

1 Introduction
Lead is naturally present in the lithosphere in negligible quantity, due to rock wind erosion or volcanic rejections [1].
Nowadays, lead is being a ubiquitous environmental contaminant due to its significant role in modern industry [2].
However, both occupational and environmental exposures remain a serious problem in many developing and industrializing countries [3].
Effectively, lead constitutes the most abundant nonessential element in the human organism, due to its dispersion in ambient air, in many foods, in drinking water, and in dust. Its toxicity is closely related to its accumulation in certain tissues and its interference with the bioelements, whose role is critical for several physiological processes. It has many undesired effects, including neurological [4,5], behavioural [6,7], immunological [8–11], renal [12–14], hepatic [13], and especially haematological [15,16] dysfunctions.
However, biochemical and molecular mechanisms of lead toxicity are poorly understood [2]. Various mechanisms were suggested to explain them: inhibition of the calcium-pump, a transport protein [3], disturbances in mineral metabolism, inactivation of several enzymes, demyelinisation of nervous tissues, etc. Recently, oxidative stress has been proposed as another possible mechanism involved in lead toxicity. According to many workers, lead-induced damage may result from disturbance of the prooxidant and antioxidant balance that is found in cells [15].
Generation of highly reactive oxygen species (ROS) such as the hydroxyl radical [OH°], hydrogen peroxide [H2O2], superoxide anion and lipid peroxides [LPO°] after lead exposure has been reported [5].
Based on the above considerations, this study was carried out to investigate the effects of dose-dependent exposure to lead on oxidative stress parameters in rat blood.
About 99% of the lead present in the blood is bound to erythrocytes. They have a high affinity for lead and contain the majority of the lead found in the blood stream, which makes them more vulnerable to oxidative damage than many other cells. Moreover, erythrocytes can spread lead to different organs of the body [16]. Therefore, we tried to investigate the effects of dose-dependent lead exposure on an end-product of lipid peroxidation known as MDA in hepatic cells.
2 Materials and methods
2.1 Chemicals
Lead (II) acetate was purchased from Prolabo (France). Butylated hydroxytoluene (BHT), 2-thiobarbituric acid (TBA), Coomassie G250 and bovine serum albumin (BSA) were obtained from Sigma Chemicals Co. (Germany). All other chemicals were of the highest grade available.
2.2 Animals
All experiments were performed with ‘Wistar’ male rats weighing 150–200 g, which were obtained from the Tunisian Society of Pharmaceutical Industries.
The animals were housed in clean plastic cages and allowed to acclimatize in the laboratory environment for a week ( with 12-h dark/light cycle and ventilation system).
In the experimental period, animals had access to food and water ad libitum.
2.3 Experimental design
The animals were randomly assigned into three groups of 16 individuals and treated for seven days. The first group [T] served as control and received daily 15 mg Na+/kg b.w. i.p. as sodium acetate solution.
The second [Pb 5] and third [Pb 15] groups received daily, respectively, 5 mg and 15 mg Pb2+/kg b.w. i.p. as lead acetate solution for the same period.
Throughout the experience, the rats were daily weighed.
At the end of the treatment, 24 h after the last dose of lead, animals were killed by decapitation without preliminary anaesthesia, and arteriovenous blood was taken quickly. The liver was removed by transverse abdominal incision and kept frozen at −80 °C.
2.4 Preparation of plasma and tissue extract
Blood collected on heparin was taken and let rest for 30 min at room temperature. Plasma was obtained by centrifugation at 1000 g for 10 min at 4 °C, and stored at −80 °C in aliquots until use.
Livers were excised immediately, washed with ice-cold physiologic saline solution (0.9%), blotted dry, and weighed. Tissues were homogenized for 30 s in 10 volumes of ice-cold 10 mM phosphate buffered saline (PBS, pH 7.4). Homogenates were centrifuged at for 10 min at 4 °C to remove cell debris, nuclei, and mitochondria. Resulting supernatants served for malondialdehyde (MDA) measurements.
