

1 Introduction
Deux des cancers hormonaux dépendants, le cancer du sein chez la femme et le cancer de la prostate chez l'homme, constituent en France, par leur fréquence croissante et leur gravité, un problème de santé publique très préoccupant [1] : les influences des hormones stéroïdes sexuelles, estrogènes et progestérone, d'une part, androgènes, d'autre part, présentent des points communs. Ce sont des agents promoteurs de tumeurs qui, en activant des récepteurs nucléaires spécifiques, vont stimuler la croissance et la division cellulaire dans leur tissu cible. De plus, les inhibiteurs spécifiques de leur synthèse ou de leur action sur leur récepteur se sont avérés efficaces et sont largement utilisés au premier rang des traitements des cancers ciblés sur des molécules spécifiques intervenant dans leur genèse.
Chez la femme, au moins trois cancers (sein, endomètre, ovaire) sont sensibles à des degrés divers aux hormones ovariennes. Le cancer de l'endomètre, très stimulé par les estrogènes, est également induit par le tamoxifène, mais il est protégé de l'effet mitogène des estrogènes par les progestatifs. De même, si les estro-progestatifs des contraceptifs oraux peuvent légèrement augmenter le risque de cancer du sein en prise prolongée chez la jeune femme, ils protègent du risque de cancer de l'endomètre et de l'ovaire [2] (Fig. 1). Les bases de la spécificité tissulaire d'action de ces hormones sont encore mal comprises, bien que le niveau des divers co-modulateurs des récepteurs doive y jouer un rôle essentiel [3]. Les mécanismes de la cancérogenèse hormonale sont complexes. Je me limiterai au cancer du sein, qui est le plus fréquent et de loin le plus étudié, grâce en particulier aux modèles constitués par les lignées de cancer du sein humains.
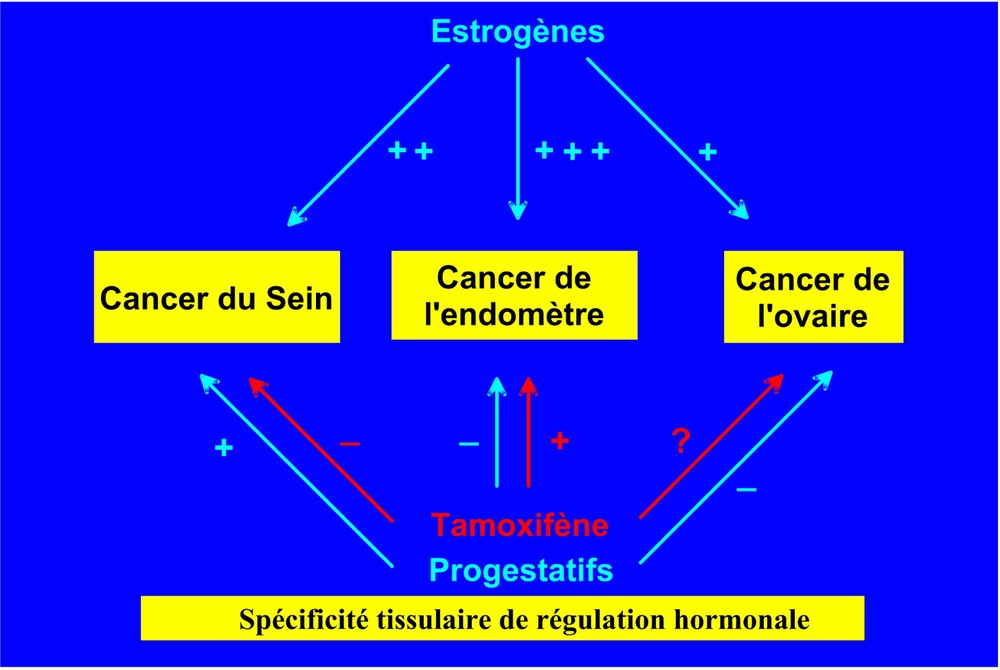
Effet variable des hormones ovariennes et de leurs antagonistes selon le tissu d'origine des cancers chez la femme. Les mécanismes de la spécificité tissulaire de régulation hormonale ne sont pas élucidés. On évoque la balance entre divers co-modulateurs et différentes formes de récepteurs hormonaux, qui transmettent l'effet des hormones sur la croissance de ces cancers.
