are consistent and demonstrate that the repressor bends this operator fragment and the deformation of the operator requires both a complete operator site and more than just the HTH motif of the headpiece.
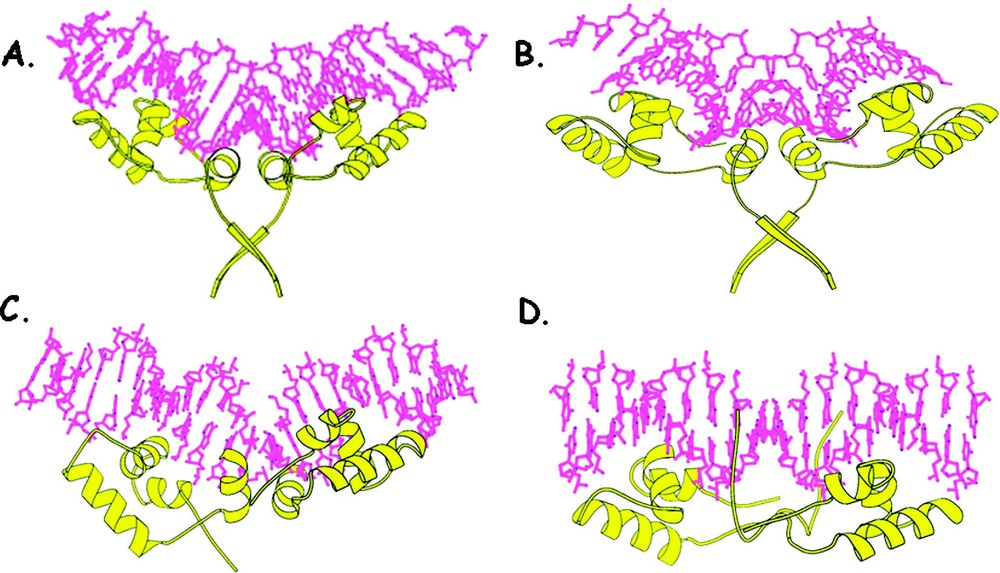
The lac repressor–DNA complex (residues 1 to 68) is shown in yellow. (A) illustrates the binding to the repressor to the symmetric operator. The HTH motif fits snugly in the major groove of the operator DNA and the hinge helices bind and expand the minor groove of the operator. (B) shows the binding to the repressor to the natural operator as seen in the crystal structure. (C) shows the binding of the repressor to the natural operator, as seen in solution. (D) illustrates the binding to the repressor to a non-specific sequence, as seen in solution.
The repressor forms a network of interactions with the operator. The headpiece domains of the repressor form specific interactions with bases in the major groove as well as electrostatic interactions with the phosphate backbone. Residues on the first helix of HTH motif participate in numerous sequence-specific contacts in the left half site but not in the right half site. The second helix of the HTH motif is essential for specificity. Tyr17 and Gln18 are key residues and form hydrogen bonds directly to the bases. In addition, the side chain of Arg22 interacts favorably with a particular base, and there are a number of interactions towards the ends of the operator involving His29, Ser31, and Thr34 [34, Spronk, 1999 #724]. The repressor also interacts with the bases in the minor groove of the operator. When the dimeric repressor binds to this operator, the hinge region forms an α-helix and the helices self-associate to form a structural unit that binds to the minor groove of the operator. Gln54 and Asn50 contact the phosphate backbone of the operator and form nonspecific electrostatic interactions. The most notable feature of the binding of hinge helices are a pair of leucine residues at position 56 (one from each monomer) that make direct contacts with the bases in the minor groove of the operator. These leucine residues are in close proximity to the center of the operator DNA and appear to work as a lever to pry open the minor groove. The repressor binds to the major groove of the operator by recognizing a specific sequence using key amino acids that are localized to the HTH motif. Additional interactions are formed by the hinge helix in the minor grove. The repressor interacts with its operator by recognizing bases in both the major and the minor grooves and it appears that insertion of the hinge helices is responsible for distorting the conformation of the operator.
The stability of the repressor–operator complex is further increased by interactions between the headpiece and the core. There are extensive protein–protein interactions between the N-terminal subdomain of one monomer and the DNA-binding domain of the dimer related monomer [34]. At this interface, the short loop connecting the headpiece domain and hinge helix of one subunit of the repressor, residues 46–51, contacts the dimer related molecule, residues –, which forms the end of helix six in the N-terminal subdomain (Fig. 12). More than 1700 A2 of surface are buried at the interface between the headpiece and the core. Mutations of residues at this interface, in particular Arg118, produce repressor mutants that have an phenotype (non-operator binding) and suggests that these interactions are important in stabilizing the operator-bound conformation. In addition, these interactions may also be important to the allosteric mechanism since they are responsible for the orientation of the N-terminal domain and the conformational transition between the induced and the repressed states.
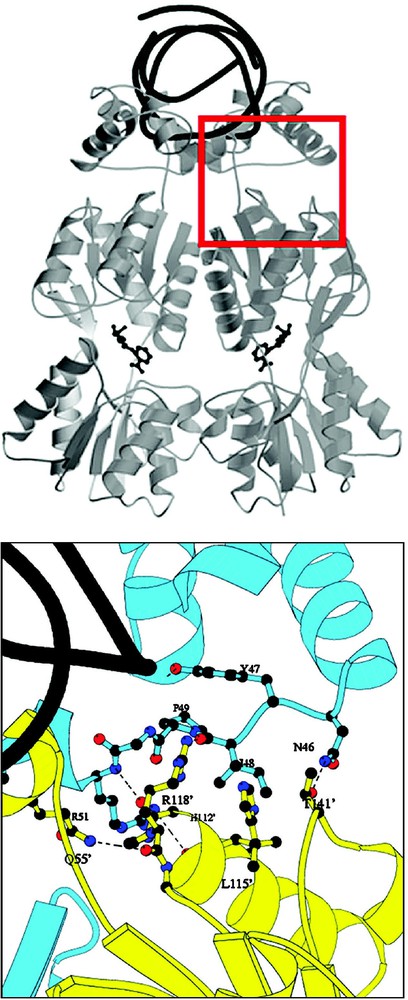
Interactions at the interface between the DNA-binding domain and N-terminal subdomain of the core. One subunit of the repressor is colored yellow, while the other subunit is colored blue. Atoms of the repressor involved in important interactions are shown in ball-and-stick. Hydrogen bonds are shown as dotted lines the interactions between the DNA-binding domain and NH2-subdomain of the core. The operator DNA is shown as black ribbon. Mutations of Arg118 and other residues at this interface disrupt the affinity of the repressor for operator. Not visible in the figure (behind helix 6) is the interaction between Arg51 and Asn113.
Is the structure of the repressor bound to this synthetic idealized operator relevant? It has been argued that the binding of the repressor to the ‘ideal-operator’ site is artificial and is not representative of the true operator repressor complex [35]. In the natural operator, the two half sites are not perfectly symmetric; in addition there is an insertion of an additional G–C base pair between the two half sites. Although the half site sequences of the natural and the idealized operator are similar, the two half sites are out of register with one another. If the operator adopted a canonical B-form DNA conformation, the two half sites of the symmetric operator would be spaced 3.4 Å closer along the DNA axis, and rotated by 36° relative to the natural operators. For the repressor to accommodate binding to both sequences, it would require either altering the conformation of the repressor so as to reorient one or both of the headpiece domains, or altering the conformation of the operator by changing the degree of bending or unwinding. From the structures of the repressor bound to the symmetric operator, it would appear that lac repressor could accommodate binding to the natural operator either as a rigid protein dimer with somewhat different recognition of the left and right half-sites, or by altering the dimer conformation so as to recognize both half-sites in a similar fashion. The latter would entail a change in conformation in one protein monomer with respect to the other. Intuitively, one would imagine that the repressor would adopt an altered conformation and bind the right operator in the same fashion as it binds the tighter left half site. Although the structures determined by X-ray crystallography and NMR provided similar pictures for the repressor binding to the ‘ideal operator’, the structures of the repressor bound to the natural operator are surprisingly different.
