

Abridged English version
T cells genetically manipulated to express fluorescent proteins (e.g., the Enhanced Green Fluorescent Protein, GFP) have become prime probes to unravel the complex mechanisms that are involved in the generation of human and experimental disease. In particular the approach provides insight into the pathogenic steps not only during clinical disease, but also into the important phases before and thereafter.
Using conventional confocal microscopy, and subsequent molecular analysis of brain infiltrating Green T cells from autoimmune brain lesions, we had learned before that millions of the pathogenic effector T cells invade the brain at the onset of EAE. In fact, in these early stages of disease, the overwhelming majority of invading T cells are, green, i.e. are effector cells. Further, it has been clear that invasion by these cells must have occurred rapidly, within a few hours.
This speed is remarkable, considering that the brain white matter is formed by tightly packed neuronal processes, axons, which are enwrapped by the isolating multilayer of myelin substance. The gaps left between the packed myelinated axons are filled completely by glia cells, astrocytes and microglia.
To explain such a rapid invasion of such a dense tissue we used the new optical technique of 2-photon microscopy. The technology makes used of two laser beams, which scan through the distinguishable structures of living tissues. The method offers an unprecedented depth of penetration into the tissue with extremely high structural resolution. Further, the imaging is unusually gentle, with remarkably low phototoxic effect on the tissue.
We now have followed autofluorescent green effector T cells on their way from the inside of blood vessels into the brain tissue, and noted a discrepant behaviour. Many of the cells (about 60–70%) cruise through the nervous tissue with unexpected speed. They navigate without respecting any visible structural barriers. Other cells (30–40%), however, become attached at a particular place and begin to toss around some central object.
What enables the T cells to slip that fast through the packed brain tissue, which signals drive them through the CNS, and what arrests them all of a sudden? The first two questions remain unanswered to date. We assume that the T cells release molecules – enzymes, presumably – that loosen the tissue structure and thus create space for the migrating T cells. But this does not explain the forces required for the migration, and the signals that must be required for proper guidance. We postulate that the cells follow signals (e.g., chemokines) emanating from local tissue cells that tell them to move on, but other mechanisms are possible, too. Further we speculate that the migrating cells are on the search of their brain autoantigen, which is known to be presented by local ‘presenting cells’. In the case of the arrested cells, there is now good evidence that these cells have found their autoantigen, that their arrest reflects presentation and recognition of the antigen.
Several observations bolster our interpretation. First, as mentioned, MBP specific, autoreactive T cells migrate and then may be arrested locally. In contrast, ovalbumin specific T cells, which are not supposed to recognize their target antigen in the brain tissue, only migrate, but are never stopped. Arrest correlates with potential antigen recognition. Second, we recently established a correlation between the pathogenic potential and the migratory behaviour of encephalitogenic T cells. T cells with a high pathogenic potential, as the MBP specific ones, display a relatively high proportion of arrested cells, while in the case of their correlates with low pathogenic potential (e.g., specific for other brain auto-antigen such as S100β), the proportion of arrested cells is significantly lower. This further correlates with in situ activation as assessed by transcription and production of activation markers, pro-inflammatory cytokines and chemokines. Interestingly, about one third of all infiltrating MBP specific T cells show marked down-modulation of their T cell receptor complex, an indication of active antigen recognition. Finally, preliminary observations suggest that blockade of MHC class II antigens, which bind and present antigenic peptides to T cell receptors, cancels capture of T cells, but does not interfere with their migration.
1 Introduction
La sclérose en plaques, l'encéphalopathie la plus importante chez des jeunes adultes du monde « développé », passe communément pour une maladie auto-immune causée par des lymphocytes T qui, stimulés en dehors du système nerveux central (SNC), franchissent la barrière hématoencéphalique (BHE) et se nichent au sein du cerveau et de la moelle épinière [1]. Selon ce concept, la reconnaissance de l'auto-antigène spécifique par les cellules effectrices intruses déclenche une réponse inflammatoire, qui aboutit à la destruction des gaines myélinisées et des axons.
Cependant, il faut souligner que, même si à l'heure actuelle cette pathogénie reste à être confirmée formellement, elle est renforcée par un certain nombre d'arguments convergents. D'abord, l'histologie suggère un processus immuno-pathologique. Dans leur état actif, les plaques présentent typiquement des foyers inflammatoires, avec des cellules immunes (lymphocytes et macrophages) qui enveloppent les vaisseaux sanguins, formant des « manchons », et qui pénètrent dans le tissu nerveux voisin. De plus, on a trouvé dans des lésions de SEP des cellules T spécifiques d'auto-antigènes de myéline. Enfin, le succès clinique des thérapies anti-lymphocytaires suggère fortement une pathogénie auto-immune [2].
Reste une énigme capitale : étant donné l'isolement naturel du système nerveux, tissu strictement séquestré de la circulation sanguine, comment les lymphocytes T auto-réactifs peuvent-ils franchir la BHE imperméable à la plupart des cellules et macromolécules sanguines ? De plus, comment peuvent-ils fonctionner effectivement dans un milieu réputé immunosuppressif ? Énigme de l'arrivée, énigme centrale de la SEP.
La recherche neuro-immunologique a largement bénéficié d'un modèle expérimental, l'EAE (encéphalomyélite auto-immune expérimentale), qui représente fidèlement des processus cardinaux de la pathogénie dans le SNC. L'EAE est induite chez un animal expérimental par l'immunisation contre du tissu nerveux homologue (dérivé de la même souche de souris ou rat). Cette formule d'immunisation produit des effets neurologiques et histologiques très semblables à ceux de la SEP. Mais dans le cas de l'EAE, la nature auto-immune de l'affection est évidente, les lésions sont créées par de cellules T auto-réactives [3].