Les cancers du sein sont hétérogènes. Les cancers héréditaires liés à des mutations germinales de gènes suppresseurs de tumeur à forte pénétrance BrCa1 et BrCa2 ne représentent que 5 à 7% des cancers du sein. Même dans ce cas, le mode de vie (facteurs nutritionnels, hormones, facteurs liés à la reproduction, etc.) intervient, la pénétrance ayant augmenté chez les femmes nées après 1940 (80% de cas à 55 ans) par rapport aux femmes nées avant 1940 (55% de cas à 55 ans) [4]. De plus l'ovariectomie prophylactique diminue d'environ 50% l'incidence des cancers du sein avec mutation BrCa1, suggérant que les hormones ovariennes favorisent ce cancer à des étapes initiales, bien que leurs récepteurs aient disparu au stade de cancer invasif [5].
Pour les 90% des cancers du sein dits sporadiques, les mécanismes initiaux restent très mal connus. Cependant, le rôle favorisant des hormones ovariennes, suspecté depuis longtemps [6], est maintenant bien établi, ce qui ouvre la possibilité d'une prévention. En effet, les facteurs de risque mis à part, les antécédents familiaux et le retard à une première grossesse sont essentiellement liés au temps d'exposition naturelle ou thérapeutique aux hormones ovariennes [7].
C'est la confrontation des résultats des études épidémiologiques, cliniques et scientifiques aux laboratoires qui a abouti à la notion que les hormones favorisaient chez la femme le développement des cancers hormonodépendants (Fig. 2). Comme d'autres tumeurs solides [8], un cancer hormonodépendant évolue très lentement en plusieurs étapes : il peut être initié 10 à 15 ans avant de se révéler cliniquement. Celui-ci peut même ne pas se révéler avant le décès de l'individu pour d'autres causes, comme le montre la fréquence à l'autopsie des cancers de la prostate infra-cliniques après 70 ans et celle des cancers du sein in situ chez la femme [9]. La définition des facteurs favorisant la cancérogenèse peut ainsi être différente pour un médecin et pour un biologiste moléculaire et cellulaire. Le médecin considère les facteurs favorisant l'apparition clinique ou radiologique d'un cancer, alors que le biologiste considère les altérations du génome conduisant à une perte du contrôle des mécanismes de division cellulaire, d'apoptose et de réparation, qui aboutit à une prolifération anarchique des cellules cancéreuses, ne respectant plus les tissus voisins. D'ailleurs, les hormones utilisées dans les traitements substitutifs (THS) de la ménopause [10,11], et à un degré moindre en contraception orale, viennent d'être classées par l'Agence internationale de recherches sur le cancer (WHO/Lyon) comme des agents cancérogènes de classe 1 pour les cancers du sein et du foie, essentiellement du fait de leur activité de promoteur de tumeurs [12].
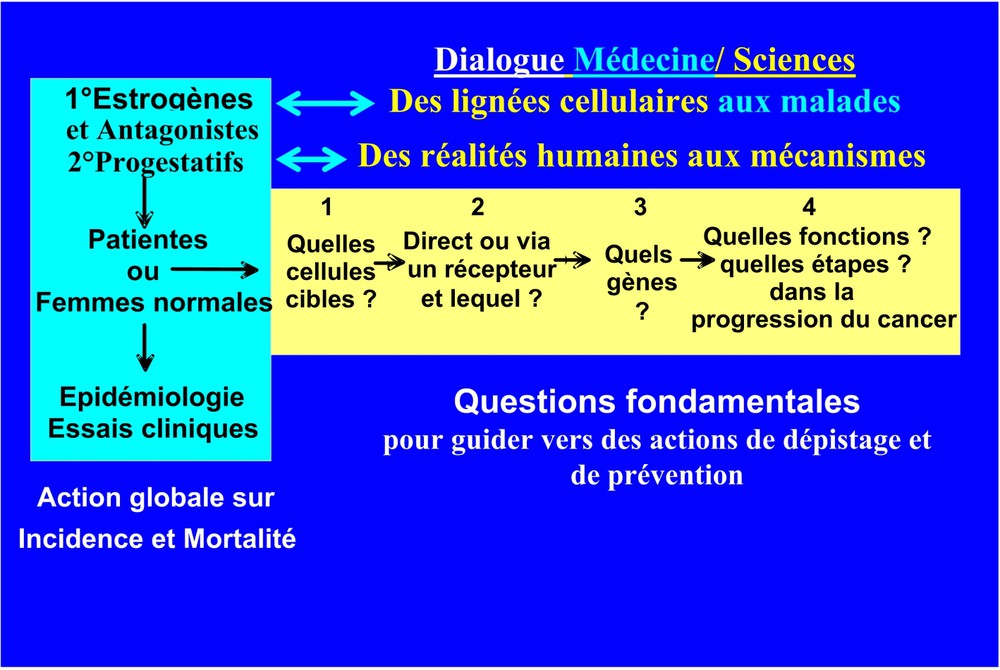
Des lignées cellulaires aux malades et des malades aux mécanismes. Le rôle des hormones ovariennes dans les cancers génitaux chez la femme, vu sous l'angle intégré par les cliniciens et épidémiologistes et analysé dans leur mécanisme par les biologistes cellulaires et moléculaires. Pour les estrogènes dans les cancers du sein, il y a une très bonne cohérence entre les deux approches et les études fondamentales ont facilité les applications thérapeutiques. Ce n'est pas le cas pour les progestatifs, où les résultats cliniques relancent d'autres études fondamentales.