The headpiece binds to the right half site of the natural operator differently in the crystal than it does in solution. In the crystals the repressor binds to the natural operator as it binds to the symmetric operator, without major structural rearrangement [36]. As the natural operator has an additional base-pair in the center of the binding site, the headpiece interacts with the right half site by recognizing bases that are shifted one base pair along the DNA. In other words, the headpiece binds to different bases in the right half site compared to the dyad-related bases. In solution the headpiece domains change their orientation so they can recognize each half site similarly [37]. The headpiece binds to the left half site of the natural operator as it binds the symmetric operator. However, for the headpiece to make the similar interactions with the right half site, the second headpiece is translated by one base pair further away from the center and undergoes a 48° rotation relative to the headpiece bound to the left half site. The reason for the discrepancy between the two structures is unclear and may reflect that the repressor binds to the right half site less efficiently than it binds to the left half site of the operator [38]. The differences may be attributed to the difference in the binding affinity of the two sites. The most highly constitutive operator mutations occur in the left half of the operator suggesting that repressor binds less tightly to the right half of the operator than the left [10]. If the repressor binds weakly to one half site, then the headpiece could easily be influenced by additional constraints and the core domain could be the origin of the differences. As described above, the core domain of one monomer makes extensive contacts with the headpiece domain of the dimer related monomer in the crystal structure. These interactions are far from tenuous and may be responsible for orienting the headpiece domains in the structure of the intact repressor. The importance of the interactions between the headpiece and the core were demonstrated by inserting a glycine linker into the repressor immediately after the hinge helix. This modified repressor binds to the operator with a substantial drop in affinity [39]. The binding of the headpiece domains to the operator are subjected to additional constraints; as a consequence, it is very difficult to form any definite conclusions about the binding of the repressor to its natural operator from the low resolution crystal structure or the high resolution solution structures of the artificially constructed dimeric headpieces. Irrespective of the detailed interactions, both the NMR and X-ray structures observed that the conformation of the natural operator DNA is bent and the hinge helices are inserted in the minor groove of the operator. The conformation of the DNA with the ‘ideal operator’ and the natural operator are qualitatively quite similar and the hinge helices are responsible for distorting the conformation of these operators in similar ways.
5 The binding of the inducer and anti-inducer to the repressor
Inducers and anti-inducers bind to the same site on the repressor, but interact with the repressor differently. These effector molecules associate with the repressor molecule, forming a ternary complex that either decreases or increases the affinity of the repressor for the operator. Each repressor monomer has a single binding site and there are specific interactions that stabilize the complex. The effector molecules bind to a pocket that is located at the interface of the NH2-terminal and COOH-terminal subdomains of the repressor. The binding sites for these effector molecules are approximately 40 Å from the HTH motif and the operator binding site. So how do these effector molecules bind to the repressor and alter the operator binding affinities?
The effector molecules are galactosides that are both chemically and structurally related; however, these molecules do not bind in an identical fashion to the repressor [34]. Fig. 13 shows the binding to the inducer and the anti-inducer in the effector binding site. The inducer molecule, IPTG, forms hydrogen bonds to the amino acid side chain of Asn246, Arg197, and Asp149, as well as van der Waals interactions with a hydrophobic surface created by Leu73, Ala75, Pro76, Ile79, Trp220, and Phe293. The anti-inducer, ONPF, binds to the same pocket but in a different conformation. The anti-inducer also forms van der Waals interactions with the residues creating the hydrophobic pocket. The galactose ring of IPTG and the fucose ring of ONPF, which differ only at the substituent, are bound quite differently to the repressor. The nitrophenyl ring of ONPF is stacked over the indole ring of Trp220, but also contacts Pro76, Ala75, and Leu73. In addition to being a competitive inhibitor, ONPF increases the apparent binding of the repressor to the operator. The binding of this anti-inducer shifts the equilibrium in favor of operator binding and prevents the repressor from adopting the induced conformation.
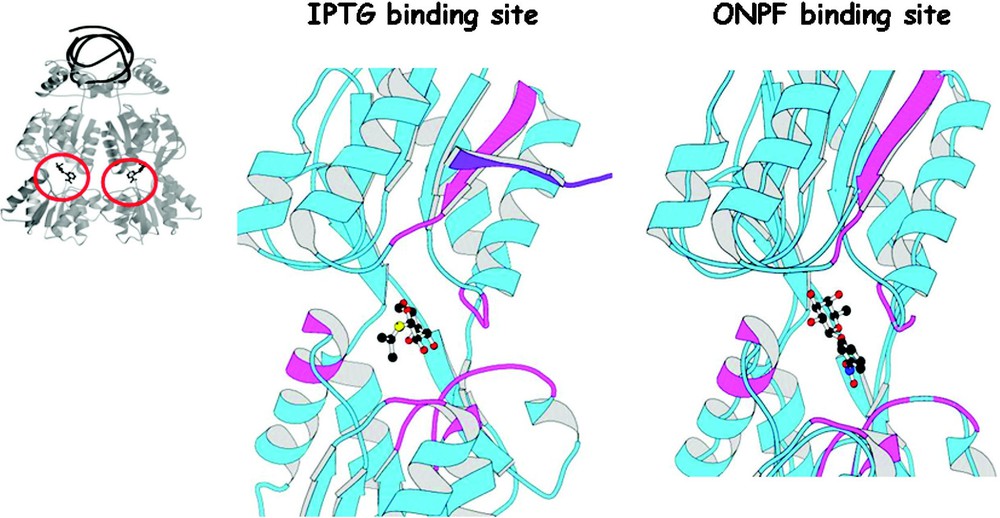
The binding of the inducer, IPTG, and the anti-inducer, ONPF, to the core of the repressor. A ribbon diagram of a monomer of the repressor is shown in blue. The IPTG and the ONPF molecules are shown in stick representation. The location of mutants are shown in violet color. The dimer mate of the strand B is shown in purple. The figure is adapted from Lewis et al. [30].
The mutant repressor molecules, classified by an phenotype, bind to the operator DNA with wild type affinity but are incapable of induction. These substitution are either defective in sugar binding and/or cannot transmit the allosteric signal to the DNA binding domain. The position of the point mutations cluster in five general locations with respect to the linear sequence and include residues 70–80, 90–100, 190–200, 245–250 and 272–277. When these mutations are mapped onto the protein, as is illustrated in Fig. 14, most of the mutations are in close proximity to the effector binding site. Altering the side-chains in the effector binding pocket directly alters the affinity of the inducer.