2 Le SNC intact, terre aride pour les cellules immunitaires
En 1948, Sir Peter Medawar, prix Nobel en 1960, observait que des coupes de peau de lapin, transplantées sur la peau d'un autre lapin, sont bien rejetées ; pourtant ces mêmes tissus, transplantés à la surface du cerveau de l'animal receveur, sont tolérés [4]. Cette observation marquait l'origine du concept du « privilège immunologique » du SNC [5]. Cette théorie postule que certains tissus dans l'organisme sont exclus de la surveillance immunologique qui protège nos organes contre des microbes envahissants et également contre des cellules cancéreuses apparues fraîchement. Donc, à quoi bon ce privilège douteux qui exposerait nos tissus nerveux à des infections et à des tumeurs ? Quelles structures seraient responsables de l'absence de réponse immune ?
Au contraire de la plupart des autres tissus, le SNC est séparé de la circulation sanguine par une barrière quasi imperméable pour les macromolécules et les cellules, la barrière hémato-encéphalique (BHE). Dans le SNC normal, les vaisseaux sanguins sont enveloppés par des cellules endothéliales reliées par des jonctions étroites (tight junctions) [6]. Cela implique que les cellules immunitaires qui circulent dans les vaisseaux sanguins sont exclues du SNC, et, en conséquence, que le cerveau et la moelle épinière sont privés de surveillance immunologique classique.
Le système nerveux se distingue des autres tissus par une autre particularité. La plupart des tissus sont drainés par un réseau de vaisseaux lymphatiques, qui transportent des structures antigéniques, mais aussi des cellules dendritiques, cellules présentatrices d'antigènes, aux ganglions lymphatiques locaux [7]. Pourtant, le système nerveux ne possède pas un tel système de drainage lymphatique bien organisé. En effet, hormis une connexion entre le cerveau frontal et les ganglions lymphatiques cervicaux, qui permet l'exportation de certaines molécules, on ne trouve pas de vaisseaux lymphatiques bien organisés dans le système nerveux [8]. Ainsi, l'échange humoral entre les systèmes nerveux et immunitaires est limité au strict minimum, ce qui renforce la spécificité du SNC.
L'absence des vaisseaux lymphatiques est expliquée par des recherches récentes. Des études réalisées sur des souris transgéniques démontrent que le développement des vaisseaux lymphatiques est la conséquence d'interactions complexes entre des facteurs inducteurs solubles, comme des chimiokines, des membres de la famille VEGF et les cellules progénitrices [9]. Les facteurs angiogéniques sont produits de préférence au cours d'une réaction inflammatoire ou immunitaire. Il semble que le SNC, qui normalement exclut des réponses inflammatoires, ne produise pas la gamme des facteurs inducteurs requis pour la néoformation de vaisseaux lymphatiques.
Le troisième aspect caractéristique « anti-immunitaire » du SNC normal est le déficit quasiment total en molécules du complexe majeur d'histocompatibilité (CMH), voire en cellules dendritiques. Ni les molécules CMH de classe I ubiquitaires, qui s'expriment continuellement sur les surfaces cellulaires de toutes les lignées cellulaires, ni les molécules de classe II, dont l'expression est induite par des cytokines pro-inflammatoires (interféron gamma, TNF-alpha), ne se trouvent dans le SNC normal [10]. En effet, alors que l'on trouve des molécules de classe I sur les membranes endothéliales des vaisseaux cérébraux, elles sont absentes des autres cellules locales, neurones, astrocytes, oligodendrocytes et microglie inclus.
La carence en molécules CMH ou leur absence dans le SNC trouve plusieurs explications. D'abord, tandis que, sans doute, les milieux nerveux conservent en principe la capacité à produire des cytokines et chimiokines pro-inflammatoires, les taux de ces facteurs libérés sont bas dans les tissus normaux, et la gamme des facteurs produits dans le SNC est limitée. En effet, il y a certaines cytokines qui sont produites dans le SNC sain, comme, par exemple, le « facteur de nécrose des tumeurs alpha » (TNF-α), l'interleukine-1 et l'interféron gamma, les cytokines pro-inflammatoires les plus importantes. Il semble qu'elles jouent un rôle physiologique dans des fonctions comme le sommeil, l'anorexie, la plasticité et le développement neuronal [11]. Mais leurs niveaux dans les tissus nerveux sont bas, bien plus bas que dans les réactions inflammatoires du cerveau, ou dans les organes lymphoïdes périphériques.
Point très important, ce n'est pas seulement la production insuffisante des molécules « immunitaires » qui est responsable de l'absence des produits CMH dans le système nerveux. En effet, le SNC a développé des mécanismes de régulation négative, qui suppriment activement la production des molécules CMH. Spécifiquement, ce sont les neurones intacts qui suppriment la production des molécules CMH par des cellules gliales voisines [12]. Cette observation résulte d'expériences utilisant des cultures de neurones dérivés de l'hippocampe de jeunes rats. Durant une période de deux ou trois semaines, il se forme dans ces cultures un réseau de neurones supporté par une couche de cellules gliales, des astrocytes. Ayant atteint un certain degré de maturation, les neurones commencent à échanger des signaux électriques par des contacts synaptiques. Ni dans ces cultures, ni dans le tissu cérébral in vivo, les cellules gliales (astrocytes et même la microglie) ne produisent de molécules CMH. Elles résistent même à leur induction par l'interféron gamma. Cette suppression est levée de façon abrupte après une paralysie des neurones, par exemple, par le blocage des canaux sodiques avec de la tétrodotoxine (poison du poisson-ballon, Fugu). En présence de neurones paralysés, les cellules gliales répondent rapidement aux signaux de l'interféron gamma par la synthèse de novo et l'expression de CMH [13]. Par ailleurs, non seulement la paralysie des neurones favorise l'induction de la synthèse des molécules CMH dans les cellules gliales voisines, mais encore les neurones paralysés eux-mêmes se mettent à synthétiser des protéines CMH sous l'action de l'interféron gamma (vide infra).