Il est davantage discuté que ces hormones interviennent dans les étapes initiales comme agents mutagènes initiateurs de tumeurs [13]. L'influence initiale des estrogènes sur des cellules normales et un génome non muté reste débattue pour les estrogènes naturels. Le cas est différent pour les estrogènes de synthèse, tels que le diéthylstibestrol, qui induit des mutations de la lignée germinale à l'origine de cancers de l'endomètre et du vagin chez les filles de femmes traitées par cet estrogène avant les années 1970 [14]. C'est pourquoi l'administration d'antiestrogènes de synthèse non stéroïdiens chez une femme jeune doit être accompagnée de moyens contraceptifs. Les estrogènes naturels peuvent, sans interagir avec leur récepteur, se lier directement à l'ADN après leur métabolisme oxydatif en dérivés 2- et surtout 4-hydroxylés. Les quinones correspondantes vont former des liaisons covalentes avec les bases puriques de l'ADN et entraîner des mutations somatiques pouvant être pathologiques. Ces mutations, obtenues in vitro en culture de cellules et in vivo chez l'animal à fortes doses, ne sont ni démontrées, ni totalement exclues chez la femme [15]. Une alternative à l'effet mutagène direct des estrogènes passe par leur activité mitogène, transmise par leur récepteur qui, en raccourcissant la durée du cycle cellulaire selon des mécanismes bien étudiés en lignées, interférerait avec les mécanismes de réparation de l'ADN.
Excluant les rares cancers du sein dus aux gènes de susceptibilité connus (BrCa1/2), j'envisagerai uniquement l'effet des hormones ovariennes comme agent promoteurs des cancers du sein sporadiques, en évoquant successivement :
- (a) le mécanisme d'action des estrogènes comme agent promoteur et mitogène, montrant que les études fondamentales mécanistiques ont facilité le transfert vers la clinique ;
- (b) celui des progestatifs, moins bien connu, mais dont l'effet délétère sur le sein est démontré par les essais cliniques et épidémiologiques des THS de la ménopause ;
- (c) les étapes initiales de la cancérogenèse encore bien mystérieuses, en discutant la piste constituée par les cellules souches ;
- (d) notre approche in situ par immunohistochimie des lésions mammaires prolifératives précancéreuses et des cancers in situ ;
- (e) enfin, les actions de prévention des cancers du sein abordées par les médecins et les scientifiques.
2 Les estrogènes
Concernant les estrogènes, les approches intégrées sont en totale cohérence avec les études moléculaires et cellulaires.
Le transfert, depuis les recherches fondamentales jusqu'à la prise en charge des malades, a été complet et constitue un succès, grâce à l'endocrinologie fondamentale. Il a abouti à l'utilisation, en pratique médicale courante, du dosage des récepteurs hormonaux dans la tumeur, puis à l'utilisation de l'antiestrogène tamoxifène comme premier exemple d'une thérapie ciblée d'un cancer sur une protéine, le récepteur des estrogènes (RE) intervenant dans sa pathogénie.
On connaissait déjà, il y a plus de 20 ans, l'importance des estrogènes dans la progression des cancers du sein, à la suite de la démonstration du fait que ces hormones stimulaient directement et spécifiquement la croissance de lignées humaines de cancer du sein et que les antiestrogènes inhibaient cette croissance, si toutefois ces cellules exprimaient les RE (revue dans [16]).
Les biologistes moléculaires ont permis de disséquer les divers domaines fonctionnels des RE [17], les chimistes et physiciens ont précisé les modifications de la structure dans l'espace du domaine de liaison d'un ligand, selon que celui ci est agoniste ou antagoniste [18,19]. Les mécanismes d'action des hormones stéroïdes sur l'initiation de la transcription de certains gènes et l'ouverture de la chromatine sont compris dans leur grande ligne grâce à la recherche fondamentale. La nature des gènes endogènes responsables du contrôle de la croissance et de la prolifération cellulaire, de l'apoptose et de l'invasion tumorale est plus difficile à préciser et leur liste continue à croître.
