

1 Introduction
Low temperature is one of the most important limiting environmental factors for plants, which can cause a significant reduction in growth and in the yield of many agriculturally important crops including rice [1]. Plants exposed to cold temperatures generate reactive oxygen species (ROS), which interact with several cellular components, such as DNA, proteins, lipids and pigments [2]. Although the underlying mechanism has not yet been clearly defined below the proteomic level, plants have been shown complementary protective responses to cope with the cold stress [3]. Gene or protein identification in response to biotic or abiotic stresses is the basic step for the understanding of the molecular mechanism, or thus the production of enhanced tolerance transgenic plants to that particular stress. Two-dimensional polyacrylamide gel electrophoresis (2-DE) is frequently used to investigate protein expression patterns or to identify the specific protein under several biotic [4] or abiotic stresses in including low temperature [3,5]. Proteomics, or the systematic analysis of the proteins expressed by the genome, is not only a powerful molecular tool for describing complete proteomes at the organelle, cell, organ or tissue level [6] but also, for comparing proteomes affected by different physiological conditions, such as those resulting from the exposure to cold or several other stressful environmental factors. Although 2-D electrophoresis coupled with mass spectrometry is likely to provide an extremely useful tool to identify abundant proteins, it is of limited use for the identification of low-abundance proteins from leaf samples [7].
Recently, a great of effort has been taken to address the protein expression pattern in rice leaf in response to cold stress [3]; however, 35.3% of the identified proteins were photosynthesis related proteins, such as Rubisco or its derivatives. It must be an obstacle to the identification of the low-abundant or rare proteins from the leaf samples. In leaf proteins, Rubisco is the most abundant protein (about 50% of total soluble protein) that has hindered high resolution 2-DE. Due to the abundance of Rubisco proteins in leaves, it is still difficult to isolate rare proteins that may be differentially expressed.
To overcome this problem, we previously developed a protein extraction and polyethylene glycol (PEG) mediated pre-fractionation method, which provided the enrichment of low-abundant proteins in rice leaves [8]. In this present study, rice seedlings were exposed to extreme (5 °C) or moderate (10 °C) low temperatures and samples were collected after different durations. Proteins were extracted and fractionated by our previous method [8] and analyzed by 2-DE, which resolved a total of 14 proteins which were showing a constant, or at least at one time point, an up-regulation in response to cold stress. The possible roles of the identified proteins in response to cold stress are discussed.
2 Materials and methods
2.1 Plant materials
Rice (Oryza sativa L. cv. Dongjin) seeds were used as primary explants. Seeds were collected from the Yeongnam Agricultural Research Institute Milyang, Korea. Seedlings were grown hydroponically for three weeks in a control growth chamber at 28 °C under fluorescent light with a 16-h photoperiod. Three weeks old seedlings were subjected to cold stress by incubating in two separate growth chambers with a temperature setting of 5 or 10 °C. The humidity was maintained at 60%. Samples were harvested after 12, 24, and 36 h from the 5 °C, and after 24 and 72 h from the 10 °C treatments. After each treatment, the 3rd and 4th leaves were collected and immediately frozen in liquid nitrogen, and kept at until use.
2.2 Protein extraction
Proteins were extracted from the leaf tissue with Mg/NP-40 buffer and fractionated with PEG 4000, essentially following the method described by Kim et al. [8]. Nowadays, this method is widely used as a potential protein extraction method for rice samples for 2-DE analysis [4,8]. Briefly, the treated leaf samples were placed in liquid nitrogen, transferred to a pre-chilled mortar, and ground to fine powder with a pestle in liquid nitrogen. One gram of each sample was extracted with 12 ml Mg/NP-40 buffer containing 0.5 M Tris-HCl (pH 8.3), 2% v/v NP-40, 20 mM MgCl2, 2% v/v β-mercaptoethanol, 1 mM phenyl methyl sulfonyl fluoride and 1% w/v polyvinyl polypyrrolidone, followed by centrifugation at for 15 min. The proteins in the supernatant were subjected to PEG fractionation by adding a 50% w/v PEG stock solution. The extract was adjusted to a final concentration of 15% w/v PEG. The mixed sample was incubated on ice for 30 min and then centrifuged at for 15 min. The supernatant was recovered and precipitated with acetone and stored in 1 ml aliquots at until required. The acetone-precipitated sample was used for 2-DE analysis. Protein content was measured according to the procedure of Lowry et al. [9].
