

1 Introduction
Palaeogenomics seeks to reconstruct ancestral genomes from the comparison of the gene content and structure of modern species, thereby allowing the identification and the detailed description of evolutionary mechanisms such as duplications, translocations, inversions and fusions. Paleogenomics can be either based on fossil DNA sequence analysis or, when not available, on large-scale comparative genomics analyses aiming at identifying shared chromosomal structures and shuffling events for ancestor modeling. During the last decade, the constant improvement in the construction of genetic/physical maps as well as in sequencing technologies led to the establishment of numerous plant genome drafts to perform synteny-based modeling of extinct ancestors in cereals. The aim of the present article is to review these recent findings.
2 Synteny-based paleogenomics inference
Several flowering plant genome sequences (grape, cucumber, soybean, Medicago truncatula, poplar, Arabidopsis thaliana, castor bean, papaya, rice, Brachypodium, maize, sorghum, apple; http://synteny.cnr.berkeley.edu/wiki/index.php/Sequenced_plant_genomes) including four grass genomes (rice, maize, sorghum and Brachypodium; see Fig. 1A for genome features) are presently available, while several others are expected to be released soon (e.g., tomato, potato, cassava, banana, peach, date palm, foxtail millet...). Furthermore, the number of high-resolution gene-based genetic maps such as for the Triticeae for example (see Fig. 1A for genome features) is also increasing thereby enabling to perform evolutionary comparative genomics studies even with unsequenced genomes (for review see [1]). The first comparative genetic mapping studies performed in the 1980s were based on restriction-fragment-length-polymorphism (RFLP) markers. They indicated that despite large differences in ploidy level, chromosome number and haploid DNA content the linear order of markers remained largely conserved between grass species over 50–70 million years of divergent evolution [2]. The deduced level of synteny between grasses was however largely overestimated due to artificial redundancy created by undetected intra-genome duplicated loci based on such markers [3]. Development of new alignment parameters and the use of statistical tests [4] allowed identifying and distinguishing orthologous and paralogous gene sets between sequenced genomes and to characterize duplications shared between different genomes and perform ancestor modeling [5].
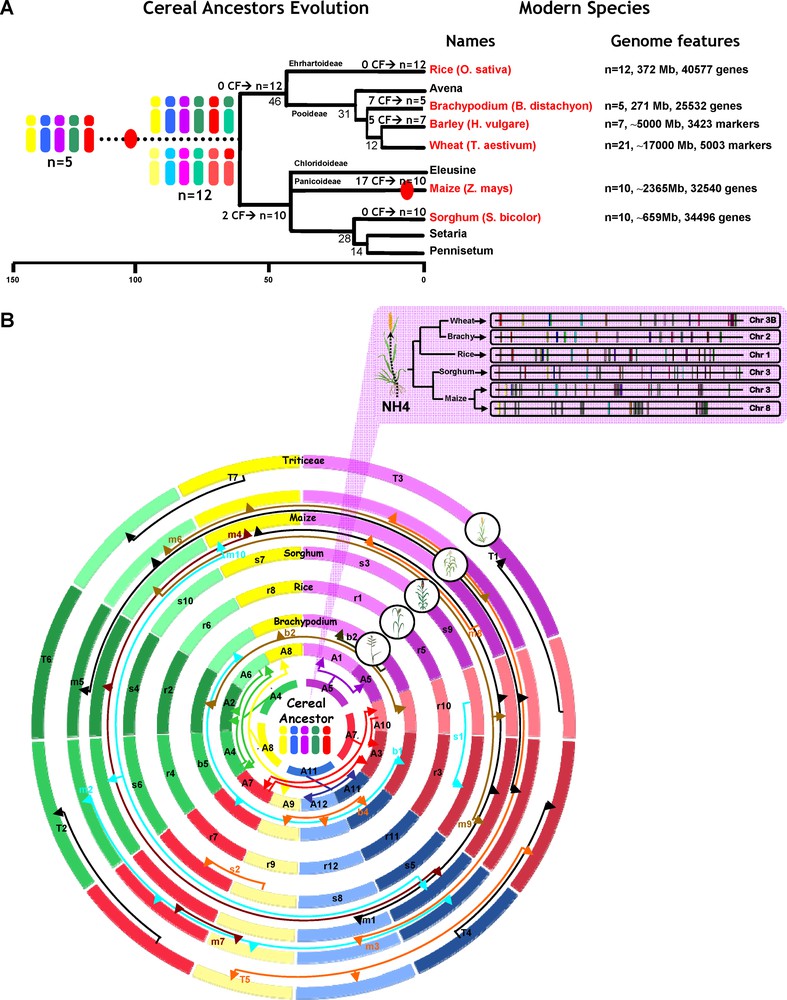
Cereal paleogenomics (updated from [8]).