Les études sur lignées de cancer du sein ont permis de comprendre les effets in vivo des estrogènes chez les femmes ménopausées sur des cellules mammaires âgées ayant subi les premières mutations ou ayant même déjà développé des microcancers ou des cancers in situ. Le mécanisme de l'effet mitogène directe des estrogènes a été précisé sur les diverses lignées de cancer du sein RE positives généralement établies à partir de métastases pleurales [20,21]. C'est ainsi que plusieurs protéines mitogènes ont été décrites, qu'elles soient sécrétées (facteurs de croissance, protéases telle que la cathepsine D) ou intracellulaires (c-myc, c-fos, cycline D1 et cycline E). Ces protéines relaient les estrogènes comme facteurs mitogènes autocrines (après leur sécrétion) ou intracrines (sans être sécrétés) (revue dans [22,23]).
Ces dernières années, l'extrême complexité de ces régulations s'est accentuée du fait de la multiplication des partenaires interagissant avec un récepteur et de leur combinaison. Citons la découverte d'un deuxième récepteur, le RE β [24], le nombre croissant de co-modulateurs (plus de 15 activateurs et répresseurs différents) qui, couplés à des activités enzymatiques, permettent de comprendre les modifications de la chromatine, induites différemment par les estrogènes ou leur antagonistes [25]. Le schéma s'est complété avec les interférences transcriptionnelles des récepteurs hormonaux sur d'autres facteurs transcriptionnels (AP1, sp1, etc.) et l'étroite interrelation entre la voie des récepteurs nucléaires et la voie membranaire des facteurs de croissance et des cytokines stimulant une cascade de phosphorylation. Le tout aboutit à une accélération de la transition G1 → S du cycle cellulaire, via les cyclines et leurs kinases, et explique en partie les mécanismes de contrôle de la division des cellules cancéreuses en culture. De même, en clinique, les indications des antiestrogènes se sont précisées, d'autres molécules que le tamoxifène, telles que les antiestrogènes purs, le raloxifène, agissant également sur les RE, ou les anti-aromatases, inhibant la synthèse d'estrogènes à partir des androgènes surrénaliens, permettent d'affiner les traitements [26]. Ainsi l'effet promoteur de tumeur des estrogènes lié à leur activité mitogène et l'efficacité des antiestrogènes et anti-aromatases dans le traitement des cancers du sein RE positifs sont confirmés et compris dans leurs grandes lignes.
3 Le cas de la progestérone et des progestatifs
L'étude de l'effet des progestatifs sur les lignées de cancer du sein en culture n'a pas permis de comprendre leur effet globalement néfaste sur le sein au cours des traitements substitutifs prolongés de la ménopause, où ils sont nécessaires pour s'opposer au fort effet mitogène des estrogènes sur l'endomètre. Les études américaines WHI [10,27], britanniques du Million Women Study [28] et de la cohorte française des femmes de la MGEN [29] ont clairement démontré le risque supérieur observé avec les progestatifs par rapport aux estrogènes seuls chez les femmes hystérectomisées. Dans ce cas, ce sont les observations faites en situation réelle chez les femmes qui réorientent les recherches fondamentales, afin de comprendre les mécanismes de ces effets délétères sur des modèles moins simplistes que les lignées cellulaires. En effet, la progestérone ne stimule pas directement la prolifération des cellules mammaires cancéreuses in vitro, mais, de par son activité antiestrogénique, est plutôt antiproliférative [30]. Les difficultés s'expliquent en partie par la grande variété des progestatifs utilisés, lesquels peuvent interagir et agir, non seulement sur les récepteurs de la progestérone (RP), mais aussi, en fonction de leur structure, sur les récepteurs des glucocorticoïdes et des androgènes. Cependant, l'étude des lignées a permis de décrire plusieurs protéines induites via les RP telles que l'acide gras synthase [31], le récepteur de l'EGF, le VEGF retrouvés in vivo chez les femmes dans les lésions précancéreuses et les cancers et qui pourraient expliquer l'effet délétère des progestatifs in vivo. De plus, le RU486, un antagoniste de la progestérone, inhibe la croissance des cancers du sein en culture via les RP, ce qui a conduit à proposer les antiprogestatifs comme thérapie ciblée complémentaire des antiestrogènes pour les cancers du sein exprimant les RP [32].
4 Le rôle des estrogènes et de la progestérone dans les étapes initiales de la cancérogenèse mammaire n'est pas élucidé
4.1 Les difficultés sont grandes
Chez la femme, le début du processus cancérigène est silencieux ; il peut avoir été initié 10 à 15 ans avant le diagnostic si l'on tient compte du temps de doublement d'une cellule et des périodes de quiescence.