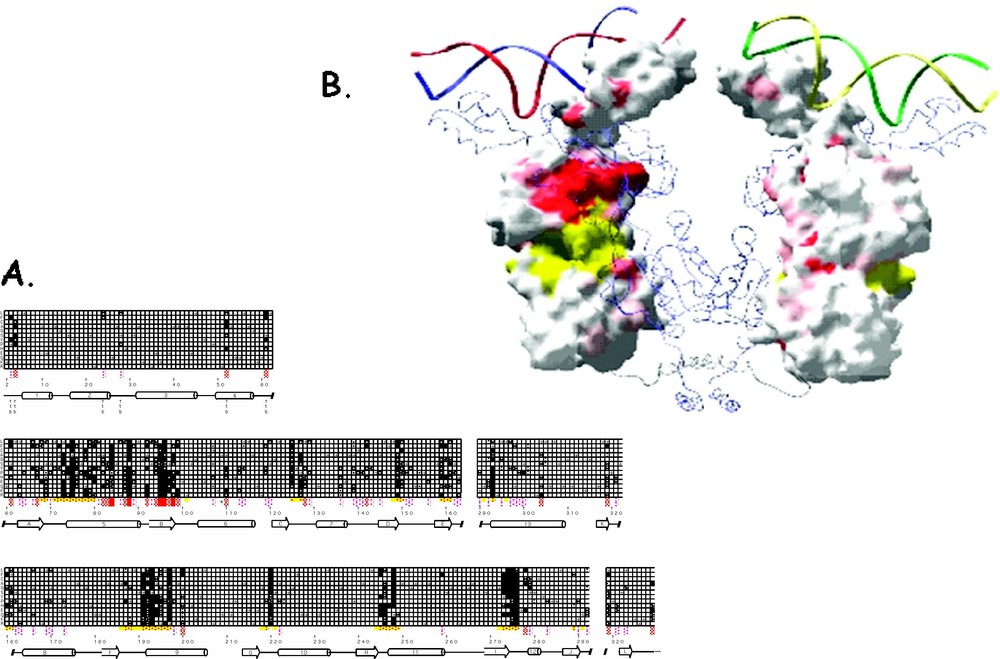
A summary of the genetic data for mutations that produce the phenotype. (A) The checkerboard summarizes the genetic data as described in Fig. 12. Each amino acid replacement was tested for β-galactosidase activity in the presence of IPTG. Repressor molecules that have phenotype are represented by black squares, dark gray for weak phenotype and white if there was no change in the phenotype. (B) The genetic data is mapped on the surface structure of lac repressor. Locations of mutants are shown in yellow and red. Yellow areas are within 8 Å of the IPTG molecule. The figure was made using GRASP [77].
6 The structural basis for the allosteric transition
There are two distinct conformations of the repressor that correspond to the induced and repressed states. The repressor adopts a conformation in the presence of the operator that is subtly different from the structure of the repressor bound to inducer. The change in conformation was illustrated when crystals of the lac repressor bound to lac operator were exposed to an allosteric effector; they immediately shattered (Fig. 15) [40]. This of course is reminiscent of the shattering of crystals of deoxyhemoglobin when exposed to air [41]. Interestingly, the crystal structure of the repressor in the absence of any ligand more closely resembles the structure of the repressor bound to the inducer, even though the two conformations are in equilibrium.

Crystals of the lac repressor bound to a 16 base-pair symmetric operator fragment. (A) A picture of a crystal of the repressor bound to the symmetric operator sequence. (B) is also a crystal of the repressor bound to the operator. The inducer molecule, IPTG, was added to the crystal solution, causing the crystals to shatter almost immediately.
The allosteric signal is transmitted through the dimer interface. By comparing the structure of the repressor in the induced and the repressed states, it becomes clear that the N-terminal and C-terminal subdomains to not change conformation. The structures of these domains are essentially invariant; however, there is a significant difference in their orientation. The change in orientation of the N-terminal subdomain relative to the C-terminal subdomain can be described as a small hinge motion. This change in structure alters both the intramolecular interactions between of the N-terminal and C-terminal subdomains of the monomer and the intermolecular interactions between the two N-terminal subdomains. When the repressor binds to its operator DNA, the two N-terminal subdomains rotate compared to when the inducer is bound. In the induced conformation, the N-terminal domains separate from each other in the repressed state, while preserving the two-fold axis of the dimer. The hinge motion does not alter the conformation of the C-terminal subdomains or the interface between the two C-terminal subdomains of the dimer. The C-terminal subdomain dimer appears to be the rigid scaffolding that is necessary to maintain the dimeric repressor. The mutational data illustrates clearly that the allosteric signal is transmitted through the dimer interface. The mutations that produce the phenotype and are not directly involved with inducer binding cluster at the monomer–monomer interface between the N-terminal subdomains (Fig. 16). From the position of the mutations, it is likely that a signal is transmitted from the effector site, through the dimer interface to the hinge helices and the DNA binding domains. Binding of the inducer causes a subtle structural change in the N-terminal subdomain, which is sufficient to destabilize the repressor–operator complex and reducing the repressor's affinity for the operator by several orders of magnitude.
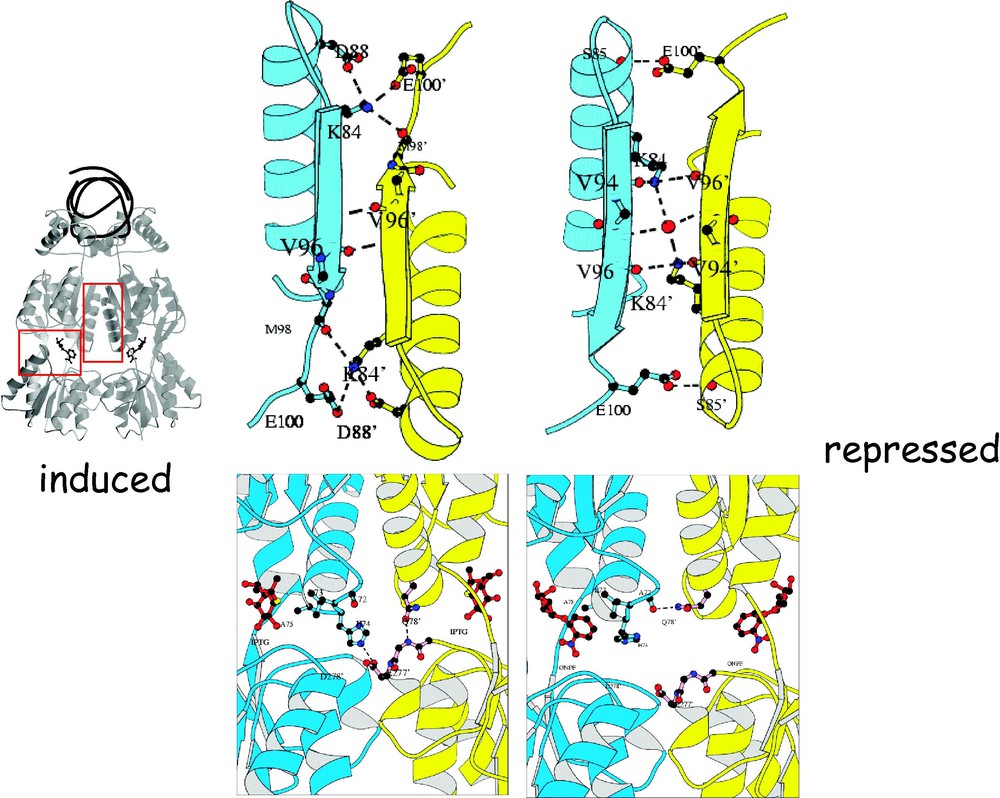
The figure highlights the change in structure observed in the induced and the repressed conformation. (A) Comparison of interactions at the dimer interface involving Lys84 in the IPTG-bound (left) and operator-bound (right) structures. The view is looking down the molecular 2-fold symmetry axis, from the point of view of the operator. For clarity, only the secondary structural elements of the repressor at the dimer interface are shown. Notice that the interactions of Lys84 change dramatically. (B) Comparison of interactions near the inducer-binding pocket in the structures of the repressor bound to IPTG (left) and operator/ONPF (right). The ONPF and IPTG molecules are shown in red in ball-and-stick. Notice that the His74-Asp278 ion pair formed in the IPTG-bound conformation is broken in the operator-bound conformation, and that the interactions of Gln78 are also altered.