Les mécanismes moléculaires contrôlant la suppression par les neurones des gènes CMH dans les cellules gliales voisines restent encore à déterminer. À l'heure actuelle, on discute l'idée selon laquelle l'activité « électrique » de la membrane neuronale pourrait délivrer des signaux suppresseurs, peut-être via un contrôle des niveaux de calcium intracellulaire. Plus probablement, les neurones actifs communiquent leurs signaux suppresseurs via des facteurs humoraux [12]. On connaît, par exemple, des neuropeptides et des cytokines qui sont libérés de neurones actifs et qui suppriment l'expression du CMH. Peut-être plus importants sont les neurotrophines, facteurs trophiques sécrétés par des cellules neurales, qui contrôlent la croissance, la spécialisation et la communication des neurones, mais, de plus, régulent l'activité des cellules qui sont hors du SNC, notamment celles des cellules du système immunitaire. La neurotrophine classique, le NGF (nerve growth factor), libérée par des neurones, supprime fortement l'expression des gènes CMH dans les cellules gliales in vitro [14]. De plus, in vivo, la neurotrophine supprime les réponses inflammatoires de l'EAE [15]. Il semble que les neurotrophines soient intégrées dans un réseau de facteurs solubles contrôlant l'homéostase inflammatoire, non seulement dans le SNC, mais également dans le reste de l'organisme. En particulier, par la voie du récepteur « de basse affinité », p75, membre de la famille des récepteurs TNF [16], les neurotrophines suppriment des réponses inflammatoires, qu'elles soient allergiques ou auto-immunes [17].
On se demandera pourquoi le SNC, tissu extrêmement vulnérable, se priverait de la surveillance immunitaire en général et de l'expression de molécules CMH en particulier ? On a spéculé sur le fait que la fragilité du parenchyme nerveux exigerait que le nombre des réponses inflammatoires ou immunitaires se limitât à un minimum essentiel. Évidemment, chaque réponse inflammatoire causerait des dommages « collatéraux », aboutissant à l'activation pathologique des cellules gliales et à la dégénérescence des neurones locaux.
Selon une théorie alternative, mais pas exclusive, la fonction du CMH dans le SNC ne se limite point à la réponse inflammatoire. On a trouvé des membres de la famille CHM dans l'organe voméro-nasal, où ces protéines participent à la reconnaissance des phéromones [18]. De plus, des produits CMH pourraient jouer un rôle dans le développement des neurones immatures [19].
3 Le SNC dégénératif : milieu propice aux réactions immunitaires ?
Bien qu'à l'état normal, le SNC présente un milieu supprimant les cellules immunitaires et ses réponses, on connaît tout une gamme d'états pathologiques présentant sans doute des réactions inflammatoires ou immunologiques. On citera, à titre d'exemple, les encéphalites auto-immunes cliniques ou expérimentales, des infections virales, des tumeurs et, de manière plus étonnante, des processus neurodégénératifs. Il est bien accepté que la plupart des maladies neurodégénératives, incluant les maladies d'Alzheimer, de Parkinson et de Huntington, présentent des réponses inflammatoires importantes [12]. Les lésions dégénératives sont caractérisées par l'expression « ectopique » des molécules CMH, particulièrement sur les cellules microgliales, et par des infiltrations cellulaires inflammatoires. Apparemment, la dégénérescence neuronale crée un milieu favorable aux réponses inflammatoires, un milieu « pro-immunitaire ».
Quels sont les facteurs transformant les tissus cérébraux de l'état anti-immunitaire à l'état pro-immunitaire ? Un modèle expérimental, l'axotomie unilatérale du nerf facial, a été particulièrement informatif [20]. Ce modèle s'appuie sur la section unilatérale du nerf facial, nerf exclusivement moteur, d'une souris ou d'un rat. L'interruption du nerf périphérique provoque une dégénérescence rétrograde, qui progresse le long du nerf pour gagner le noyau moteur facial niché dans le tronc cérébral et hébergeant les somata des nerfs faciaux. Ainsi, après axotomie unilatérale, le tronc cérébral contient un noyau dégénératif (du côté de l'axotomie) et un noyau intact de l'autre côté, une situation très favorable à la comparaison des effets de la dégénérescence neuronale sur le milieu cérébral.
Le modèle de l'axotomie unilatérale du nerf facial a été utilisé dans une pléthore d'études visant à examiner les effets de la dégénérescence neuronale sur la structure immunologique des tissus nerveux. En combinant des techniques immunochimiques et moléculaires, les travaux entrepris ont montré que la perte de fonctions des neurones moteurs faciaux atteints résulte dans la production de novo d'une gamme de gènes « immunitaires », parmi lesquels les gènes CMH (classe I et classe II), des cytokines et chemokines, des molécules de co-stimulation ou d'adhésion cellulaire requises pour un fonctionnement correct des cellules immunitaires [21].
La preuve formelle de la transformation pro-immunitaire du tissu neurodégénératif est clairement apportée par le système de l'EAE transférée. Les cellules T encéphalitogéniques injectées par voie intraveineuse chez un rat sectionné unilatéralement montrent une affinité quasi-exclusive pour le noyau facial dégénératif. Tandis que des légions des cellules T envahissent le noyau lésionné, les mêmes cellules négligent le noyau controlatéral intact. Cette migration discordante renforce l'idée selon laquelle le milieu dégénératif est devenu un terrain attractif et favorable aux cellules auto-immunes, et qu'il promeut l'immigration et la réactivité des cellules T [22,23].