Il y a peu de modèles satisfaisants permettant d'étudier la cancérogenèse humaine. Les modèles de rongeurs sont différents, avec prédominance des sarcomes et non des épithéliomas, vie plus courte et cancer d'apparition plus rapide, faisant intervenir des oncogènes dominants et des régulations hormonales différentes [33]. Cependant, les systèmes chimériques constitués de greffe de tissu mammaire humain dans des souris immunodéprimées sont des approches prometteuses [34].
Les cellules mammaires humaines précancéreuses sont peu accessibles et nécessitent des méthodes effractives, telles que les biopsies mammaires, plus difficiles à mettre en œuvre que pour d'autres cancers [8].
Enfin, le rôle cancérigène de l'apport extérieur de molécules à activité estrogénique dans l'environnement ou l'alimentation (xénoestrogènes, phytoestrogènes, etc.) n'est pas démontré. Malgré une affinité pour les RE, la quantité de ces apports exogène est-elle suffisante pour interférer avec les sources endogènes des estrogènes naturels ? Les produits sont variés, on manque de données épidémiologiques, mais la prudence s'impose.
4.2 Observation directe de la glande mammaire normale par double immunofluorescence
L'observation directe de la glande mammaire normale par double immunofluorescence a cependant montré que l'effet mitogène des estrogènes faisait intervenir plusieurs types de cellules [35]. En effet, dans les glandes mammaires normales et contrairement au cas des cancers du sein, ce sont les cellules qui ne contiennent pas de REα qui se divisent et qui sont marquées par le Ki 67, marqueur indicateur des cellules en réplication. Cela suggère des régulations intercellulaires et l'intervention de facteurs sécrétés agissant à distance (facteurs paracrines) ou au contact (facteurs juxtacrines) sur les cellules voisines.
4.3 Cellules souches et cancérogenèse mammaire
L'étude du développement des glandes mammaires chez la souris a permis de montrer l'existence de cellules souches conduisant à des précurseurs de la série épithéliale, qui ont conservé la possibilité de former une glande mammaire constituée de canaux et d'alvéoles après stimulation hormonale. Ces cellules, localisées dans l'unité ductulo-lobulaire terminale considérée comme le lieu de départ de la cancérogenèse, se divisent très lentement, mais, du fait de leur immortalité, elles sont exposées à l'accumulation de mutations au cours d'une vie entière. Certaines cellules souches expriment également des récepteurs des estrogènes et de la progestérone, suggérant que les hormones ovariennes, en amplifiant cette population au cours des cycles menstruels, pourraient favoriser le développement de cancers hormonodépendants. Une grossesse précoce pourrait cependant permettre d'éliminer la majorité de ces cellules immortelles avant leur mutation. Deux types de cellules précurseurs contenant ou non des RE seraient à l'origine des deux voies principales de la cancérogenèse mammaire, aboutissant aux cancers hormonodépendants ou hormono-indépendants de grade histopronostique plus élevé [36,37].
5 Notre approche rétrograde de la cancérogenèse mammaire humaine
Avec mes collègues pathologistes, j'ai étendu aux lésions « à risque », définies selon les critères de Dupont et Page [38], la stratégie consistant à caractériser, dans les tumeurs prélevées chez les malades, certaines des protéines régulées par les hormones et décrites dans les lignées cellulaires humaines. L'analyse directe, sur des prélèvements biopsiques de mastopathies proliférantes et de cancers in situ, de gènes et de protéines dont le niveau d'expression est modifié par rapport au tissu normal adjacent, constitue une première approche pour préciser la signification de ces protéines dans les étapes initiales de la cancérogenèse. Nous avons ainsi précisé par immunohistochimie sur coupes de tissus, fixées au formol et incluses en paraffine, les types de lésions « à risque » dans lesquelles l'expression de plusieurs protéines liées à l'action des hormones ovariennes (récepteurs et protéines hormono-induites) étaient modifiées par rapport au tissu mammaire normal adjacent (Fig. 3).