There are specific interactions between the core and the headpieces that stabilize the complex with the operator and are therefore important for the allosteric signaling. If the allosteric mechanism of the repressor involved a simple displacement of the hinge helix as a consequence of the N-subdomain reorientation, then the insertion of the glycine spacer between the hinge helix (residues 50–58) and the core domain (residues 62–330) would be expected to de-couple the propagation of the allosteric signal, resulting in repressor molecules that do not respond to IPTG. However, contrary to this notion, the insertion of the glycine spacer dramatically decreased the repressor's affinity for the operator, while the allosteric response to IPTG remained intact [39]. The glycine linker apparently interferes with the extensive network of interactions between the N-terminal subdomains and the DNA-binding domains of the repressor. These interactions are important for stabilizing the operator bound conformation of the repressor. In an analogous fashion, the conformational change, caused by IPTG binding disrupts the network of interactions between the N-terminal subdomains and the DNA-binding domains, and thereby destabilizes the operator bound conformation. Thus, rather than an allosteric mechanism involving a simple pulling of the hinge helix from the minor groove of the operator, it appears that IPTG-binding disrupts inter-subunit interactions that are required for stabilizing the operator-bound conformation. Adding glycine residues alters the interactions between the N-terminal subdomain and the DNA-binding domain that are critical for stabilizing the operator-bound conformation. Of course, the added residues could affect the repressor structure in other ways, such as by increasing the entropic cost of hinge-helix binding, affecting the folding of the hinge-helix, or changing the orientation of the DNA-binding domains relative to the operator [39].
The equilibrium between the induced and repressed conformations of the repressor can be altered by changing particular amino acids at the dimer interface in the repressor. For example, mutating the side-chain of the amino acid at position 110 of the repressor dramatically shifts the equilibrium [42]. A repressor with an A110T substitution has a higher affinity for the inducer (IPTG) and a lower affinity for the lac operator than the wild-type repressor, while the A110K mutation has just the opposite phenotype; it binds to the operator with higher affinity than the wild-type repressor, but has a decreased affinity for the inducer. The amino acid at position 110 is located on helix six of the repressor, which is at the dimer interface between the N-terminal subdomain. Substitutions at this amino acid position alter the equilibrium by affecting the conformation of the repressor, which indirectly affects the inducer binding, as well as the repressor's binding of the operator. Although there is no detailed structural data on these two mutants, the position of the substitution is consistent with the notion that the allosteric transition is propagated through the monomer–monomer interface of the N-terminal subdomains and that the equilibrium between the induced and repressed conformations is established by residues at the dimer interface. By changing a single amino acid, it is possible to shift the equilibrium between the high operator affinity, low inducer affinity conformation and the low operator affinity, high inducer affinity conformation.
The allosteric transition is similar to the one observed in hemoglobin [43]. Hemoglobin exists in two distinct conformations that are referred to as the R and T states. In the presence of oxygen, hemoglobin adopts a conformation where the interactions that form the hemoglobin tetramer are ‘relaxed’. In contrast, the association of the hemoglobin tetramer in the deoxy state is ‘taute’. The conformation of the repressor bound to DNA is analogous to the oxy form of hemoglobin or the R state, while the repressor bound to inducer corresponds to the T state or the deoxy form. By analogy, the repressor adopts a ‘relaxed’ conformation when bound to the operator as hemoglobin does when bound to oxygen. The inducer molecule, IPTG, performs the same role in the repressor as the allosteric effector, 2,3 bisphophoglycerate, does in hemoglobin, stabilizing the T conformation. In the induced state, the quaternary structure of the repressor, like deoxyhemoglobin, forms a specific set of electrostatic interactions across the dimer interface. Switching between the induced and repressed conformations alters the monomer–monomer interface and the specific interactions (Fig. 17). For example, in the induced conformation, an ion pair is formed between Lys84 of one subunit and of the other subunit but in the repressed conformation, this ion pair is broken, and different interactions are formed. As illustrated in Fig. 17, Lys84 is wedged between two β-strands in the induced conformation but in the repressed conformation this lysine contacts the carbonyl oxygen's of Val94 and . Lys84 plays a key role in the allosteric transition of the repressor and mutations of the lysine have a profound effect on the function of the repressor, resulting in either the or the phenotype, depending upon the particular substitution.
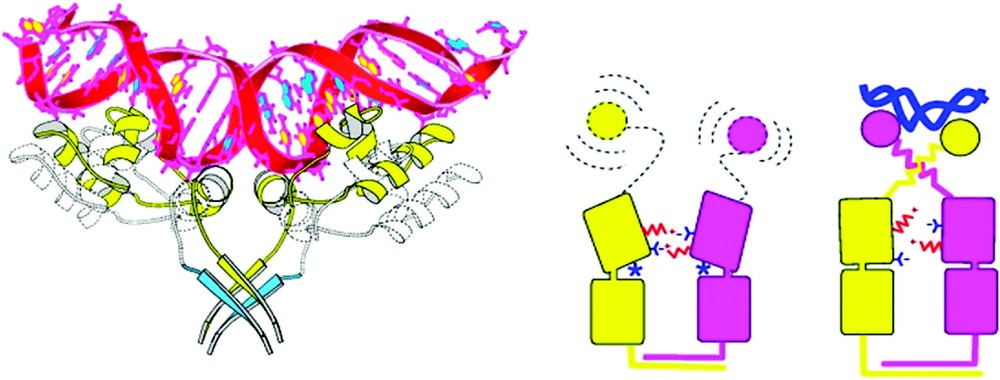
The allosteric signal is transmitted from the N-terminal subdomains to the DNA binding domains of lac repressor. (A) The figure suggests changes in the DNA binding domains when going from the induced to the repressed conformation. The DNA complex is shown in yellow, while the IPTG complex is shown in blue. The addition of IPTG causes a change in the NH2-terminal subdomains of the dimer, which causes the hinge helices in repressor to move apart. This movement disrupts the dimerization of the helices and the helices become disordered. The HTH motifs move out of the major groove binding sites (B). Cartoon that summarizes the allosteric change for lac repressor. The left side shows a dimer of lac repressor bound to IPTG (asterisk). A number of salt bridges exist between the N-terminal sub-domains of the core, and the N-terminal sub-domains are rotated and translated apart compared to the DNA-bound form. The hinge helices are not formed when the repressor is bound to IPTG; the entire HTH headpiece is completely mobile and is therefore disordered. The right side shows a dimer of the lac repressor–DNA complex. The DNA is represented by a black double coil. The salt bridges that exist in the IPTG-protein complex are broken by the movement of the N-terminal sub-domains of the core. The hinge helices are present in the DNA-protein form, ordering the entire HTH DNA binding domain. Adapted from [30].