2.5 Haematological parameters
2.5.1 Haemoglobin concentration
Haemoglobin was measured colorimetrically according to Drabkin and Austin's method [17] through its transformation to cyanmethemoglobin under the action of potassium ferricyanide and potassium cyanide. Haemoglobin concentration was expressed in g/l of blood.
2.5.2 Haematocrit content
Haematocrit measurements were carried out in capillary tubes centrifuged with HEMATOCRIT 20 Hettich for 15 min at 1000 rpm.
2.6 Assay of SOD activity
The superoxide dismutase (SOD) (E.C.1.15.1.1) level in erythrocytes was determined using the Ransod Kit supplied by the Randox Laboratories (Ardmore, Northern Ireland, UK).
Xanthine and xanthine oxidase were used to generate superoxide anion, which reacts with 2-(4-indophenyl)-3-(4-nitro-phenyl)-5-phenyl tetrazolium chloride (INT) to form a red formazan dye. Changes in the absorbance were determined at 505 nm during the first three minutes of the reaction. Enzyme activity in the sample was calculated from a standard curve. One unit of SOD is defined as the amount that inhibits the INT reaction by 50%. Specific activity was defined as Units/g haemoglobin.
2.7 Assay of GPx activity
Glutathione peroxidase (GPx) (E.C.1.11.1.9) level in erythrocytes was based on that of Paglia and Valentine [18]. After the addition of cumene hydroperoxide, the conversion of NADPH to NADP+ by glutathione reductase and generated oxidized glutathione was continuously monitored spectrophotometrically at 340 nm for 2 min. GPx activity was expressed in units/g of haemoglobin.
2.8 Plasma total bilirubin assay
Total bilirubin concentration was measured colorimetrically according to Jendrassik and Grof [19]. Total bilirubin is determined in the presence of caffeine, which releases albumin bound bilirubin, by the reaction with diazotised sulphanilic acid.
Changes in absorbance were read at 578 nm against their samples blank. Total bilirubin was expressed in mg/dl of plasma.
2.9 Lipid peroxidation in liver
The lipid peroxidation product in livers was determined by TBARS (thiobarbituric acid reactive substances) expressed as the extent of malondialdehyde (MDA) production [20,21]. The samples were suspended in PBS pH 7.4, mixed with butylated hydroxytoluene (BHT)-TCA solution (1% w/v BHT dissolved in 20% TCA), and centrifuged at 1000 g for 10 min. The supernatant was then mixed with 0.5 N HCl and 120 mM TBA (2-thiobarbituric acid) in 26 mM Tris, and heated in a water bath at 80 °C for 15 min. After cooling, the absorbance of the resulting chromophore was determined at 532 nm using a BIORAD UV-visible spectrophotometer (Smart Spec 3000), and MDA production was determined by using an extinction coefficient of , and expressed in nmol MDA/g of proteins.
2.10 Protein assay
The protein content of supernatants was spectrophotometrically estimated according to the method of Bradford [21] using bovine serum albumin as standard.
2.11 Statistical analysis
All results were expressed as deviation. Comparisons between the groups were performed by one-way ANOVA followed by Student t-test. Differences were considered significant at .
3 Results
3.1 Body weight of rats
There was a steady decrease in body weight of rats of the two treated groups compared to the respective values of the control group () (Table 1).
Body weight of controls and Pb-exposed rats treated for seven days by i.p. administration of lead acetate (5 mg and 15 mg Pb/kg body weight)
| Control (n=16) | Pb 5 (n=18) | Pb 15 (n=18) |
| Body weight (g): | ||
| Initial weight (g): 174.94±10.77 | 167.35±4.65 | 181.25±7.09 |
| Final weight (g): 191.69±10.44 | 166.53±4.36a | 155.38±5.46a/b |
a , compared to the corresponding value of control group.
b , compared to the corresponding value of group [Pb 5].
Statistical analysis of growth rates is shown in Fig. 1. For the two groups treated with different doses of lead acetate, we noted a significant decrease () in growth rates with increasing the dose of lead. However, growth suppression in group [Pb 15] was considerably greater than that observed in group [Pb 5] ().