2.3 One-dimensional SDS-PAGE and 2-DE
For one-dimensional SDS-PAGE, 25 μg of crude proteins (after extraction by NP-40 buffer), total soluble proteins, PEG fractionated supernatants, or PEG fractionated pellets were separated on 12% polyacrylamide gels at a constant current of 20 mA. 2-DE was performed as described by Kim et al. [4]. The IEF gel mixture consisted of a 4.5% w/v acrylamide solution. 9.5 M urea, 2% v/v NP-40, and 2.5% v/v pharmalytes (pH 3–10 : pH 5–8 : pH 4–6.5 = 1 : 3.5 : 2.5; Amersham BioSciences, CA, USA). Each sample (150 μg of protein for silver staining, 350 μg of protein for CBB staining) was mixed with the sample buffer and then loaded on an IEF gel (18 cm tube gel). SDS-PAGE in the second dimension was carried out as described by Laemmli [10]. The 2-DE gels were silver-stained or CBB-stained as described previously [4].
2.4 Image and data analysis
Image and data analysis of the gels were performed using the PDQuest software (Version 7.2; Bio-Rad, Hercules, CA, USA). CBB-stained gels were selected for the profile analysis. To compensate for subtle differences in sample loading, gel staining, and destaining, the volume of each spot (i.e., spot abundance) was normalized to the relative volume. Spots were detected and quantified with Bio-Rad PDQuest software on the basis of their relative volume, that is, the spot volume divided by the total volume over the whole set of gel spots. After automated detection and matching, manual editing was carried out. The intensities of induced protein spots from the control and treated samples were recorded as digitalized images using a high-resolution scanner (GS-710 Calibrated Imaging Densitometer, Bio-Rad). At least 3 gels were used for each sample and the SD was calculated. Only those with significant and reproducible changes were considered to be differentially expressed proteins.
2.5 In-gel digestion and MALDI-TOF or ESI-MS/MS analysis
Protein spots were excised from the CBB-stained gel and washed with 50% v/v ACN in 0.1 M NH4HCO3, and vacuum-dried. The gel fragments were reduced for 45 min at 55 °C in 10 mM DTT in 0.1 M NH4HCO3. After cooling, the DTT solution was immediately replaced with 55 mM iodoacetamide in 0.1 M NH4HCO3. After washing with 50% ACN in 0.1 M NH4HCO3, the dried gel pieces were swollen in a minimum volume of 10 μL digestion buffer containing 25 mM NH4HCO3 and 12.5 ng/μL trypsin (Promega, WI, USA) and incubated overnight at 37 °C. Tryptic-digested peptides were extracted according to the protocol described by Kim et al. [4]. The extracted peptides were analyzed using a Voyager-DE STR MALDI-TOF mass spectrometry (PerSeptive Biosystems, Framingham, MA, USA) according to Kim et al. [4]. All obtained peptide mass fingerprints were analyzed by using Protein Prospector (http://prospector.ucsf.edu).
For protein samples not successfully identified by MALDI-TOF MS or for further confirmation, ESI-MS/MS was performed using a QSTAR pulsar-i MS system (AB/MDS Sciex, Toronto, Canada) equipped with a nano-electrospray ion source (MDS Protana, Odense, Denmark) according to Lee et al. [11]. Briefly, the ion-spray voltage was set to a potential of 850–900 V and Scandata of tryptic peptides were acquired over the range 400–1200 Da in the positive mode. MS/MS mode was operated over the range 80–2000 Da with manually optimized collision energy settings for each peptide. The resulting peptide sequence was submitted to the Protein Info (http://service.proteomics.com/PROWL/proteininfo.html) against in the NCBInr (update on 11 June 2005) database for identification and MASCOT (http://www.matrixscience.com) was utilized for searching and interpreting the raw MS/MS data. A mass accuracy of in the masses of both precursor and product ions was selected for searching the product ion spectra.