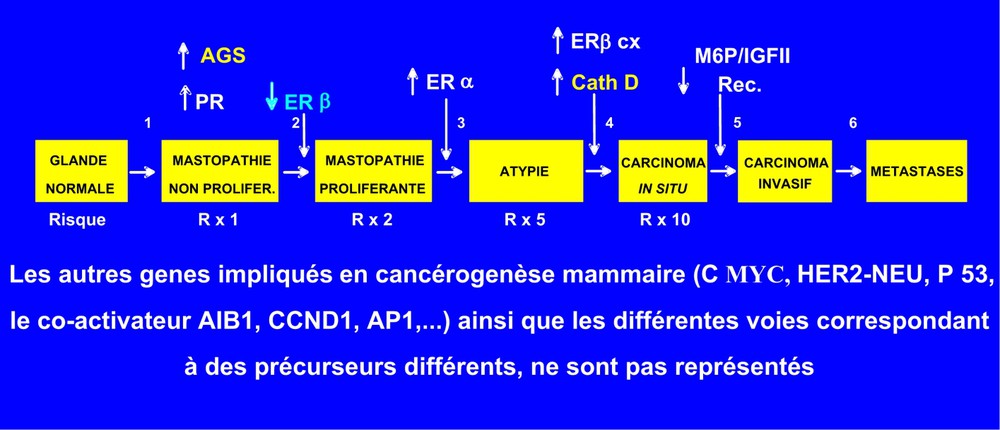
Variations du niveau de certaines protéines liées à l'action des hormones ovariennes au cours de la cancérogenèse mammaire. Les variations d'expression des récepteurs des hormones stéroïdes ovariennes et, en particulier, une diminution du rapport REβ/REα et une augmentation des récepteurs de la progestérone (RP) dans les hyperplasies mammaires suggèrent fortement une hypersensibilité précoce aux estrogènes et aux progestatifs. La surexpression de deux enzymes induites par les facteurs de croissance, l'AGS également induite par la progestérone et la cathepsine D, également induite par les estrogènes, est en accord avec le rôle synergique de ces hormones en cancérogenèse mammaire. Le récepteur du mannose 6P et de l'IGFII (M6P/IGFII) qui transfère les cathepsines et l'IGFII vers les lysosomes a été proposé comme gène suppresseur de tumeur.
5.1 Niveau d'expression des deux RE
C'est ainsi que le niveau d'expression des deux RE évolue en sens opposé dans les mastopathies à risque par rapport aux glandes normales. Le REα est exprimé faiblement et exclusivement dans les cellules épithéliales luminales des glandes normales (∼5% des cellules) ; il augmente jusqu'à 55% dans les lésions proliférantes avec atypie, pour rester élevé dans les carcinomes in situ de bas grade (39%). Il est cependant très bas (0,7%) et souvent nul dans les carcinomes in situ de haut grade (ou comédo carcinomes) [37]. Inversement, le REβ est très largement réparti dans les cellules normales (85%), à la fois dans les cellules luminales et basales et également dans les cellules stromales et inflammatoires. L'expression de la protéine REβ baisse ensuite significativement dès les mastopathies proliférantes sans atypie (34%), pour demeurer à un niveau bas dans les cancers in situ de bas grade (37%) et être très bas (3%) dans les carcinomes in situ de haut grade [39]. Cette baisse du REβ dans les étapes précoces de la cancérogenèse évoque une fonction de protéine suppresseur de tumeur, en totale cohérence avec les études sur lignées et chez l'animal, indiquant que le REβ, en s'hétérodimérisant avec le REα, atténue ses activités transcriptionnelles et mitogènes induites par les estrogènes [24,40]. L'ensemble de ces variations aboutit à une augmentation du rapport REα/REβ et suggère une augmentation de la sensibilité aux estrogènes, qui pourrait expliquer la persistance de l'action des estrogènes formés par aromatisation des androgènes après la ménopause ainsi que l'efficacité en prévention des antiestrogènes et des antiaromatases. Ces résultats indiquent des régulations très différentes pour les 2 RE, également montrées sur lignées cellulaires et dans la glande mammaire normale [41]. Pour le REβ, un blocage épigénétique par méthylation de l'ADN sur îlots CpG du promoteur ON interviendrait [42], comme pour d'autres gènes suppresseurs de tumeur. Des variants d'épissage des RE sont également exprimés à des niveaux variables au cours de la cancérogenèse. C'est ainsi que l'expression du REβcx est augmentée dans les cancers, in situ alors que celle du REβ diminue [43]. Récemment, sur la même population de lésions pré-invasives, nous avons observé une augmentation de l'expression du récepteur de la progestérone, atteignant un maximum pour les lésions prolifératives atypiques [44], ce qui suggère également une augmentation précoce de la sensibilité aux progestatifs.
Ces résultats suggèrent que les hormones ovariennes interviendraient également précocement dans le processus, lent et à plusieurs étapes, de la cancérogenèse mammaire.