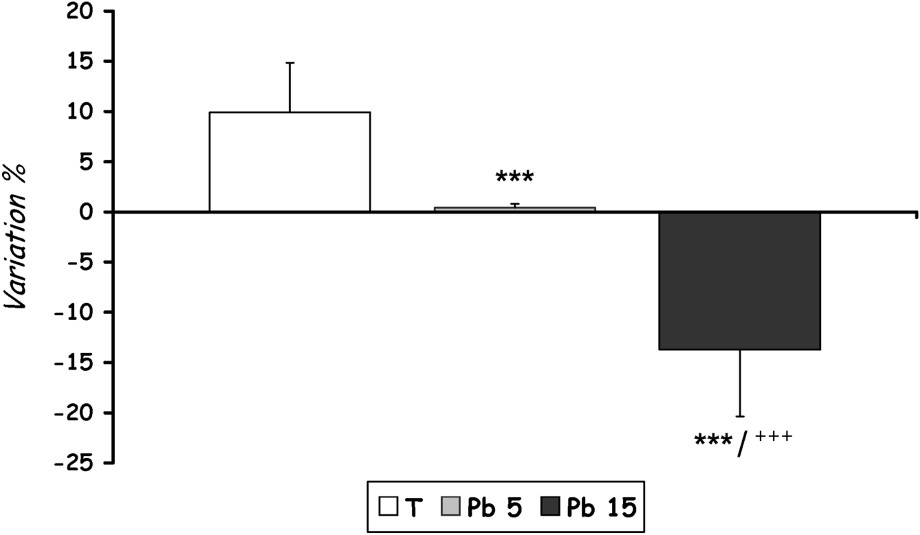
Growth-rate evolution of controls and Pb-exposed rats treated for seven days by i.p. administration of lead acetate (5 mg and 15 mg Pb/kg body weight). Results are expressed as mean±SD. N=16 rats for each group. 0.01 compared to the corresponding value of group [Pb 5].
3.2 Haematological parameters
As is shown in Figs. 2 and 3, lead administration induces, in both treated groups [Pb 5] and [Pb 15], a marked decline in haematocrit and haemoglobin levels () when compared to control.
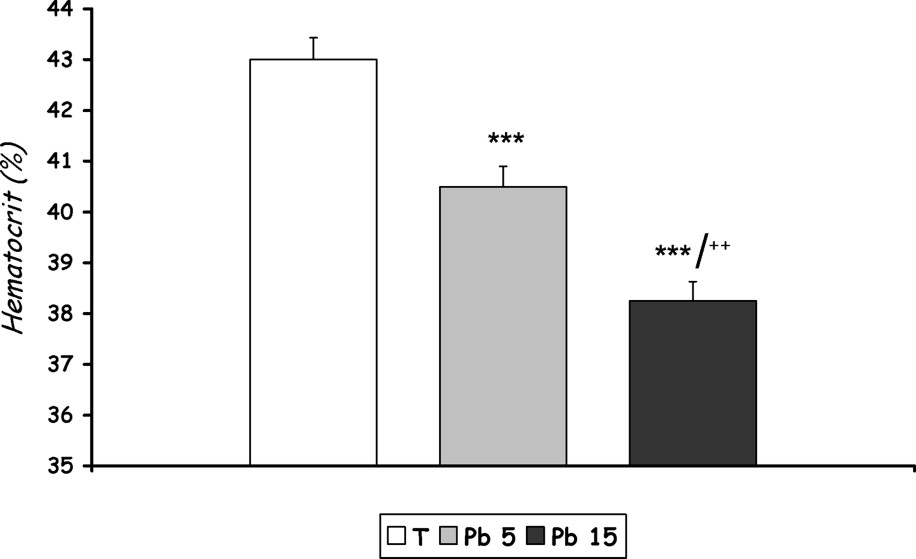
Haematocrit level of controls and Pb-exposed rats treated for seven days by i.p. administration of lead acetate (5 mg and 15 mg Pb/kg body weight). Results are expressed as mean±SD. N=16 rats for each group. compared to the corresponding value of group [Pb 5].