(A) Cereal genome evolution. Schematic representation of the phylogenetic relationships between grass species. Divergence times from a common ancestor are indicated on the branches of the phylogenetic tree (in millions years, as the underneath scale). Sequenced genomes are indicated in red. Whole genome duplication (WGD) events are illustrated with red circles on the tree branches. The evolution of chromosome numbers of modern species from the ancestral genome structure is indicated with the number of chromosome fusion event (CF). Genome features regarding the six cereal genomes investigated are mentioned at the right side of the figure with the number of chromosome, the physical size and the number of annotated unigenes. (B) Cereal circles. The Triticeae (wheat, barley), maize, sorghum, Brachypodium and rice chromosomes are represented as concentric circles according to their genome size with Brachypodium as the smallest circle at the centre. The five chromosome colors refer to the five ancestral chromosomes (A5 = purple, A7 = red, A11 = blue, A8 = yellow, A4 = green) and the colored arrows indicate the relationships between the 12 intermediate and the five ancestral chromosomes after the whole genome duplication event. The two inner circles represent the n = 12 (A1 to A12) chromosome intermediate ancestor and the n = 5 (A4-5-7-8-11) chromosome ancestral grass genome. The Triticeae, maize (double circle), sorghum, Brachypodium chromosomal fusions are symbolized by colored arrows. The Top inset illustrates a micro-colinearity relationship between wheat (chromosome 3B), rice (chromosome 1), Sorghum (chromosome 3), maize (chromosomes 3-8) and Brachypodium (chromosome 2) for a conserved nitrogen (NH4) use efficiency (NUE) locus. Conserved orthologous genes are illustrated with the same color code and non-conserved genes are shown in grey.
Recent paleogenomic modeling of cereals led to the identification of an ancestral grass karyotype with a minimal physical size of 33.6 Mb that is structured in five protochromosomes and comprises a minimum of 9138 protogenes [6]. The characterization of seven paleoduplications and of the relationships between different conserved regions allowed identifying evolutionary events that shaped the grass genomes since their divergence from a putative ancestor with five chromosomes (Fig. 1A). Fifty to ninety million years ago (mya), the n = 5 ancestor went through a whole genome duplication (i.e. WGD shown as red dot in Fig. 1A) followed by two interchromosomal translocations and fusions that resulted in an n = 12 ancestor intermediate (5 + 5 + 2= 12 chromosomes). The cereal genomes derived from this n = 12 ancestor intermediate were: (i) the rice genome that retained this original chromosome number of 12; (ii) the maize and sorghum genomes, which evolved from the 12 intermediate ancestral chromosomes through two chromosomal fusions (CF in Fig. 1A) and that resulted in a Panicoideae ancestor with n = 10 (5 + 5 + 2-2) chromosomes; (iii) the Triticeae ancestral genome that underwent five chromosomal fusions resulting in a basic number of n = 7 (5 + 5 + 2-5) for the wheat and barley genomes; and (iv) the Brachypodium genome that evolved through seven chromosomal fusions resulting in a basic number of n = 5 (5 + 5 + 2-7) chromosomes. The maize ancestor genome underwent a recent specific whole genome duplication event, resulting in an intermediate with n = 20 chromosomes followed rapidly by at least 17 chromosomal fusions leading to a modern genome structure with ten chromosomes (n = 10 = (5 + 5 + 2–2) × 2–10).
Paleogenomics data between grass genomes allowed extending the model pioneered by Mike Gale's group [7] and to arrange these chromosomes into concentric ‘cereal circles’ with a representation of synteny blocks consisting of five independent and non-redundant linkage groups representing the ancestral cereal genome structure [8]. Thus, including the ancestral genomes as the inner circles and proposing a reconstruction of monocot genome colinearity from ancestors with n = 5 and 12 chromosomes, it becomes possible to identify for any radius of the ‘cereal circles’ the ancestral relationships and origins (e.g., whole genome duplications, breakages, chromosomal fusions) of the different chromosomes in each of the four modern genomes using a simple color code (Fig. 1B). Despite a global conservation of gene content and order between cereal genomes, intergenic regions have been subject to different rates of repeat sequence invasion (Fig. 1B, top inset) so that no orthologous sequences are found in such non-genic sequences.