2.6 Western blot analysis
Twenty-five μg of protein samples from crude protein, total soluble protein, PEG supernatant fraction, or PEG pellet fractions were separated by SDS-PAGE, and transferred onto nitrocellulose membrane (Amersham Biosciences) using a semidry electrophoretic apparatus (EBU-4000, C.B.S. Scientific Co Inc. CA, USA). The blotted membranes were blocked for 1 h in TTBs (50 mM Tris-HCl, pH 8.2, 0.1% v/v Tween 20, and 150 mM NaCl) containing 5% w/v nonfat dry milk and subsequently incubated with an anti-Rubisco large subunit antibody (Anti-Rubisco, RbcL, R4404, Sigma) at 1:10 000 dilution for 1 h. The membranes were washed () in TTBs, and a secondary anti-chiken IgG (A9046, Sigma) antibody conjugated with peroxidase diluted 1:1000 in TTBs was used for immunodetection. After the blots were washed with TTBs, the immunoblot signals were detected using ECL (Perkin Elmer Life Sciences, Boston, MA, USA) and visualized on X-ray films (Fuji Medical X-ray film, 100 NIF, Japan).
2.7 Statistical analysis
Results of the each spot intensity were analyzed by analysis of variance (ANOVA) and the Student's t-test to compare the different treated and control samples. The average of the three samples with SD was calculated. Significance was determined at the levels.
3 Results and discussion
3.1 One-dimensional SDS-PAGE and 2-DE analysis
To confirm whether the fractionation technique successfully eliminated the Rubisco from the rice leaf protein samples, PEG supernatant, crude protein, total soluble protein, and PEG pellet protein samples were applied to SDS-PAGE analysis. As shown in Fig. 1, PEG supernatant protein sample was resolved into a wide range of distinct bands, excluding the Rubisco subunits. In contrast, the large and small subunits of Rubisco were clearly detected in the crude, total soluble or PEG pellet protein samples. In addition, relatively lesser band intensity other than Rubisco subunits and smaller number of bands were detected in the crude, total soluble and PEG pellet proteins compared to the PEG supernatant fraction (Fig. 1A). Elimination or reduction of Rubisco proteins from the leaf samples mediated by the PEG fractionation method were further confirmed by the Western blot analysis using antibody raised against Rubisco large subunit. As shown in Fig. 1B, the large subunit of Rubisco was not detected in the PEG supernatant fraction, indicating that prefractionation of leaf protein samples with PEG successfully eliminates the Rubisco proteins. The superiority of the PEG fractionation method was described extensively in our previous reports [4,8]. In support of our result, recently, it has also been reported that a 16% PEG fractionation method was precipitating predominantly the Rubisco large subunit from the Arabidopsis leaves, which largely increases the total number of proteins – up to five-fold – compared to the non-fractionated protein samples [12]. These results suggest that PEG prefractionation can increase 2-DE resolution, which may enrich the low-abundant proteins in the samples. In addition, it also indicate that the PEG fractionation can be used as a versatile application method for the reduction of the most abundant leaf protein, Rubisco, from all kinds of plant species. To investigate the temporal changes of the low-abundant proteins in rice leaf in response to cold stress, we therefore used the PEG fractionated protein samples for 2-DE analysis. High resolution of 2-DE gel patterns in the pI range between 4–7 were detected by silver staining for data analysis or by CBB staining for MALDI-TOF MS and/or ESI-MS/MS. Using the PDQuest software, differences in the intensity of protein spots between the control and cold treated samples were compared (Fig. 2). Thirty protein spots were found to be differentially expressed in response to cold treatments. Among these, 14 up-regulated proteins spots (Fig. 3) were excised from the gels and further analyzed by MALDI-TOF MS and/or ESI-MS/MS analysis.