Haemoglobin level of controls and Pb-exposed rats treated for seven days by i.p. administration of lead acetate (5 mg and 15 mg Pb/kg body weight). Results are expressed as mean±SD. N=16 rats for each group. compared to the corresponding value of [T]. compared to the corresponding value of [T]. compared to the corresponding value of [Pb 5].
3.3 Antioxidant enzymes
- – A significant decrease in SOD activity was recorded in group [Pb 15] compared with control (), but the activity of this enzyme did not decrease significantly in group [Pb 5] (Fig. 4).
- – Lead exposure caused a light increase in GPx activity in RBCs of rats treated with 15 mg Pb/kg b.w., which remains non significant compared to control. The group treated with 5 mg Pb/kg b.w. did not record changes in the activity of GPx compared to control (Fig. 5).
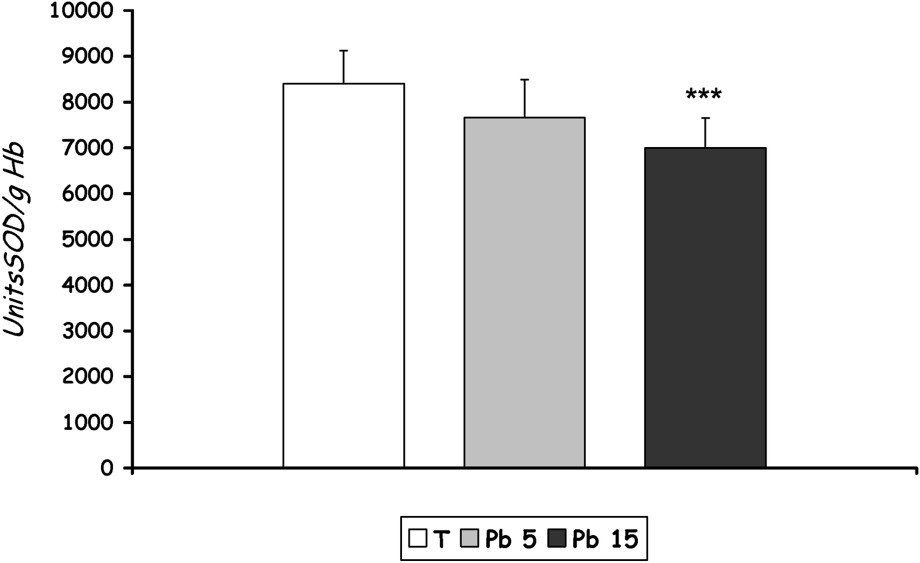
SOD activity in erythrocytes of controls and Pb-exposed rats treated for seven days by i.p. administration of lead acetate (5 mg and 15 mg Pb/kg body weight). Results are expressed as mean±SD. N=16 rats for each group. compared to the corresponding value of [T].
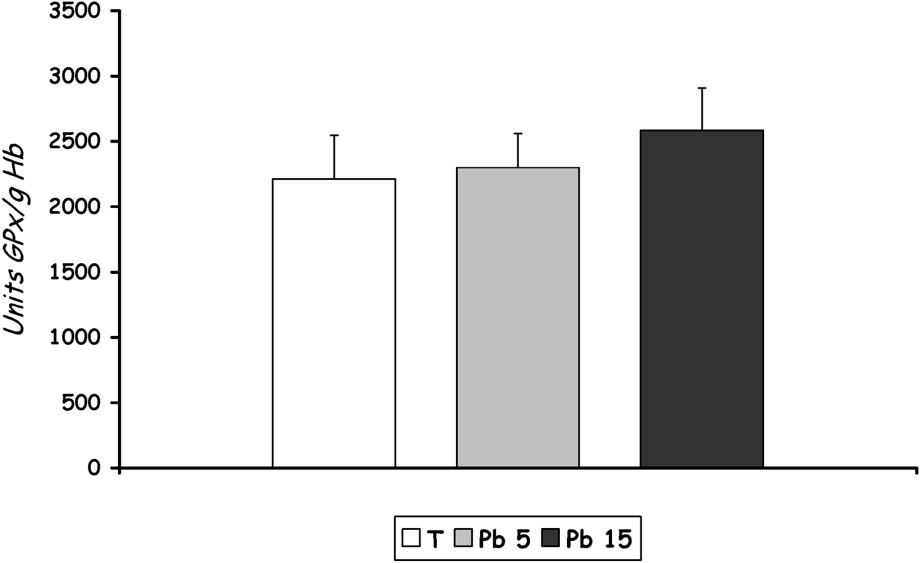
GPx activity in erythrocytes of controls and Pb-exposed rats treated for seven days by i.p. administration of lead acetate (5 mg and 15 mg Pb/kg body weight). Results are expressed as mean±SD. N=16 rats for each group.