Another aspect of the molecular structure of the repressor that is analogous to hemoglobin is a specific slat bridge. In hemoglobin a salt bridge forms between an aspartic acid and a histidine that is responsible in part for the Bohr effect. In the repressor, His74, which lies at the bottom of the N-terminal subdomain near the inducer-binding pocket, forms distinct interactions in both conformations. In the induced conformation, His74 forms an ion pair with of the C-terminal subdomain of the other subunit. This is the only interaction between the N-terminal subdomain of one monomer and the C-terminal subdomain of the other monomer, and thus could help define the relative subdomain orientations in the inducer-bound conformation of the repressor. When the repressor is bound to the operator, the ion pair between His74 and is broken and these residues become solvent exposed. If this ion pair is critical for allosteric signaling, then mutations of H74 or D278 should result in a repressor with a diminished response to inducer. While this was the case for all mutations of D278, mutations of H74 resulted in different effects on operator and inducer binding, depending on the mutation [44]. Although the H74–D278 ion pair may not be essential for the allosteric transition, these two residues are important for repressor function. Quite unexpectedly, mutating these residues, which are at the monomer–monomer interface of the repressor, has uncovered remarkable features of the repressor that lends insight into the relationship between the structure and the function. As described below, the D278L mutation changes the specificity of dimerization, such that repressor molecules bearing this substitution can dimerize with each other, but not with wild type repressor molecules [45].
7 Non-specific binding
The repressor must be able to find its operator by ‘searching’ through thousands of bases of non-operator DNA in order to function as a molecular switch. While this seems like a daunting task, the repressor is able to discriminate between operator and non-operator DNA. The non-operator DNA accelerates the rate that the repressor finds its operator by correctly orienting the repressor, allowing it to affectively ‘slide’ or ‘hop’ along the DNA before arriving at the target site [46]. How the repressor protein can discern the specific DNA sequences is still a mystery. Clearly, the specificity of repressor binding relies on the operator's unique chemical and structural signature that is accessible in the major and minor grooves of the DNA. What constitutes the difference between specific and non-specific binding and how does the repressor bind non-specifically to the DNA?
The structure of the lac headpiece bound to an 18-base-pair long fragment of DNA illustrates how the repressor may bind non-specifically [47]. In solution the repressor headpiece undergoes a conformational change when presented with non-operator DNA. The non-specific complex is created by an extensive electrostatic network of interactions. In this structure, residues that provide specificity through interactions with the base pairs in the major groove when bound to the operator, shift and twist so as to hydrogen bond and/or form electrostatic interactions with the phosphates on the DNA backbone (Fig. 11D). In contrast to the specific complex, the hinge region is disordered, and as a consequence there are no minor groove contacts. The central kink or bending of the operator observed in the specific complex is relieved and the DNA remains in the canonical B-form. Due to the major side-chain rearrangement, a cavity is formed between the repressor and the DNA that can accommodate water molecules [47].
The structure of the headpiece domains bound to non-operator DNA provides a glimpse of a nonspecific complex and provides a model for how the repressor locates its target sites. The structure confirms that the DNA orients the repressor and thereby reduces the dimensionality of the search. If the repressor is aligned to ‘slide’ along or ‘hop’ from one region to another, the space the repressor has to search is greatly reduced and the repressor could locate its operator more efficiently. However, from the structure of the non-specific complex, it is not possible to discriminate between the two plausible mechanisms, as the structure is consistent with both the ‘hopping’ and ‘sliding’ models. It is also unlikely that the solution structure of the non-specific complex is unique since the non-specific complex was created by constructing an artificially tethered disulfide linked dimeric lac headpiece. In the absence of the cross linking, the headpiece domains do not necessarily associate and may interact with the DNA differently from what was observed. Consequently, the structure of the non-specific complex represents only one of a vast multitude of structural states that are accessible to the repressor when searching for the operator.
8 Mutant repressors with altered stability and oligomerization
Biochemical and biophysical characterization of a subset of the 4000 mutants has uncovered repressor molecules with interesting biological properties. Mutations in the repressor have been discovered that dramatically alter its stability. Most substitutions decrease the stability of the repressor, but mutations have been observed that significantly increase stability. Increasing the stability of a natural protein by introducing one or more site-specific amino acid substitutions has been well documented [48]. Increased stability can be obtained by introducing hydrogen bonds, electrostatic interactions, or by increasing the van der Waals interactions and the packing of the residues. Alternatively, stability can be increased by reducing the entropy in the unfolded structure. A dramatic increase in stability is observed when Lys84 is changed to an Ala, Leu, Met, or Ile [49,50]. Substitution of this lysine drastically increases the thermostability of the dimeric repressor by 40 °C [51].
Unfolding of the repressor is reversible, and it exhibits a single cooperative unfolding transition at ∼2.8 M urea [50]. The transition corresponds to the simultaneous disruption of the monomer–monomer interface and the monomer unfolding. At the concentration of denaturant where the transition from folded to unfolded state occurs in the native repressor, the unfolded monomers appear to remain held together by interactions at the dimer–dimer interface. In contrast, the repressor with the K84L substitution dissociates into dimers at this concentration of denaturant [50]. The single amino acid substitution stabilizes the monomer–monomer interface, which now persists to higher levels of denaturant than the dimer–dimer interface. Although the K84L substitution increases the stability of the protein, it decreases the functional properties of the repressor. Compared to the native molecule, the K84L mutant binds operator DNA with a 2-fold reduction in the apparent affinity [50]. The mutant binds inducer with the same affinity as the native protein but the association and dissociation rate constants are reduced more than 200-fold [49,50]. Apolar amino acid substitution at position 84 also reduces the in vivo induction levels approximately 10-fold for the dimeric repressor and 30-fold for the tetramer [50]. Incredibly, heating the mutant repressor to 87 °C does not alter its ability to bind the inducer, IPTG, whereas the wild type dimer loses inducer-binding activity at 40 °C [50]. Although quantitative thermodynamic data cannot be extracted from these observations, a single amino acid can markedly change functional properties of the repressor.
The amino acid at position 84 is critical for establishing the orientation of the N-terminal subdomains [52]. A comparison of the thermostable mutant repressor, K84L, with the wild-type repressor demonstrates that there are only minimal changes to the conformation of the individual subdomains (Fig. 18). The two C-terminal subdomains of the dimer are virtually identical to the wild-type structures, and the interface between these two domains is preserved. However, when the structures are overlaid by superimposing the C-terminal subdomains of the wild-type and the mutant, there are noticeable differences in the relative orientation of the N-terminal subdomains. Changing the orientation of the N-terminal domain alters the monomer–monomer interface between the N-terminal subdomains, as well as the interface between the N- and C-subdomains within a monomer. In the repressed conformation, Lys84 is positioned at the monomer–monomer interface and is stabilized by electrostatic interactions but in the induced conformation it forms an ion pair with , across the monomer–monomer interface. As seen in the structure of this thermostable mutant, the leucine moves towards the interior of the monomer–monomer interface and interacts with several apolar residues; in particular, Val80 and Val94 of the same subunit, as well as , , , and of the other subunit of the dimer. The single amino acid substitution causes the monomer–monomer interface to adopt a more tightly packed interface.