Il ne sera pas surprenant que la transformation des tissus nerveux d'un état anti-inflammatoire à un milieu pro-inflammatoire ne se limite point à notre modèle de dégénérescence du nerf facial, mais se produise dans le contexte des maladies neurodégénératives humaines. Comme cela a été signalé auparavant, les maladies d'Alzheimer, de Parkinson, de Huntington et la sclérose latérale amyotrophique possèdent toutes une composante inflammatoire. On y trouve l'activation des cellules microgliales, qui produisent et expriment des molécules CMH des deux classes, la synthèse de cytokines et chimiokines pro-inflammatoires ; souvent, on observe des infiltrations lymphocytaires.
Quel est le rôle pathogénique de la réponse inflammatoire para-dégénérative ? Selon un point de vue populaire et traditionnel, cette réaction serait franchement néfaste, avec des cellules inflammatoires sécrétant des facteurs toxiques et ajoutant ainsi à l'endommagement provoqué par le processus dégénératif intrinsèque [24]. Mais on trouve un autre point de vue, radicalement opposé. Cette dernière théorie attribue un rôle bénéfique à la réponse inflammatoire [25]. Elle se fonde sur l'observation du fait que les cellules immunitaires, en particulier les cellules T, produisent et libèrent, non seulement des médiateurs toxiques, mais aussi des facteurs régénérateurs. Ceux-ci pourraient supporter ou même sauver des cellules neurales dégénératives. En effet, il est bien connu qu'après leur activation, les lymphocytes sécrètent des facteurs neurotrophiques, qui protègent les neurones contre les effets toxiques et au même temps suppriment les réactions inflammatoires exagérées [26]. Les partisans de l'auto-immunité bénéfique, non seulement s'élèvent contre une thérapie anti-inflammatoire des maladies neurodégénératives humaines, mais se piquent même de propager une thérapie faisant appel aux réponses auto-immunes limitées.
En somme, la découverte de la régulation duale du milieu immunitaire du SNC, d'une part négative par les neurones, et d'autre part positive par les cellules immunitaires, pourrait résoudre un problème fondamental posé par la sclérose en plaques. Les plaques sont distribuées typiquement dans toute la substance blanche du cerveau et de la moelle épinière, apparemment sans règle. La découverte du contrôle de l'état immunitaire du milieu nerveux central par l'activité neuronale pourrait expliquer cette distribution. En conséquence, un endroit affaibli par la neurodégénérescence pourrait attirer les cellules T pathogènes et leur offrir un milieu propice à une attaque auto-immune.
4 L'entrée des cellules T dans les tissus nerveux centraux : deux vagues d'immigration
L'encéphalite auto-immune expérimentale (EAE) offre, sans doute, le meilleur modèle expérimental pour l'étude de l'auto-immunité dans le SNC [3]. Comme on l'a déjà constaté, l'EAE est causée par des cellules T activées. Ces cellules T « effectrices » sont auto-réactives, reconnaissant exclusivement des protéines de la myéline centrale dans le contexte de molécules CMH de classe II. Il est important de noter que, pour transférer la maladie, il faut que les cellules T soient en état d'activation maximale et que les cellules T effectrices soient également fortement activées dans les lésions cérébrales. Néanmoins, cela ne veut pas dire que les cellules effectrices activées passent directement du lieu d'injection au tissu nerveux sans changer leur état d'activité. Tout au contraire, on a appris dès les premières expériences sur l'EAE transférée qu'il existe un intervalle de temps entre l'injection et le début de la maladie neurologique de trois à quatre jours. Quel est le rôle de cette période prodromique et quel est le sort des cellules T auto-immunes pendant ce temps-là ? On verra que les cellules T subissent des changements majeurs. Non seulement elles passent par une route migratoire compliquée entre les organes lymphoïdes et le système nerveux, mais encore, durant cette migration, elles changent radicalement leur profil d'expression de gènes. Au lieu du phénotype initial d'activation maximale, elles adoptent un phénotype dit « migratoire ». Seules les cellules migratrices sont compétentes pour passer en grands nombres par la BHE. On sait bien que dans le SNC normal, les astrocytes secrètent de certaines chimiokines [27], mais il semble que leur taux soit trop bas pour les lymphocytes en général, mais suffisant pour les cellules T activées.
Il est aussi bien connu qu'au cours de l'EAE, l'invasion des tissus nerveux centraux par les cellules immunitaires se passe en deux vagues. La première vague, qui se compose d'un nombre très limité de cellules T activées, traverse la BHE dans les heures suivant la transfusion [28,29]. Ces cellules lymphoblastiques s'accrochent à la face interne des microvaisseaux cérébraux, et par des mécanismes mal connus traversent la barrière endothéliale [30]. Il faut bien noter qu'à ce moment-là, les cellules endothéliales se trouvent en état « quiescent » ou « naïf » et sont imperméables à la plupart des cellules et macromolécules sanguines. Il semble que les lymphoblastes auto-réactifs, grâce à leur activité produisent et libèrent des enzymes protéolytiques et glycolytiques qui perforent la barrière endothéliale et la membrane basale du vaisseau. Guidées par des signaux inconnus, les cellules T entrent alors dans le tissu cérébral.
Après leur entrée dans le parenchyme, les cellules T disparaissent de la vue du chercheur, et leur sort reste énigmatique. Sans preuve formelle, on présume que les cellules T auto-immunes arrivées dans le tissu continuent de sécréter des enzymes et des cytokines/chimiokines, facteurs pro-inflammatoires qui stimulent les cellules gliales et endothéliales pour produire des molécules « immunitaires », comme on l'a décrit ci-dessus.