Notre analyse des lésions à risque converge avec celle de l'analyse globale des transcriptomes des cancers du sein [45] et celle des pathologistes [46], en indiquant une très grande hétérogénéité de ces cancers, dont la genèse serait branchée plutôt que linéaire. Les 25% environ de cancers du sein n'exprimant pas de RE correspondraient à des voies de cancérogenèse différentes, pour lesquelles d'autres cibles thérapeutiques et préventives que les récepteurs hormonaux sont à trouver. D'ailleurs, les cancers surexprimant l'oncogène HER2/Neu n'expriment généralement pas REα, bien qu'ils proviennent des cellules luminales, mais ils répondent spécifiquement à l'herceptine, deuxième thérapie ciblée de ces cancers constituée par les anticorps neutralisant cet oncogène. Pour la voie conduisant aux cancers canalaires de haut grade REα négatifs et issus des cellules basales, il n'existe pas encore de thérapies ni de prévention ciblée. Cependant les cancers in situ de haut grade surexpriment plusieurs cibles potentielles, dont deux enzymes étudiées dans la même population (Fig. 3).
5.2 Cathepsine D et acide gras synthase (AGS)
Une protéase, la cathepsine D, induite par les estrogènes, et l'acide gras synthase (AGS), induit par la progestérone, tous deux associés à un risque supérieur de rechute et de métastases, sont également surexprimés avant le stade de cancer invasif, indépendamment du niveau des récepteurs hormonaux correspondants. Le niveau de l'AGS est augmenté dès les premières lésions « bénignes », puis continue à augmenter avec le risque, qui devient maximum dans les cancers in situ de haut grade [44]. La cathepsine D est augmentée plus tardivement au stade de carcinome in situ [37], ce qui est cohérent avec son association avec l'émergence clinique de métastases [47].
Ces deux enzymes (cathepsine D et AGS) ont des points communs remarquables :
- (1) elles comptent parmi les protéines les plus abondantes produites par les cancers du sein et par leurs précurseurs les plus agressifs ;
- (2) leur surexpression chez la souris facilite la progression tumorale, leur inhibition l'inhibe, elles constituent ainsi des cibles thérapeutiques potentielles ;
- (3) leur régulation par les stéroïdes ovariens est différente mais complémentaire et en accord avec la synergie des deux types d'hormones ovariennes démontrées par les grandes études épidémiologiques des traitements de la ménopause ;
- (4) bien que spécifiques de chaque classe de stéroïdes, elles sont toutes deux aussi induites par les facteurs de croissance et certaines voies d'activation des protéines kinases pouvant expliquer qu'elles soient également surexprimées dans les cancers du sein in situ et invasifs RE et RP négatifs.
6 Les essais de prévention des cancers du sein
On ne peut accepter comme une fatalité sans réagir l'augmentation récente de l'incidence des cancers du sein dans le monde occidental : en France, elle a doublé en 20 ans, et une femme sur neuf sera atteinte au cours de sa vie. Malgré les progrès des thérapies et les actions de dépistage par mammographie chez les femmes de 50 à 70 ans qui sont lancées, le cancer du sein reste de loin, chez la femme, la première cause de mortalité par cancer [1].
6.1 L'efficacité thérapeutique des antiestrogènes puis des antiaromatases
L'efficacité thérapeutique des antiestrogènes puis des antiaromatases, basée sur le rôle mitogène des estrogènes, a suscité à l'étranger plusieurs essais contrôlés de prévention des cancers du sein chez les femmes ménopausées à risque. C'est ainsi que le tamoxifène (20 mg/jour pendant 4 à 6 ans) diminue d'environ 50% le risque de cancer du sein par rapport à la prise en double insu d'un placébo [48]. Il est autorisé aux États-Unis par la FDA chez les femmes à risque. Exposant moins au risque de cancer de l'endomètre que le tamoxifène, un autre SERM, le raloxifène (60 mg/j) a été évalué. La protection pour le sein est du même ordre qu'avec le tamoxifène, et il est recommandé contre l'ostéoporose. Cependant, persistent d'autres inconvénients non négligeables pour des traitements quotidiens et prolongés : outre leur coût élevé, les SERM augmentent le risque d'accidents thrombo-emboliques veineux et artériels et pourraient faciliter l'émergence des cancers résistants aux antiestrogènes pour des applications prolongées. Dans tous les cas, les SERM ne sont concevables que sur les cancers RE positifs (∼75% des cancers du sein), tandis que d'autres moyens sont à trouver pour les cancers RE négatifs [49]. Des antiaromatases, plus récemment évalués [48], pourraient inhiber l'éventuel effet cytotoxique des métabolites hydroxylés des estrogènes. Mais ils exposent au risque d'ostéoporose chez ces femmes en carence estrogénique.
L'efficacité en prévention des antiestrogènes et des antiaromatases constitue une démonstration supplémentaire du fait que les estrogènes favorisent l'émergence clinique des cancers du sein RE+, mais elle ne permet pas de préciser à quelle étape de la cancérogenèse cette action intervient.