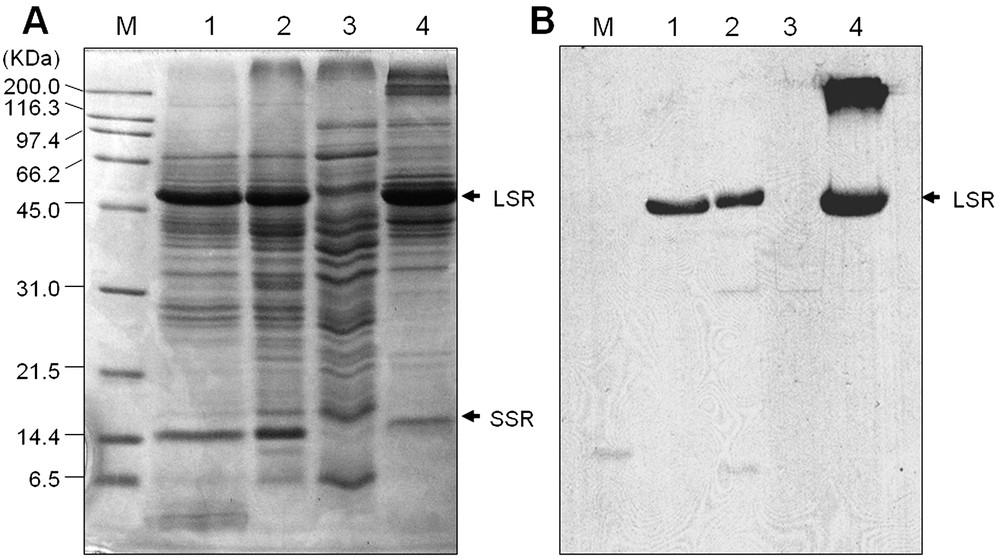
(A) SDS-PAGE analysis of rice leaf proteins. Proteins were extracted as described in Section 2. Lane 1, crude proteins; lane 2, total soluble proteins; lane 3, PEG supernatant fraction; and lane 4, PEG pellet fraction. Twenty-five μg proteins were loaded in each lane, separated on 12.5% SDS-PAGE and visualized by CBB staining. LSR and SSR indicate that large and small subunit of Rubisco, respectively. (B) Western blot analysis of Rubisco large subunits in rice leaf proteins. Proteins were extracted, loaded and separated as above and subsequently transferred onto nitrocellulose membranes. Immunodetection of Rubisco large subunit was performed using antibodies (see Section 2.6) and visualized on an X-ray film.

2-DE analysis of PEG-fractionated rice leaf proteins exposed to cold treatments. A total of 150 μg proteins were extracted and separated by 2-DE as described in Section 2, and visualized with silver-stain. Arrows are indicating the up-regulated proteins in response to cold treatments. Boxes are indicating the regions of major changes in response to cold stress. A, control; B–D indicating the gel patterns of the PEG fractionated protein samples after exposed to 12, 24 and 36 h at cold stress (5 °C), respectively.

Close-up views of the up-regulated proteins in rice leaf tissues in response to cold stresses and revealed by 2-DE analysis. A–G indicate the boxes marked in Fig. 2.
3.2 Identification of cold-responsive low-abundant proteins in rice leaf
Excised protein spots were digested with trypsin and the peptides were extracted and identified by peptide mass fingerprinting (PMF) using a MALDI-TOF and/or by sequence tag using an ESI-MS/MS. Two of the proteins (spot 8 and 9) showed no good matches while 12 were identified. The database searching results are listed in Tables 1 and 2. The spot volume means of these identified 12 protein spots is presented graphically in Fig. 4. Fig. 5 shows the spectrum of the trypsin digest protein spot 14 as a representative example of MS analysis. Among these identified proteins, five proteins such as OSJNBa0011F23.15 (spot 1), ascorbate peroxidase (APX) (spot 4), putative glutathione S-transferase (spot 5), thioredoxin h-type (Thx h) (spot 12), and nucleoside diphosphate kinase 1 (NDPK1) (spot 14) were noticed previously as cold-induced proteins [3], while the other seven proteins, such as fibrillin-like protein (spot 2), cysteine proteinase (spot 3), unknown protein (spot 6), thioredoxin peroxidase (TPx) (spot 7), drought-inducible late embryogenesis abundant protein (spot 10), RING zinc finger protein-like (spot 11), and a hypothetical protein (spot 13), were newly identified in response to cold stress. The possible role of these identified novel proteins in response to cold stress is now discussed.