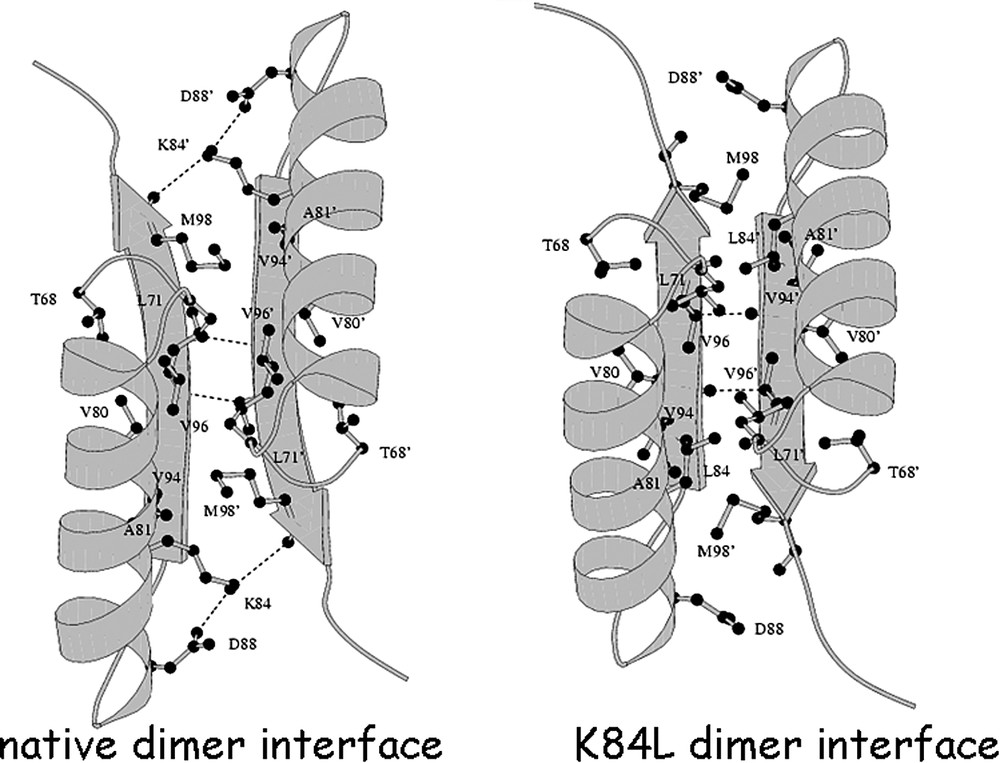
A view of the interactions at the monomer–monomer interface formed by the N-subdomains. On the left is the wild type interface and the right is the altered interface that results form the K84L substitution. The view is looking down the molecular 2-fold axis. For clarity, only the portions of the N-subdomains at the monomer–monomer interface (residues 67–101) are drawn. Hydrogen bonds are drawn as dashed lines. Notice that the inter-subunit β-sheet hydrogen bonding pattern changes, as a result of the N-subdomain reorientation.
The altered stability may be more easily rationalized by considering the K84L structure as ‘native’ and asking how de-stabilizing is the L84K substitution? Burial of an ionizable group in the interior of a globular protein is uncommon; in fact fully buried lysine residues, without compensating salt bridges or hydrogen bonds, are virtually unprecedented in nature. The high p value of a lysine assures that it resides almost exclusively at the surface of the protein, and only when the residue is deprotonated can it be incorporated in a hydrophobic core of a protein. This arrangement, of course, has an energetic cost that varies with pH but is in the range of 5–10 kcal/mol. For the lac repressor to accommodate the lysine in place of the leucine at the subunit interface observed in K84L would require the positive charge to be deeply buried and tightly packed amongst a number of other apolar residues. This process, i.e., burial of an unpaired charge in an environment of low dielectric constant, would be significantly destabilizing. Consequently, the energy that would be required to deprotonate and bury the two charged lysine residues of the dimer would be so great that the subunit interface would more likely undergo a modest rearrangement to expose the charged side chain to the solvent. The native molecule is in effect ‘destabilized’ relative to the mutant, but it is more responsive to ligand binding.
Mutations in the repressor can alter its oligomeric state in unexpected ways. A single amino acid substitution at position 278 alters the dimerization of the repressor [45]. There are over a dozen residues in the C-terminal subdomain that are responsible for creating the dimerization interface. Mutations at these positions will in some instances increase the ability of the repressor to dimerize and in other instances decrease its ability to dimerize. Most of these mutant repressors, in addition to forming homodimers, will also form heterodimers with the wild-type repressor. The mutation D278L is rather remarkable; the single amino acid change uniquely alters the interface [45]. The mutant repressor can self associate, as well as the wild-type, but will not form heterodimers with the wild-type repressors. This specific change of an aspartate to a leucine, creates a distinct interface with all of the same properties as the native structure, accept it can only self-associate. It is extraordinary that a single amino acid change can so drastically change the specificity of dimerization.
9 The LacI/GalR family of repressors
There are many repressors, which regulate the transcription of inducible genes, and have a high degree of sequence homology with the lactose repressor [53]. These proteins, referred to as the LacI/GalR family, appear to have similar structures and regulate transcription in an analogous fashion. The proteins in the LacI/GalR family have been shown by either structural studies or sequence similarities to contain headpiece domains with a HTH motif for recognizing an operator and have a core region that is responsible for effector binding and oligomerization. The allosteric effectors that regulate these repressors either act as inducers or as co-repressors.
The DNA binding domains of the proteins in the LacI/GalR family have strong sequence homology [53]. The conservation of the HTH region in this family indicates that some of the proteins may bind similar operator sites, and the amino acids that allow these proteins to discriminate between the different operators reside at a few non-conserved positions in the helix-turn-helix motif. In addition to the HTH, members of this family also use a hinge helix for binding to the operator. All of the proteins in the LacI/GalR family have a conserved leucine residue on the hinge helix, and it is likely that all these proteins pry open the minor groove when bound to operator. Consequently, in addition to the HTH motif, the hinge helix is important for operator recognition and a hallmark of this family of repressors. For example, the purine repressor, a member of the LacI/GalR family, recognizes the central portion of the operator by placing a pair of helices in the minor grove of the operator [54]. Members of the LacI/GalR family also have significant sequence homology throughout the core or effector binding domains. The members of this family bind and respond to a variety of effector molecules, such as galactose, fructose, maltose, ribulose, and b-galactosides. Related molecules also bind nucleosides or their derivatives, such as hypoxanthine and guanine. These repressors appear to have similar structures but the scaffold is draped with different amino acid side chains that create the unique specificity. There is also noticeable homology in the operators of the LacI/GalR family. The homology is particularly strong at the center of the operator such that the specificity is localized to the peripheral regions with highly conserved operator with a half site sequence -AANC at the center, although the lac repressor has a -GAGC sequence. There are only minor differences between the lactose and galactose operator sequences such that the DNA binding specificity of the lac repressor can be altered to recognize the galactose operator [55]. Simply changing the first two amino acids on the recognition helix to the gal repressor sequence alters the specificity of lac repressor to bind a gal operator.
The LacI/GalR family of proteins has sequence and structural homology with the periplasmic sugar binding proteins (Fig. 19). The periplasmic sugar binding proteins are involved in the active transport of water-soluble ligands. When these proteins bind to their ligands, they undergo a conformational change that is analogous to the changes observed in the lac repressor. The similarities in the structure and function of these proteins suggest that the repressors and sugar binding proteins share a common ancestor [56]. Not surprisingly, repressors and periplasmic sugar binding proteins with the same ligand specificity have the same or similar residues in their binding sites. Which came first, the acquisition of the DNA-binding domain or divergence of ligand specificity? Did a repressor evolve from an existing repressor or from periplasmic sugar binding proteins by acquiring the DNA-binding domain? If the divergence of ligand specificity occurred first, then the genes that code for the ancestral periplasmic binding proteins gene duplicated and one of the duplicates acquired the DNA-binding domain to evolve into a repressor. Since the sequence similarity of the N-terminal DNA-binding domain is higher than that of the C-terminal ligand–binding domain in the LacI/GalR family, it would appear that the functional divergence of ancestral periplasmic sugar binding proteins took place prior to the acquisition of the N-terminal domain [53]. This is further support by the observation that operator binding of the lac and purine repressors is very similar; they both use a hinge helix as well as the HTH structure for DNA binding [30,54]. Although the helix-turn-helix structure are found in other DNA-binding protein families, recognition of the DNA minor groove by the hinge helix is unique to the LacI/GalR family and plays a crucial role in DNA binding of the repressors. This regulatory mechanism is so elaborate that it is unlikely that such a system evolved independently for each ligand.