En effet, pendant le stade prodromique de 3–4 jours qui précède l'EAE clinique, on note des changements importants de l'endothélium cérébro-vasculaire ainsi que des cellules gliales. Pendant cette période critique, les cellules endothéliales de la BHE se mettent à produire une pléthore de molécules d'adhésion cellulaire, absentes de l'endothélium naïf, et la BHE jadis imperméable s'effondre. En même temps, des cellules gliales, en particulier la microglie, commencent à exprimer des produits du CHM, des molécules de co-stimulation et d'adhésion cellulaire nécessaires à la présentation des antigènes aux cellules T auto-immunes et au développement d'un milieu favorable aux réactions immunitaires [31]. On parle alors d'une BHE et d'un tissu nerveux « amorcés ».
Les lymphoblastes qui percent la BHE immédiatement après leur transfert représentent une faible minorité des cellules effectrices. La plupart des cellules auto-immunes restent exclues du SNC pendant la période de 3–4 jours qui précède l'attaque clinique. Durant cette période prodromique, les cellules auto-immunes subissent des changements radicaux, qui les préparent à la seconde vague d'invasion du SNC. Des études utilisant des lignées de cellules T auto-immunes exprimant la protéine verte fluorescente (voir ci-dessous) trouvaient qu'après leur transfert, les lymphocytes s'apprêtent à une migration bien réglée au travers des organes lymphoïdes périphériques. Comme première étape, ils gagnent les ganglions lymphatiques parathymiques, ganglions qui drainent les poumons et l'espace péritonéal. Après un séjour de 12–24 h, les lymphocytes quittent les ganglions pour gagner par voie sanguine la rate, où ils attendent jusqu'à l'invasion du SNC [32].
Durant leur séjour dans les organes lymphoïdes, les cellules T subissent un changement radical de leur profil d'expression génique. Les gènes marquant l'activation des cellules T (CD25, OX-40, par exemple), qui étaient fortement exprimés dans les lymphoblastes transférés, sont supprimés. En revanche, dans le même temps, une série d'autres gènes apparaît de novo, parmi lesquels se trouvent les récepteurs de chimiokines, structures essentielles pour guider les cellules durant leur migration [32]. Une analyse du transcriptome faisant appel à des microarrays témoigne d'un réarrangement profond du profil génétique des cellules T effectrices pendant leur séjour dans les organes lymphoïdes. Les facteurs contrôlant ces changements restent à présent inconnus. Il est cependant probable que ce soient des structures produites par les cellules du stroma local, facteurs solubles ou membranaires, qui transmettent aux lymphocytes des signaux suppresseurs. Par exemple, plusieurs membres de la famille de gènes B7 s'expriment sur des cellules présentatrices d'antigène, et sur d'autres cellules. Ils reconnaissent des molécules lymphocytaires comme PD-1 et répriment l'activation de ces cellules immunitaires [33]. De même, la cytokine soluble TGF-β et son récepteur complémentaire SMAD déclenchent une cascade de signaux suppresseurs [34]. Le dialogue des cellules T avec le milieu lymphoïde ne résulte pas d'une atténuation globale de l'activité immunitaire, mais plutôt cette interaction sert à remodeler leur transcriptome de façon à les rendre compétentes pour l'invasion de l'organe cible, le SNC. Le contact avec le milieu lymphoïde sculpte le phénotype « migratoire » des cellules T auto-immunes effectrices.
Pendant leur séjour dans les ganglions lymphatiques et la rate, les cellules T auto-immunes fortement activées acquièrent donc un phénotype nouveau, « migratoire », essentiel à leur invasion du SNC. Précisément 3–4 j après l'infusion des cellules T effectrices, on est en présence de millions de ces cellules en état migratoire qui, obéissant à un signal inconnu, sortent de la rate, et par la voie sanguine arrivent dans le SNC, où ils trouvent la BHE déjà stimulée.
Question décisive : quels sont les signaux dirigeant les deux vagues cellulaires à travers de la BHE ? Dans le cas des cellules T pionnières qui traversent la BHE naïve, la nature de ces signaux est complètement inconnue, l'existence même de tels signaux restant incertaine à ce jour. Il est possible que les cellules T activées répondent avec une plus grande sensibilité aux chimiokines du SNC quiescent et qu'elles reconnaissent les molécules d'adhésion sur les cellules endothéliales de la BHE. À l'inverse, les cellules T « migratoires » de la seconde vague se confrontent à une BHE et à un parenchyme neural activés. On connaît le profil particulier de molécules d'adhésion et de chimiokines pro-inflammatoires décorant la surface des micro-vaisseaux qui attire les cellules T migratoires et les fait envahir le tissu. C'est en quelques heures seulement que se produit une invasion de millions de cellules T dans le cerveau et la moelle épinière, et c'est cette invasion en masse qui marque le commencement abrupt de la maladie clinique (symptômes paralytiques et perte de poids).
5 Après l'arrivée
Les cellules T auto-immunes qui passent à travers de la BHE trouvent un milieu cérébral bien préparé pour des réponses (auto-) immunitaires. En particulier, durant la période prodromique, les cellules microgliales acquièrent de novo toutes les molécules requises pour présenter des antigènes, protéines étrangères ou auto-antigènes, aux cellules T spécifiques [35]. Pourtant la microglie ne produit pas elle-même des auto-antigènes, mais plutôt, ses cellules jouant un rôle de gardiennes du SNC, elle absorbe des protéines du liquide interstitiel (ou du liquide céphalo-rachidien) et les digère pour les présenter comme des molécules CMH. La microglie ramasse tout, aussi bien des organismes microbiens que des corpuscules de membranes ( « exosomes ») libérés des cellules neurales. Au cours de l'EAE, ce sont des vésicules de la myéline ou des protéines solubles provenant des gaines de myéline péri-axonales qui sont la source des auto-antigènes encéphalitogéniques.