3.4 Bilirubin levels in plasma
We noted a significant increase in the bilirubin level () in the group of rats treated with 15 mg Pb/kg b.w. compared to control.
The bilirubin level from rats of the group treated with 5 mg Pb/kg b.w. increased with lead treatment, but did not reach a statistical significance (Fig. 6).
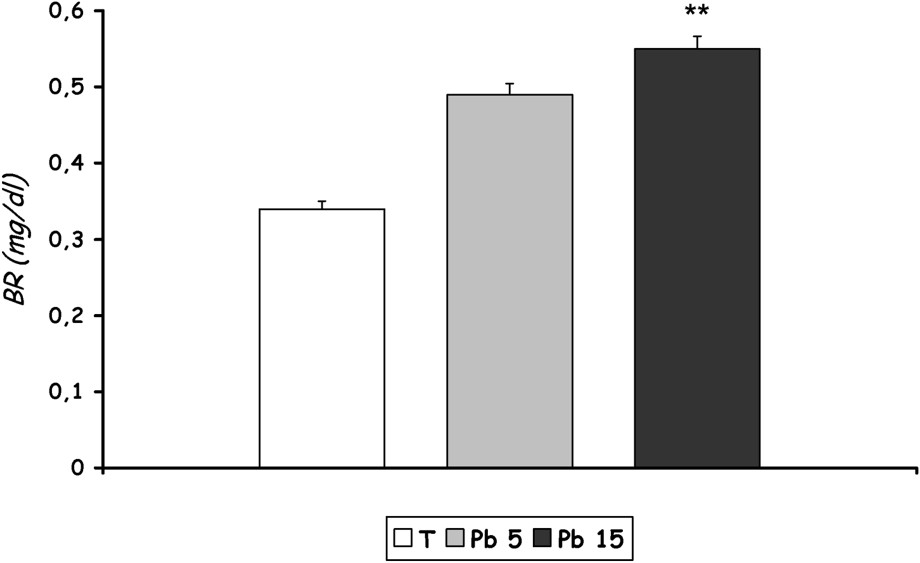
Total bilirubin level in plasma of controls and Pb-exposed rats treated for seven days by i.p. administration of lead acetate (5 mg and 15 mg Pb/kg body weight). Results are expressed as mean±SD. N=16 rats for each group. **p<0.05, compared to the corresponding value of [T].
3.5 MDA levels
The lipid peroxide level from rats of group [Pb 15] increased with lead treatment, but the effect did not reach statistical significance. No change was noted in the level of MDA in the liver of rats treated with 5 mg Pb/kg b.w. (Fig. 7).