Identification of differentially expressed proteins in rice leaves under cold stress
| Spot no. | Protein name | Acc. no.a | Species | Theoretical/observed pI | Theoretical/observed (kDa) | Score | Mb | SC (%)c |
| 1 | OSJNBa0011F23.15 | XP_474199 | Oryza sativa | 6.0/5.8 | 46.6/50.0 | 1.18e+6 | 8 | 17% |
| 2 | Fibrillin-like protein | AAO72593 | Oryza sativa | 5.0/5.1 | 33.8/35.0 | 243 098 | 6 | 28% |
| 3 | Putative Cysteine proteinase | NP_917662 | Oryza sativa | 7.6/5.7 | 38.3/32.5 | 27 454 | 4 | 18% |
| 4 | Ascorbate peroxidase | XP_479627 | Oryza sativa | 5.2/5.8 | 27.1/32.5 | 3.53e+8 | 11 | 52% |
| 5 | Putative glutathione S-transferase | AAN05497 | Oryza sativa | 6.7/7.0 | 25.7/32.0 | 11 091 | 6 | 37% |
| 6 | Unknown protein | BAD08824 | Oryza sativa | 6.8/6.3 | 25.5/6.8 | 12 200 | 5 | 27% |
| 7 | Putative Thioredoxin peroxidase | XP_464429 | Oryza sativa | 6.1/5.4 | 23.2/21.0 | 216 | 4 | 27% |
| 8 | Not matched | |||||||
| 9 | Not matched | |||||||
| 10 | Drought-inducible late embryogenesis abundant | AAL99712 | Oryza sativa | 5.1/5.5 | 20.2/18.4 | 2521 | 4 | 34% |
| 11 | RING zinc finger protein-like | XP_468174 | Oryza sativa | 6.1/5.7 | 22.0/18.0 | 926 | 4 | 28% |
| 12 | Thioredoxin h-type | Q42443 | Oryza sativa | 5.2/5.7 | 13.2/17.0 | 356 | 4 | 39% |
| 13 | Hypothetical protein | XP_493903 | Oryza sativa | 5.9/6.5 | 14.4/18.4 | 1303 | 6 | 42% |
| 14 | Nucleoside diphosphate kinase 1 | Q07661 | Oryza sativa | 6.3/6.7 | 16.8/17.7 | 5838 | 5 | 36% |
a Accession number from NCBI database.
b Number of peptides matched.
c Sequence coverage by peptide mass fingerprinting using MALDI-TOF MS.
Cold induced differentially expressed proteins identified by ESI-MS/MS
| Spot no. | Protein name | Acc. no.a | ESI-MS/MS | ||
| Sequence tag | Score | Identityb | |||
| 2 | Fibrillin-like protein | AAO72593 | TTYLDDELR | 59 | 100% |
| 4 | Ascorbate peroxidase | XP_479627 | DNSYFTELVSGEK | 95 | 100% |
| 6 | Unknown protein | AAP94211 | DGVLSTER | 46 | 100% |
| 14 | Nucleoside diphosphate kinase 1 | Q07661 | PEGLAEWR | 20 | 100% |
a Accession number from NCBI database.
b Percentage of identities with NCBI database.
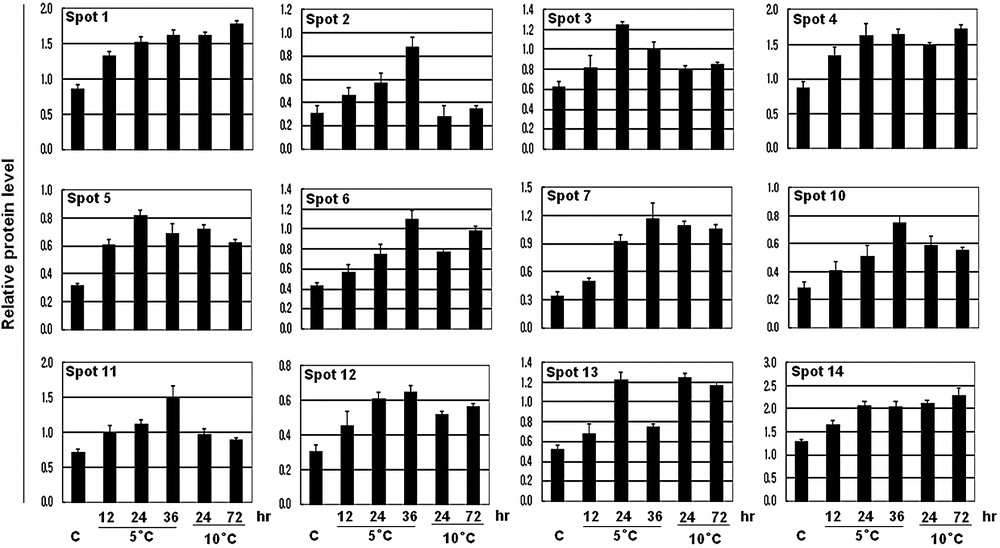
The expression levels of the 12 identified proteins compared to those of controls. Spot intensities of the 12 increased proteins were measured using a densitometer (Bio-Rad) and compared to those of controls. The average values of relative increase levels of three replicate samples are shown in the histograms.