Comment se déroule l'attaque des cellules T intruses contre ses cibles neurales ? Utilisant des méthodes traditionnelles morphologiques – soit l'immunocytochimie, soit l'hybridation in situ – on sait que la plupart des cellules T infiltrées se recrutent au sein de la classe CD4 (cellules T auxiliaires ou helper), et que ces cellules s'entremêlent avec des macrophages co-immigrés dans le SNC. Nombre de cellules inflammatoires s'arrangent autour des microvaisseaux, formant des « manchons » périvasculaires, tandis que d'autres pénètrent directement dans le tissu compact. Le séjour des cellules inflammatoires dans le SNC est éphémère, et ne dure que quelques jours. On sait bien que la plupart de ces cellules meurent d'apoptose, un processus contribuant à la résolution des lésions inflammatoires et à la terminaison des attaques cliniques [36]. Cependant, il est moins clair qu'il reste d'autres cellules T spécifiques qui survivent et regagnent le système lymphatique pour y déclencher des nouvelles rechutes [37].
Une seconde stratégie classique pour la recherche de l'attaque auto-immune utilise l'isolement des cellules inflammatoires du tissu envahi, et l'analyse par cytofluométrie de leur phénotype de surface fournit des informations sur la distribution des populations cellulaires et leur état d'activité. De plus, les cellules ainsi isolées se prêtent à des analyses fonctionnelles en culture. Ces études ont montré que les cellules T effectrices subissent une réactivation puissante au sein du tissu cérébral, suggérant fortement un processus de reconnaissance de l'auto-antigène de la myéline [32]. Mais, malheureusement, ni la morphologie, ni l'étude des cellules isolées ne peuvent fournir une image complète des événements dynamiques de la réaction auto-immune dans la lésion cérébrale. En effet, la morphologie est figée, et l'isolement des cellules rompt les interactions cellulaires avec le milieu.
Tout récemment, une nouvelle technologie microscopique, la microscopie à deux photons [38], permet l'observation directe des cellules T dans le tissu nerveux vivant. Cette méthode offre trois avantages uniques. En comparaison de la microscopie classique, la résolution des images de MDP est supérieure, la pénétration dans le tissu de plusieurs fois plus profonde, et, fait très important, la phototoxicité du rayonnement est très réduite.
L'équipe de Kawakami et Flügel a utilisé un protocole permettant le marquage des cellules T auto-immunes par l'introduction d'un gène encodant la protéine fluorescente verte (Green Fluorescent Protein, GFP), gène dérivé d'une méduse marine bioluminescente [39]. Introduit dans des cellules T auto-immunes à l'aide d'un vecteur rétroviral, le gène PFV est fortement exprimé, sans gêner la fonction (auto)immune. Donc, après le transfert, les cellules T auto-immunes fluorescentes se retrouvent dans l'ensemble de l'organisme hôte avant, durant, et après l'EAE.
L'EAE transfectée par les cellules T vertes, combinée à la microscopie à deux photons, se prête idéalement à l'analyse des événements cellulaires dans la fraîche lésion cérébrale récente. La première observation surprenante était le nombre considérable de cellules T effectrices qui entrent dans le parenchyme cérébral pendant quelques heures. Selon le point de vue traditionnel, la seconde vague d'invasion serait composée d'une minorité de cellules effectrices parmi une majorité de cellules inflammatoires non spécifiques (macrophages, cellules T non auto-immunes). Tout au contraire, les études directes des cellules T vertes démontrent qu'en vérité des millions des cellules effectrices passent par la BHE (Fig. 1), et que la seconde vague contient à plus de 90% des cellules T auto-réactives [40]. Cette proportion décroît pendant les jours suivants, parce que nombre de cellules T meurent localement d'apoptose et, de plus, parce que les cellules auto-immunes sont diluées par une armée de cellules T non auto-immunes qui infiltrent le tissu, attirées par les cellules effectrices.
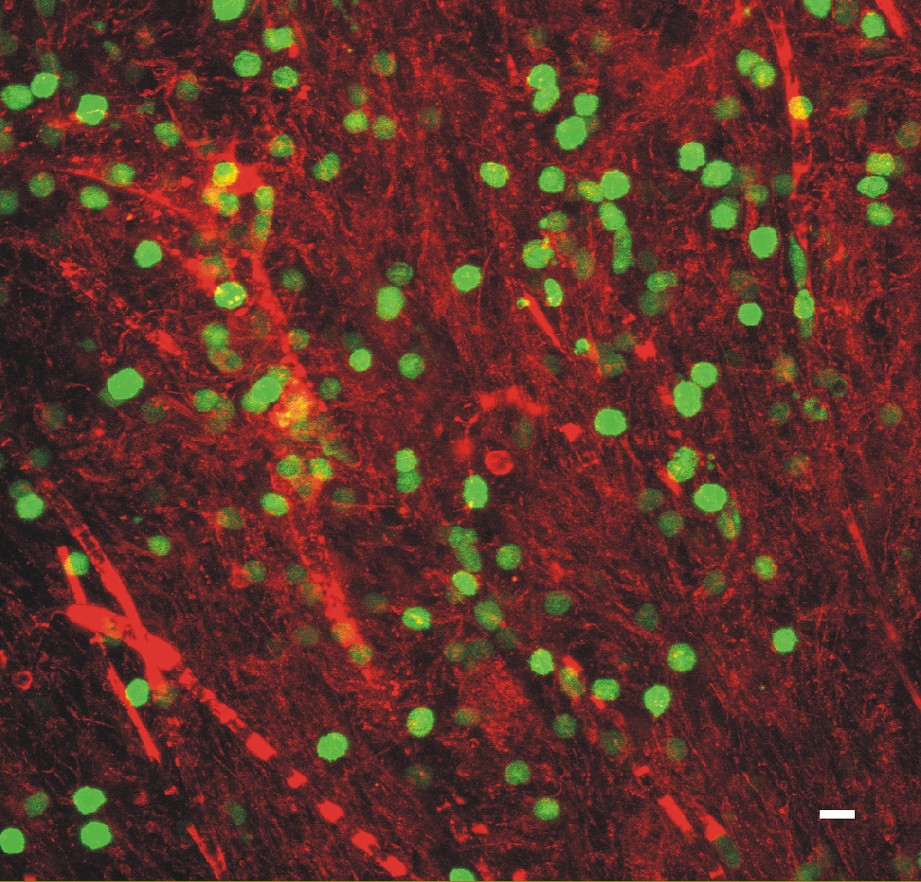
Cellules T effectrices vertes dans une lésion inflammatoire de la moelle épinière au début de l'EAE. Vert : autofluorescence de la protéine fluorescente verte de cellules T ; rouge : microvaisseaux après une infusion de dextrane marquée d'un fluorochrome rouge.