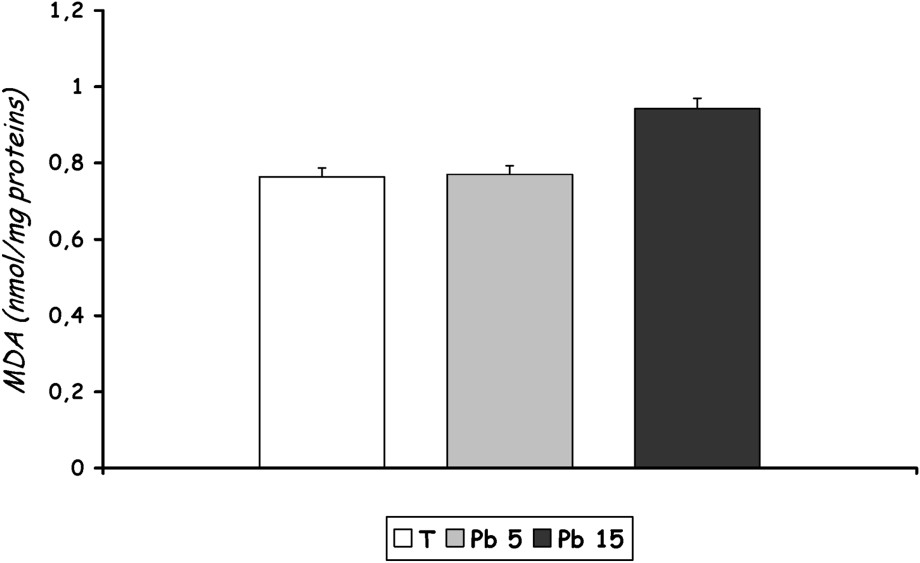
Lipid peroxidation expressed as MDA in liver homogenate of controls and Pb-exposed rats treated for seven days by i.p. administration of lead acetate (5 mg and 15 mg Pb/kg body weight). Results are expressed as mean ± SD. N=16 rats for each group.
4 Discussion
This study showed that adult male Wistar rats treated with different doses of lead acetate for seven days cause a significant decrease in body weight. This result is in agreement with several studies [22,23], which suggested that the reduced growth was due to reduced food consumption via lead effects on the satiety set-point.
The haematological system has been proposed as being an important target for lead-induced toxicity. Lead is known to interfere with haeme and haemoglobin synthesis and to affect erythrocyte morphology and survival [24].
Anaemia observed during lead poisoning in this study is thought to result from inhibition of haeme synthesis and a decreased life span of the erythrocytes [25]. This shortening of life span was observed by Teramaya [26] in experiments with rats and it is thought to be probably due to the direct toxic effect of lead upon the cell membrane [25]. Lead, furthermore, interferes with iron utilization for haeme formation in the mitochondria, and radio-iron studies showed that lead competes with iron for the incorporation into red blood cells. The precise mechanism of action of lead at stake in cellular metabolism remains largely unknown [24].
On the other hand, it has been shown that accumulation of δ-ALA induces oxidative stress in Chinese hamster ovary cells, where the induction was found to be reversed by incubating the cells with a well-known antioxidant, N-acetylcysteine [27].
Moreover, free radicals produced in the presence of heavy metals contribute to haemoglobin denaturing and precipitation, leading to anaemia [16].
These data suggest that anaemia observed in lead poisoning is caused by lead both directly and indirectly by the accumulation of δ-ALA [27].
However, much remains to be clarified about the exact nature of the mechanism by which lead induced haemolysis and shortening of the life span of circulating erythrocytes [28].
In the present study, lead is shown to alter the oxidative stress-related parameters both in erythrocytes and in liver tissue. SOD, which requires copper and zinc for its activity, was found to decrease in lead-administered rats. SOD is believed to play a major role in the first line of antioxidant defence by catalyzing the dismutation of superoxide anion radicals to form hydrogen peroxide H2O2 and molecular oxygen O2 [29]. Sivaprasad et al. [16] showed that oxidative stress created by the metal favours increased production of superoxide anions. Under oxidative stress, SOD can behave in two different ways: initially and when the stress is moderated, the cells act by suppressing the SOD, but if the stress lasts a long time and favours increased production of ROS, the enzyme is exhausted and its concentration falls [30]. Effectively, in our case, decreased SOD activity observed at only one group [15 mg Pb/kg b.w.] could be explained by the massive production of superoxide anions, which override enzymatic activity and lead to the fall of its concentration in erythrocytes. Mylorie et al. [31] suggested that this could be due to a lead-induced copper deficiency. According to Pfafferoot et al. [32], the low activity of SOD could also be due to the inactivation of the enzyme by crosslinking or damage to DNA.