Identification of NDPK1 protein by MS. The protein spot 14 was excised from the gels and spectrum of the peptides derived after tryptic digestion. A, the MS spectra. The ion 859.57 marked with an asterisk was analyzed by MS/MS. B, MS/MS spectra of ion 859.57. The y ions (y1–7) and the corresponding peptide sequence are shown. The database search resulted in its identification as NDPK1 (Accession no. Q07661).
A group of antioxidant enzymes such as, APX, GST, Thx h, and TPx was increased at least 2-fold at each time point under the cold stress. Moreover, all of these enzymes showed a gradual up-regulation under the 5 °C regime, indicating that these enzymes are highly correlated with the extreme temperature. The imposition of abiotic stresses such as drought, cold, and heat can give rise to excess concentrations of reactive oxygen species (ROS) in plant cells [13], which are potentially harmful since they initiate the peroxidation and destruction of lipids, nucleic acids or proteins [2]. Therefore, antioxidants and antioxidant enzymes function to interrupt the cascades of uncontrolled oxidation in each organelle. In a plant, APX and GST are very well functioned antioxidant enzymes that protect the cells from a wide range of biotic and abiotic stress including from ROS [14–16]. GST catalyzes the conjugation of glutathione (GSH) that arises from a wide range of toxic substrate mediated by oxidative stress, and reduces the toxicity [14]. Thioredoxin h is involved in a wide range of cellular functions, such as redox regulation by reducing disulfide bridges on the target proteins, carbon and nitrogen metabolism [17,18]. Similar to our results, up-regulation of these enzymes in rice leaves under cold stress has also been noticed in previous proteomics analysis [3,5].
Along these enzymes, TPx a low-molecular thiol-specific antioxidant enzyme, has also shown a rapid response to cold stress. It performed a gradual up-regulation with time course and increased up to 4-fold in both treatments, which indicates its higher interaction under cold stress. The role of TPx in the cellular defense against oxidative stress induced by H2O2 or singlet oxygen was noticed in microorganisms [19]. In addition, it has also been reported that TPx could function as an inhibitor of apoptosis [20]. We also identified nucleoside diphosphate kinase 1 (spot 14, NDPK1), with 36% mass fingerprinting coverage and peptide sequence tag (GEGLAEWR), which was also 100% identical to the same protein with ESI-MS/MS (Table 2). Nucleoside diphosphate kinases (NDPKs) are believed to be a housekeeping enzyme that maintains the intracellular levels of all (d)NTPs used in biosynthesis except ATP. Moreover, involvement of some developmental processes [21] and response to various abiotic stresses [22,23], NDPK2 has been considered as a multiple stress related gene.
In our investigation, we first time identified a cysteine proteinase protein in response to cold treatment in rice leaves. A significant increase of this protein was observed under 5 °C, which also showed a linear up-regulation with the time courses. However, little information is available on the induction of this protein in response to cold-stress in plants. Cysteine proteinase, one of the major groups of plant proteases, has been thoroughly studied due to its crucial role in senescence and programmed cell death [24]. Thus, we proposed that cold stress increased the ROS levels such as H2O2 or singlet oxygen in the rice leaf, initiated the senescence and programmed cell death and as a consequence cysteine proteinase was increased, and increases of TPx as well as other antioxidant enzymes protect the cells from these adverse conditions. However, to confirm this hypothesis much research should be needed.