Comment peut se dérouler l'invasion d'un si grand nombre de lymphocytes sur une si brève période de temps ? La justification de cette question tient au fait que le parenchyme cérébral est un tissu extrêmement compact. Il est bourré d'axones enveloppés de myéline et toutes les lacunes entre ces structures sont remplies de cellules gliales, astrocytes, microglies et oligodendrocytes. Donc, on pourrait penser que les cellules migratoires pénètrent lentement dans le tissu, se frayant un sentier à travers cette jungle cellulaire. Mais ce n'est pas du tout le cas. Tout au contraire, la microscopie à deux photons démontre que la plupart (à peu près 70%) des cellules T traversent le tissu à une vitesse formidable, comme les lymphocytes courant dans les organes lymphoïdes, milieux quasi fluides. Elles parcourent le tissu en toutes directions, apparemment à l'aveuglette (Fig. 2A). De plus, leur vitesse de leur migration n'est pas constante, mais plutôt hectique, avec des courses rapides suivies d'épisodes lents ou même des arrêts brefs. Assez souvent, pendant les périodes lentes, les cellules T changent de direction pour reprendre une grande vitesse [41].

Mobilité des cellules T effectrices au sein des lésions EAE [41]. Microscopie à deux photons d'une tranche vivante de moelle épinière provenant d'une ratte touchée d'EAE. On observe deux types de mouvement : des cellules T motiles (A), et des cellules T stationnaires (B). Les nombres indiquent le temps écoulé depuis le début de l'analyse (min). Les cellules représentent dix vidéos de quatre expériences indépendantes.
Les cellules T auto-immunes font penser à des explorateurs humains passant par une jungle de bambou. Quelle motivation guide les lymphocytes à travers le tissu, quels signaux les attirent, quelles forces les repoussent, et avec quelles « machettes » cellulaires se frayent-ils leurs sentiers ?
Les routes aléatoires des cellules T suggèrent un manque de flots laminaires ou de gradients de signaux chimiotactiques. On pourrait spéculer sur le fait que les cellules seraient plutôt guidées par des signaux multi-vectoriels provenant du milieu local, qui dirigeraient les cellules dans leur course erratique dans les tissus. Autrement, les cellules T pourraient suivre des signaux communiqués par des molécules d'adhésion cellulaire situées à la surface de cellules neurales ou, sur la matrice cérébrale, des signaux contrôlant la migration de type « haptotaxis ».
Cependant, les signaux chimiotactiques ou tactiles ne sont pas responsables de la vélocité de la course des lymphocytes au travers du tissu. Au-delà de ces signaux, il leur faut être extrêmement efficaces pour ouvrir un chemin entre les éléments neuraux à des enzymes protéolytiques digérant la matière extracellulaire collant les structures neuronales et gliales du SNC, mais ces mécanismes demeurent eux aussi énigmatiques. Cependant, il existe une multitude d'enzymes protéolytiques – y compris des métalloprotéinases matricielles (MMPs), des disintégrines, des granzymes et d'autres – qui s'expriment de préférence dans les lymphocytes activés [42], et qui, en principe, pourraient bien participer à la percée des routes migratoires.
Le comportement migratoire des cellules T auto-immunes suggère qu'elles parcourent le tissu cible à la recherche de leurs auto-antigènes, une exagération pathologique de la surveillance immunologique. Mais, si c'est le cas, que se passe-t-il au moment où elles rencontrent l'antigène spécifique ? Là encore, les vidéos à deux photons fournissent une réponse étonnante.
Si la majorité des cellules T traverse vivement le tissu nerveux, une minorité d'entre elles s'est arrêtée. Amarrés à un point fixe, ces lymphocytes remuent énergiquement leur corps, mais sans pouvoir s'éloigner (Fig. 2B). L'application d'anticorps monoclonaux spécifiques révèle que le pôle fixé correspond à une agrégation de récepteurs T et de certaines molécules d'adhésion cellulaire, comme le LFA (lymphocyte functional antigen). Il est bien connu que durant le processus de la reconnaissance d'antigène, ces deux molécules se concentrent sur un pôle de la membrane lymphocytaire. Elles font partie de la « synapse immunologique » qui unit la cellule T à la cellule présentatrice d'antigène pour recevoir le signal spécifique d'activation. Comme les lymphocytes de la classe CD4, dite « auxiliaire », les cellules T auto-immunes ne reconnaissent leur antigène que s'il est présenté dans le contexte des protéines CMH de classe II. En conséquence, il n'est pas trop surprenant que les synapses immunologiques touchent des membranes portant des molécules de classe II [41]. Arrêt des cellules T avec un pôle fixe, formation de synapses immunologiques et contact avec une cellule classe II+ satisfont les critères essentiels de la reconnaissance d'antigènes par une cellule T et de leur présentation par des cellules présentatrices.