Selenium-dependent GPx catalyze the reduction of a variety of hydroperoxides (ROOH and H2O2) using GSH, thereby protecting mammalian cells against oxidative damage. Hunaiti and Soud [28] showed that the activities of glutathione S-transferase, glutathione peroxidase, and reductase, as well as the content of blood GSH, had decreased with an increasing lead concentration in the blood of some occupational workers exposed to this metal. In our study, we rather note an increase in GPx activity, which remains non-significant in the group [15 mg Pb/kg b.w.] compared to controls (). Just like SOD, GPx behaves in two different ways under oxidative stress, and this result can be explained by the short duration of the treatment.
Bilirubin is also regarded as a member of the antioxidant family, even though it is known to have toxic effects at high concentrations [33]. Bilirubin has been regarded for many years as cytotoxic, mainly because of its association with neonate jaundice and its possibility of provoking irreversible brain damage at high concentrations [34]. It is only since the early 1990s that a physiological role for bilirubin as a potent antioxidant has emerged. Stocker et al. [35] noted that bilirubin possesses strong antioxidant potential against peroxyl radicals. At physiologic oxygen tension, bilirubin surpassed α-tocopherol as the most potent protector against lipid peroxidation. In plasma, it can act synergically with vitamin E to protect lipid membranes from the peroxidation initiated within the lipid phase [35,36]. The study carried out by Miréles et al. [37] showed that erythrocytes subjected to oxidative stress and incubated with high levels of bilirubin (45–60 mg/dl) with physiological levels of BSA (3 g/dl) induced: (1) a significant concentration-dependent decrease in the fluorescence decay of Cis-PnA, a polyunsaturated fatty acid that can intercalate in cell membrane, with lipid peroxidation; (2) a 50% decrease in G6PD activity and in Na+/K+ ATPase activity; (3) an increase in protein oxidation. These data suggest that bilirubin, at high levels (>30 mg/dl), may exacerbate oxidative stress, although the mechanism by which this occurs is not yet clear.
Bilirubin could be also an important cytoprotector for tissues that are less equipped for antioxidant defence, like myocardium and nervous tissues [33]. Doré et al. [38] showed that increase bilirubin formation due to activation of HO-2 (constitutive isoform of HO) protects against hydrogen peroxide-induced neurotoxicity. It has been also demonstrated that intracellular bilirubin concentrations can be locally and temporarily increased by induction of HO-1 (inducible isoform of HO) or rapid activation of HO-2, so as to resist short- and long-lasting oxidative stress [34]. It has been proposed that the specific induction of HO-1 by various forms of oxidative stress was part of the defensive mechanism mounted by cells against stress injury to decrease the levels of potential prooxidants and to increase the concentrations of active bile pigments that can act as antioxidants [39–41]. HO-1 up-regulation is followed by increased bilirubin production, altogether determining the adaptive response of cells to oxidative stress [39].
The study undertaken by Noricga et al. [42] showed that administration of bilirubin to rats reduces δ-ALA-induced lipid peroxidation, restores GSH content and the activities of the antioxidant enzymatic system to normal levels, providing evidence that bilirubin can act as an efficient antioxidant and free-radical scavenger.
Based on the above information, we adhere to the idea that bilirubin can act in vivo as efficient scavenger of ROS and that bilirubin plays a key physiological role in cytoprotection against oxidant-mediated damage. However, it is important to mention that there is a threshold of cytoprotective effects of bilirubin, as shown in the study undertaken by Miréles et al. [37].
The oxidative stress has also been implicated to contribute to lead-associated tissue injury in the liver, kidney, brain and other organs [13]. Our study showed an increase in MDA content in liver of rats treated with the highest dose of lead [Pb 15], suggesting an increase in lipid peroxidation in hepatic cells. This result is in agreement with the study undertaken by Patra et al. [13], who recorded an increase in MDA content in the liver and the brain of rats subjected to subchronic exposure to lead for a period of four weeks. Pagliara et al. [43] showed that lead-induced liver hyperplasia was followed by apoptosis mediated by oxidative stress in Kupffer cells. The group treated with 5 mg Pb/kg b.w [Pb 5] recorded no changes in MDA content in liver compared to controls, suggesting that this may be due to the short duration of the treatment.