D'autres molécules présentant les avantages des antiestrogènes actuels sans leurs inconvénients sont à l'étude : elles reposent sur une meilleure connaissance des bases moléculaires de la spécificité tissulaire d'action hormonale et sur une étroite collaboration entre diverses disciplines scientifiques et médicales.
6.2 Autres pistes de prévention
D'autres pistes de prévention sont proposées, telles que la protection par des ligands agonistes du REβ, la réexpression génique du REβ par des agents de déméthylation des bases de l'ADN ou l'utilisation de phytoestrogènes. Celles ci n'ont pas encore fait leurs preuves.
Il serait particulièrement intéressant de reproduire chez la jeune femme le fort effet protecteur d'une première grossesse précoce (avant 25 ans) [2]. C'est en effet avant l'apparition d'altérations du génome, certaines induisant une instabilité génétique, que l'on serait le plus efficace. Au cours de la grossesse, l'afflux considérable d'hormones (la concentration d'estradiol est multipliée par 100), suivie de leur chute brutale à la délivrance, pourrait favoriser par apoptose l'élimination de la majorité des cellules souches. Un traitement hormonal court induisant une différentiation mammaire à ce stade permettrait une protection prolongée. Des essais chez les rongeurs à base de hCG [2] ou d'associations estroprogestatives à très fortes doses [50] sont prometteurs. Il n'existe pas encore d'études réalisées chez la femme reproduisant l'effet naturellement protecteur d'une grossesse précoce. Est-il socialement possible de renverser la tendance croissante, dans les pays développés, aux premières grossesses de plus en plus tardives ?
7 Conclusions
Malgré l'explosion des connaissances en biologie et dans les mécanismes de cancérogenèse et de signalisation et l'introduction des nouvelles technologies, les pratiques médicales évoluent trop lentement. Le cancer du sein reste de loin en France la première cause de mortalité par cancer chez la femme (aux États-Unis, c'est devenu le cancer du poumon). Il est amplement démontré que les estrogènes, via leur récepteur, sont des agents promoteurs liés à leur effet mitogène et qu'il est efficace d'inhiber leur action ou leur production. Cependant, les mécanismes initiaux de la cancérogenèse mammaire restent à préciser.
Pour les cancers du sein, le transfert vers la clinique a cependant été initié et réussi assez tôt grâce à l'endocrinologie fondamentale, qui a permis en pratique médicale le dosage des RE dans les tumeurs, puis l'utilisation des antiestrogènes dans le traitement ciblé de certains cancers du sein.
À ce jour, deux types de recommandations me paraissent devoir être proposés pour diminuer l'incidence des cancers du sein et des autres cancers hormonodépendants.
(1)Au niveau des pouvoirs publics, il est essentiel de ne pas négliger la recherche fondamentale, domaine dans lequel la France a démontré sa compétitivité. Il faut en priorité comprendre les mécanismes pour mieux les prévenir. Le cas des cancers du poumon est différent car, ayant identifié le carcinogène principal, il suffit d'abaisser la consommation du tabac pour être efficace. Pour les cancers hormonodépendants et en particulier les cancers les plus fréquents, ceux du sein et de la prostate, les progrès passeront à la fois par la recherche fondamentale et par une recherche « de transfert », conduisant à des applications cliniques sur des critères uniques d'excellence et d'originalité. Il faut pouvoir appliquer les résultats de la recherche à la pathologie humaine et les confronter à ceux des études épidémiologiques et des essais thérapeutiques contrôlés. Cela nécessite un dialogue constructif entre médecins et chercheurs des organismes de recherche. Les exemples précédents nous ont montré qu'il faudra vraisemblablement du temps pour démontrer, puis choisir et transférer à la clinique certaines des hypothèses passionnantes récemment proposées par les scientifiques.
(2)En pratique médicale et dans l'immédiat, les malades et leurs médecins ne pouvant attendre, il faut dépister les femmes à risque si elles le demandent ; des consultations d'oncogénétique ont été créées à cet effet. Dans le cas des femmes porteuses de mutations des gènes BRCa1/2, outre la surveillance, des méthodes de prévention plus agressives telles qu'une ovariectomie, voire une mastectomie bilatérale prophylactique peuvent être envisagées, car on ne dispose pas encore de chimioprévention démontrée, mais il est urgent de mieux faire.
Dans la majorité des cas, on doit à ce jour se contenter de conseiller une meilleure hygiène de vie permettant de se prémunir des risques évitables, tels qu'un THS prolongé après la ménopause, l'abus d'alcool, l'obésité et la sédentarité, ce qui présente également le mérite de protéger contre les maladies cardiovasculaires.
Remerciements
Je remercie les membres de l'unité 148 de l'Inserm et tous mes collègues et maîtres, en particulier Étienne Baulieu et Maurice Tubiana.