En conclusion, la microscopie à deux photons a permis pour la première fois l'observation en temps réel de l'attaque des lymphocytes T auto-immuns contre leurs cellules cibles dans le SNC. Elle nous a appris que, dans la phase initiale de l'attaque, des millions de cellules effectrices auto-immunes envahissent le SNC en quelques heures, et également que des millions de ces lymphocytes se mettent à reconnaître et à réagir contre l'auto-antigène retrouvé.
6 La phase terminale : l'attaque
L'invasion du SNC par les lymphocytes auto-immuns correspond exactement au début précipité de la manifestation des signes cliniques. Néanmoins, l'attaque des cellules T effectrices contre la myéline n'est pas directe, pour les raisons suivantes. D'abord, les lymphocytes auto-immuns, qui appartiennent à la lignée CD4 « auxiliaire », ne sont pas des cellules cytotoxiques munies de granules lytiques contenant de la perforine, des granzymes et d'autres facteurs toxiques pour des cellules cibles. De plus, leurs cellules cibles – les oligodendrocytes et leurs gaines de myéline – ne produisent pas de protéines CMH de classe II et ne peuvent donc pas présenter les peptides de la myéline en contexte reconnaissable par les cellules T. Les auto-antigènes sont plutôt présentés par des cellules présentatrices spécialisées – cellules de la microglie, macrophages, cellules dendritiques. Les mécanismes de l'attaque auto-immune sont plus indirects, plus subtils et probablement divers.
Tout d'abord, les cellules T effectrices pourraient endommager le tissu nerveux lors de leur migration. La course de millions de cellules auto-immunes à travers le tissu pourrait bien rompre le contexte intercellulaire entre les neurones et les éléments gliaux, un processus ayant sans doute des conséquences fonctionnelles. Mais il faut dire qu'il y a des types de cellules auto-immunes qui diffèrent sensiblement dans leur capacité encéphalitogénique et qui, néanmoins, parcourent le tissu avec les mêmes intensité et vitesse [40].
Par ailleurs, l'activation des millions de lymphocytes reconnaissant l'auto-antigène aboutirait à la production et sécrétion massive de facteurs pro-inflammatoires – cytokines, chimiokines et d'autres – dispersés dans tout le tissu nerveux. Évidemment, les conséquences pour la fonction nerveuse d'une telle inondation inflammatoire seraient énormes. Il est connu que certains médiateurs inflammatoires agissent directement sur les neurones ou leurs axones [43], alors que d'autres procèdent d'une manière indirecte, soit par la stimulation les cellules gliales, soit en recrutant des macrophages dans le tissu.
Plusieurs arguments soulignent qu'en effet les macrophages activés sont les amplificateurs les plus importants de la réponse auto-immune. Argument plus direct, la déplétion des macrophages par des liposomes toxiques protège l'animal de l'EAE clinique sans prévenir l'entrée des cellules T dans le SNC [44]. De plus, il est bien connu que, dans les différents modèles d'EAE, la sévérité clinique est directement liée au nombre de macrophages activés présents dans les lésions cérébrales. Dans le cas de l'EAE classique du rat Lewis, les cellules T spécifiques de la protéine basique de la myéline, qui produisent une encéphalite très aiguë, sinon létale, causent des lésions riches en macrophages activés. À l'inverse, d'autres cellules auto-immunes, reconnaissant la protéine astrocytaire S-100b, pénètrent en grand nombre dans le SNC, mais sans altérations cliniques importantes [45]. Chez ces animaux, on ne trouve que peu de macrophages, et ces rares phagocytes sont essentiellement en état quiescent.
7 Épilogue
Grâce à des progrès récents, conceptuels et technologiques, l'énigme de l'arrivée des cellules immunitaire jusqu'au SNC est en cours de résolution. On a appris que, bien que les tissus nerveux, cerveau et moelle épinière, jouissent d'un état immunitaire privilégié qui exclut toute surveillance immunitaire effective, ce dernier privilège est soumis à conditions. Des études utilisant des modèles d'auto-immunité cérébrale ont démontré que le SNC permet des réactions immunes sous deux types de conditions :
- – d'abord, il faut que le tissu nerveux central soit modifié pour permettre des réactions immunitaires. Ceci s'accomplit par l'application de forts stimulus pro-inflammatoires, qui induisent des molécules immunitaires (par exemple CMH, molécules d'adhésion cellulaire, cytokines et chimiokines), ou par une atténuation de l'activité neurale, qui supprime la réactivité immunitaire dans le SNC normal ;
- – ensuite, il faut que les cellules immunitaires acquièrent un phénotype particulier qui leur permet d'envahir le SNC en passant à travers une barrière hémato-encéphalique activée. Ce phénotype est acquis dans les organes lymphoïdes périphériques à la suite d'une interaction entre des cellules T activées et des cellules stromales. Il est caractérisé par la réduction des gènes d'activation et, en même temps, par l'induction de récepteurs de chimiokines et d'autres structures requises pour la migration contrôlée.
Évidemment, le nouveau concept de la réactivité immunitaire du SNC influera sur la thérapie des maladies auto-immunes du SNC. Par exemple, au lieu d'une immunosuppression globale, on tentera de moduler les milieux immunitaires du SNC afin de rétablir l'état immuno-réfractaire. De même, on cherchera des marqueurs spécifiques des cellules migratoires, cibles idéales pour l'interception des cellules T avant leur pénétration dans le cerveau.
Remerciements
L'auteur souhaite exprimer sa reconnaissance toute particulière aux membres du « Groupe neuro-immunologique », dont les travaux forment la base des concepts discutés et au Pr. Roland Liblau (Toulouse), pour son assistance linguistique.