Comparison of the lac I family monomers and periplasmic amino acid binding proteins. Ligands bound to each monomer are shown in ball and stick representation. The amino acid binding protein with bound arginine [78] is shown in the upper left panel. The ribose binding protein [79] is shown in the upper right panel with the bound ribose molecule. A monomer from the dimer PurR-DNA-hypoxanthine complex [80] is shown in the bottom left panel. A monomer from the intact tetrameric lac repressor–DNA complex [30] is shown in the bottom right panel. No ligand is bound to the DNA form of lac repressor. The ligand binding domains of both PurR and lac I repressors are more closely related to the periplasmic sugar binding proteins than the amino acid binding proteins.
10 Plasticity and the formation of dimers
One of the fundamental differences between the repressor family and the periplasmic binding proteins is the oligomeric state. Repressor molecules are dimeric or tetrameric while the periplasmic binding proteins are all monomeric. For the repressor to have evolved from the periplasmic binding proteins the oligomeric interface must be pliable. If the structures of these oligomeric proteins in the LacI/GalR family evolved from a monomeric periplasmic binding protein then, they must have evolved a dimeric interface. One of the early mutations isolated in the lac repressor, designated T41 [57], is a point mutation that exhibits wild-type inducer binding properties but is monomeric [58]. This mutated repressor, Y282D, although incapable of binding DNA, still maintains sufficient structure to bind effector molecules. If the Y282D mutant could serve as a model for the primordial repressor, then the plasticity of the dimer interface could be accessed by looking for second-site revertants.
Twenty-two second-site mutations were identified that compensate for the Y282D mutation and produce fully functional repressor molecules capable of binding DNA [59]. The mutations that compensate for the dimerization defect cluster into discrete regions with respect the 3-dimensional structure (Fig. 20). Many of the revertants that were characterized are in close proximity to the Y282D mutation and are likely to reestablish the dimer interface between the C-terminal subdomains. Other mutations appear to indirectly compensate for the mutation by creating altered repressors that exhibit higher affinity for DNA. The first group of nine mutations appeared at six different positions: M223I/T, N246S, Q248R, D274N/G, T276A/I, and P284S, which are in the immediate vicinity of the original mutation. These revertants allow for a local adjustment to accommodate the original mutation. If one assumes that there is not significant rearrangement of the repressor, then these mutations are predominately at the interface of the C-terminal subdomain.
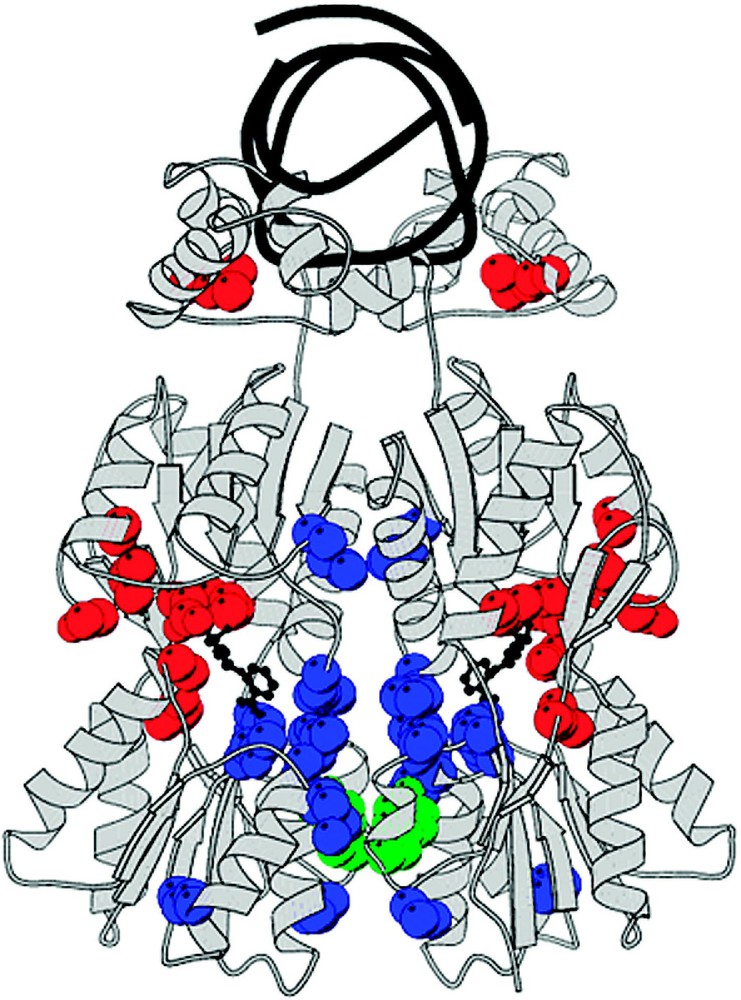
The plasticity of the repressor suggests that the monomeric periplasmic binding proteins would have evolved to form higher ordered structures. The position of second site mutations are displayed on the repressor dimer bound to the operator. The mutation Y282D that prevents dimer formation is shown in green. The second-site mutations that restore dimer formation are highlighted as space filling to illustrate their position with respect to the structure. One group of mutation, colored blue, is at the dimer interface. The second site mutations colored red are primarily at the interface between the N-terminal and C-terminal subdomains.
The second group of mutants: A133T/V, D149N, V150I, S151P, S191F, L296M, and V321I are quite surprising. These residues appear at the interface between the N-terminal and the C-terminal subdomain of the repressor monomer and are also directly involved in inducer binding. It is not intuitively obvious how these mutations reverse the phenotype back to wild-type and form stable dimers in a background where the C-terminal subdomain can no longer self associate. The revertants may alter the conformational equilibrium and stabilize the DNA-bound conformation, which could allow the detection of extremely low levels of assembled dimers, not perceptible by in vitro characterization. Some of the second site revertants were even more surprising. For example, the M42I mutation is located in the headpiece of the repressor and most likely affects DNA binding directly. Stabilizing the DNA-binding domain indirectly creates a repressor with a higher affinity for DNA than the wild-type. The increased affinity for the operator may shift the equilibrium sufficiently to allow the detection of the Y282D oligomer [59]. The ability of so many amino acid substitutions to potentially restore the native phenotype suggests that there is sufficient plasticity in the structure such that a repressor could have easily evolved from a periplasmic binding protein. Changing just a few critical side chains on the surface of the periplasmic binding protein would be sufficient to convert a monomer to a dimer. The large number of second site revertants suggests that only minimal changes in the periplasmic binding proteins may be necessary to facilitate oligomerization. From the diversity of these sites it appears that the oligomeric interface is remarkably flexible and there are many different ways to establish higher ordered structures.