A fibrillin-like protein (spot 2) was identified with a 28% mass fingerprint coverage, and peptide sequence tag (TTYLDDELR) which was also 100% identical to same protein with ESI-MS/MS. Interestingly, its response to cold temperature was found only to 5 °C but not at 10 °C. Fibrillins are ubiquitous lipid-binding proteins of plastids that are induced under several abiotic stress conditions including cold [25]. Abscisic acid (ABA) plays a major role in regulating plant growth and development and mediating adaptations to environmental stress such as cold, drought, and salinity [26]. Recently, it has been shown that ABA treatment induced the fibrillin accumulation, which enhanced the tolerance of photosystem II toward light stress-triggered photoinhibition in Arabidopsis [27]. These results lead us to propose that the induction of fibrillin-like protein in rice leaf under cold stress is due to increases of endogenous ABA synthesis.
Spot 10 was matched to a drought-inducible late embryogenesis abundant protein with 34% mass fingerprint coverage. Late embryogenesis abundant (LEA) proteins represent several families of proteins that are accumulated under water deficit conditions [28]. LEAs are also highly induced a late in embryogenesis, and in nearly all the vegetative tissues during normal growth conditions and in response to stress leading to cellular dehydration such as drought, low temperature and salinity [29] or exposed to exogenous ABA [30]. Based on these previous reports, it could be speculated that up-regulation of drought-inducible late embryogenesis abundant proteins in rice leaf under cold stress are likely to lead to a reduction of water status in leaves [3] or to increases of the endogenous ABA.
We identified a RING zinc finger protein-like (spot 11) protein, which showed a gradual increasing pattern to 5 °C and was markedly increased up to 3-fold after exposure at 36 h. However, no significant changes were observed at 10 °C, indicating that this protein is highly responsive to extreme temperature. Consistent with our results, it has also been found, by transcriptome or microarray analyses, that in rice [31,32] or soybean [33], a C2H2-type zinc finger protein is highly responsive to extreme cold stress, and overexpression of this gene into plants provided better tolerance to cold stress [33]. In this study, we for the first time identified that a RING zinc finger protein-like protein is also highly responsive to extreme cold temperature. The RING (a really interesting new gene) finger motif was defined as a novel zinc-finger domain [34], which has important regulatory roles in the development of a variety of organisms or involved in various signal transduction pathways in plants [35]. In addition, the RING motif is a protein–protein interaction domain that has been implicated in a range of diverse biological processes [36]. Based on the above discussion, it could be speculated that RING/zinc finger domain containing proteins may be used as a marker protein for extreme cold temperature.
Spot no. 1 matched to OSJNBa0011F23.15 of the rice database. A BLAST search of this protein was assigned as a glutamine synthetase shoot isozyme (GS), a chloroplast precursor. Recently, it was reported that a transgenic rice with a GS2 gene exhibited enhanced tolerance to salt stress [37] and a GS shoot isozyme, chloroplast precursor was increased in rice leaf to cold stress [3], which indicates that GS might have a pivotal role in abiotic stresses. Spot no. 6 and 13 were matched to unknown protein and hypothetical protein with 27 and 42% mass fingerprint coverage, respectively. Peptide sequence tag (DGVLSTER) from spot no. 6 also was 100% identical to same protein with ESI-MS/MS. However we are unable to address these proteins functions in response to cold stress.
4 Conclusions
In this study, to gain a better understanding about the cold responsive low-abundant proteins in rice leaves, total proteins were extracted from the treated samples and fractionated by 15% PEG. The elimination of the Rubisco from the PEG fractionated samples was confirmed by Western blot analysis. To achieve the identification of the low-abundant proteins, PEG fractionated protein samples were further analyzed by 2-DE. Of the fourteen up-regulated proteins, 12 of them were identified by MALDI-TOF mass spectrometry or ESI MS/MS analyses. Using this technique, we successfully identified some novel proteins that showed a higher sensitivity to cold stress and that have yet not been reported in the earlier cold proteomic analyses of rice leaves. These results may indicate a new insight into cold stress and also provide genes of interest for transgenic research.
Acknowledgements
This work was supported by a grant from the BioGreen 21 program, Rural Development Administration, Korea, and by Korea Research Foundation Grant. D.-G. Lee and S.-H. Lee are supported by the scholarship from the BK21 program, Ministry of Education and Human Resources Development, Korea.