11 DNA looping and the function of auxiliary operators
With the sequencing of the lac operon, two short lengths of DNA were discovered that resembled the lac operator [10,60]. One of the sequences, named O2, was found to be 401 bp downstream of the primary operator, and the other O3 was 92 bp upstream of O1. While these two sequences were quite similar to the primary operator, constitutive mutations were never found in either O2 or O3 and they did not appear to be of any biological significance. As a consequence, they were referred to as pseudo-operators. However, these pseudo-operator sites, which are distant from the promoter, increase repression of the lac promoter [61] and destruction of either pseudo-operator decreases repression two- to three-fold. Moreover, destruction of both pseudo-operators decreases repression 70-fold [62]. Clearly, these pseudo-operators play a role in regulating the operon and were renamed auxiliary operators. The tetrameric repressor, in principal, is ideally suited to bind simultaneously two operators and create repression loops [63].
When a single lac repressor tetramer binds two operators that are separated by 93 or 401 base-pairs, a continuous piece of DNA must bend to form a repression loop [64]. The formation of a repression loop depends upon the physical properties of DNA as well as the length of the intervening loop [65]. The first direct observation that DNA looping actually occurs and plays a functional role was demonstrated in the arabinose system [66]. There are two plausible mechanisms for looping the DNA that are consistent with the architecture of the lac repressor tetramer. The two subunits of the tetramer can bind to the primary operator site and the other dimer subsequently associates with an ancillary operator. Alternately, free repressor dimers could bind to separate operators and a loop would occur when the dimeric repressors associate into a tetramer. Like the hinge region (helix 4), the C-terminal helices undergoes a coil to helix transition upon tetramer formation with no detectable intermediates [67]. By either mechanism, the repressor acts like a double clamp bringing two operators that are separated in linear sequence close together. The order of events is dictated by the concentration of the repressor in the cell, the dimer–tetramer equilibrium [68,69], and the binding affinity of dimeric repressor molecules to the operators [70]. Both mechanisms are plausible and depend on the precise physiological conditions.
When the repressor tetramer binds to two operators, the DNA could ‘wrap toward’, ‘wrap away’, or form a ‘simple loop’ [28]. All three models are credible and consistent with the observed quaternary structure of the repressor. However, given the dimensions of the repressor and the length of the loop between O1 and O3, it is unlikely that the DNA would wrap around the molecule. The ‘wrapping away’ model is extremely attractive, since in the structure of the repressor bound to the symmetric operator, the DNA adopts a conformation that is curved away from the repressor and is more consistent with ‘wrapping away’ from the molecule. Shortly after the structures of the repressor were determined, it was observed by electron microscopy that the conformation of the repressor observed in the crystal structure was not unique [71]. Approximately 56% of the negatively stained repressor molecules have the ‘V’ shape and adopt a conformation that is similar to that observed in the crystalline state. The other 44% of the repressor molecules are in an extended conformation with the DNA binding sites at opposite ends of the molecule. In this conformation, the repressor could quite easily bind to distant operators and adopt a topology that is consistent with the ‘simple loop’ model. Given that the intermolecular contacts observed in the crystal structure are tenuous at best, it is not surprising that the tetrameric repressor can adopt alternate conformations. If the repressor is not a static structure and confined to the ‘V’ shape, then there are likely to be a large number of geometrically and topologically different DNA loops that can form and it is less likely that the shape of the loop will be an important parameter in the regulation.
The distance between the operators is extremely important for loop formation. The level of repression decreases with increasing separation of the operators. When the operators are separated by more than 1000 base-pairs there is no noticeable increase in repression [72]. For shorter separations, the exact spacing is crucial. Repression of the operator is strong when the upstream operator is placed 59, 70, 81 and 92 bp upstream of O1 [72]. At these specific spacing the operator sites are centered on the same face of the DNA. In contrast, when the operators are separated by some intermediate spacing, repression drops to the level of a single operator. To form loops using DNA of these lengths demands flexibility of the DNA molecule as well the protein. A tethered dimer that is arranged with a variable ‘V’ shape is entirely consistent with these data.
12 The lac operator–repressor system is functional in the mouse
The lac genetic switch has been adapted to regulate transcription of gene expression in mammalian cells [73]. The repressor was not only capable of repressing the reporter genes, but the gene could be reactivated by the addition of inducer into the culture medium [74]. More recently, the lac repressor and operator have been shown to regulate gene expression in the mouse [75]. Two lines of transgenic mice were generated; one line of transgenic mice expresses the lac repressor and the other line expresses the tyrosinase gene under the control of the lac operator. The tyrosinase enzyme catalyzes the first step in melanin biosynthesis and is part of the operon that controls the color of the mouse [76]. To ubiquitously express a functional repressor, the codons of the bacterial repressor gene were changed to resemble a mammalian coding sequence. Then the lac operator sequence was integrated into the promoter of the reporter tyrosinase gene. When the transgenic mice were crossed, the double transgenic mouse contained a regulated operon. The lac repressor binds to the operator sequences located in the tyrosinase promoter and blocks the transcription of tyrosinase. The coat of the double transgenic mouse is unpigmented and indistinguishable from that of a nontransgenic albino mouse. Introducing lac regulation into the mouse prevented the tyrosinase from being expressed. When the double transgenic animal is given IPTG in the drinking water, tyrosinase expression is derepressed, resulting in a phenotype indistinguishable from the wild-type brown pigmented mouse.
The tyrosinase transgene expression is fully reversible. The mouse strain expresses tyrosinase only in the presence of IPTG. Thus binding of the inducer by the repressor regulates the expression of the genes that control pigmentation of the mouse. Once IPTG is depleted, tyrosinase is again turned off and the albino phenotype returns. Interestingly, these effects occur in both adult mice and in embryos that are exposed to IPTG via the mother's drinking water. Reversible regulation of pigmentation in the transgenic mouse by elements from the lac operon of E. coli is the first successful demonstration that bacterial sequences can be used to create regulated operons in mice and the switch functions in the mouse as it does in their bacterial counterparts.
13 Conclusion and future directions
Regulating gene expression is a fundamental process of life and is essential for controlling metabolic events, development, and disease. Although the specific details for controlling regulation can be extraordinarily diverse and complex, the concepts put forward by Jacob and Monod were revolutionary and serve as the foundation of all gene regulation. Over the past half century, the details of the operon have been elucidated using genetic, biochemical, and structural techniques, yet the principles that were originally put forward have only been slightly altered and refined. The structure of the repressor is a beautiful punctuation to the theory providing a detailed molecular model for picturing how the operon functions. The three-dimensional structure of the repressor allows us to see how the repressor binds to its operator and how inducer molecules bind and effect its conformation. The structures make it easier to understand at the molecular level how this system functions.
The ability of the lac system to control the transcription of genes in the mouse is incredibly exciting. In the future it is likely that the lac repressor could be used to regulate a variety of promoters, which would move the system to the next level. Endogenous loci could be switched on and off to create models of disease and development. By modifying both the target promoter and the gene encoding the lac repressor, the lac system was able to control the transcription of genes so that they can function analogously in the complex environment of the mouse. Specific loci can be switched on and off repeatedly to create reversible models of human disease and normal development in the mouse. Assuming that the lac operator can be successfully incorporated into a given promoter, then it should be possible to regulate virtually any mammalian gene. As the mouse is the most widely used experimental system to model human disease and development, the ability to regulate genes using the lac repressor and operator will greatly broaden the range of biological questions that can be addressed experimentally. The implications are of course far-reaching and the ability to regulate genes will allow the modeling of human disease and development [75]. Monod would not have been at all surprised to see lac in mice, as he once wrote, “anything that is true of E. coli must be true of elephants, except more so.”