3 Impact of paleopolyploidy on gene structures and functions
Recent evolutionary studies provide an opportunity to get insight into the genes that operated during the construction of modern plant species, especially those that have been structurally retained during evolution after WGD and that are referred to as ‘deletion-resistant genes’ by Andrew Paterson's group [9]. ‘Deletion-resistant’ gene families correspond to transcriptional regulators that are retained more significantly after whole genome duplication events and for which paralogous copies are maintained, leading to copy number variation. This is in contrast with ‘duplication-resistant’ genes for which one paralogous copy is systematically lost to return to a diploid state. Thus, additional copy number variations of ‘deletion-resistant’ genes with altered/modified functions would continually appear and be selected for during evolution [6].
More than thirty years ago, based on protein sequences from vertebrates Susumu Ohno proposed polyploidisation as a major source of de novo biological pathways inherited from duplicated gene copies [10]. Paleogenomic analyses in cereals confirmed this conclusion [10], leading to the identification of a polyploid common ancestor showing that the actual species have been shaped through several rounds of whole genome duplication followed by numerous chromosomal fusion (Fig. 1A) events leading to the reduction of chromosome number [1,6] in modern species. Yves Van de Peer's group [11] proposed that this paleotetraploidy event, usually considered as a rare and evolutionary dead end phenomenon, might have been the basis for species diversification and survival during the Cretaceous-Tertiary extinction period, 65 mya.
Duplicate genes that persist in multiple copies may diverge by differentiation of sequence and/or function. This process is affected by factors including pathway redundancy and modularity, as well as dosage of gene expression. Overall, recurrent gene or genome duplications generate functional redundancy followed either by pseudogenization, concerted evolution, subfunctionalization or neofunctionalization during the course of genome evolution. The derived functional divergence either from subfunctionalization or neofunctionalization processes between duplicated genes has been proposed as one of the most important sources of evolutionary innovation and plasticity in cereals [12]. Finally, the consequence of polyploidization (reciprocal gene loss, paralogous gene copies, acquisition of novel functions…) could explain how whole genome duplications favored the emergence of new cereal species.
4 Molecular mechanisms driving chromosome number reduction
The comparison of today's species chromosome numbers with their common reconstructed paleoancestor described in the previous sections has led to intense speculation on how chromosomes have been rearranged over time in grasses. Based on the detailed paleogenomics inference of the grass genomes paleohistory from n = 5 to 12 ancestral grass karyotypes, in terms of gene content (i.e. 9138 protogenes catalog), order (i.e. 6045 protogenes with ancestral positions) and rearrangements (duplications, inversions, deletions), sequence intervals were delineated comprising a complete set of synteny break points of orthologous regions from rice, maize, sorghum and Brachypodium genomes [13]. By focusing on these sequence intervals, this work showed that chromosome number variation/reduction from the n = 12 common paleoancestor was driven by non-random centric double strand break repair events. It appeared that centromeric/telomeric illegitimate recombination between non-homoeologous chromosomes led to chromosomal fusions and synteny break points [13].
This analysis suggests that cereal chromosome number reductions from n = 12 to 5 (Brachypodium) and 10 (sorghum and maize) were due to recurrent series of insertions of a chromosome into the centromeric region of another chromosome, i.e. nested chromosome fusions. As the insertion of a complete chromosome into a centromeric region is likely to result in a dicentric chromosome, the derived composite chromosome could only be maintained as functional provided one centromere became either inactive or lost and then identified as a centromere remnant in today's species. This nested-chromosome-fusion pattern of evolution seemed to have involved centromeric/telomeric repeats and was independent of the gene content because there was no evidence that homoeology caused by paleotetraploidy of cereals contributed to or accelerated this process [13]. Finally, rapid and massive structural (i.e. nested chromosome fusions and derived duplicated gene loss) and functional (i.e. neo- or subfunctionalization) changes following whole genome duplications might provide the ability of polyploids to quickly adapt to survive environmental conditions, not tolerated in their diploid ancestors.
Polyploidization followed by diploidization provided a new dynamic pathway for extensive chromosome reshuffling based on chromosome fusions resulting in reduced numbers of chromosomes in today's grass species compared to their common paleotetraploid ancestor. Chromosome fusion boundaries correspond in modern species to regions of abnormal recombination due to mutation and repair activities. These regions could be considered as ‘fragile’ genomic structures as they became hotspots of chromosomal rearrangements such as inversions and repeats invasions. A comparison of the structure of intervals comprising chromosome fusion points allowed proposing that these regions correspond to: (i) meiotic recombination hotspots; (ii) high sequence turn over loci through repeat invasion; and (iii) hotspots of evolutionary novelty that could act as a reservoir for producing adaptive phenotypes [13]. These regions then became preferential sites for additional structural adaptations due to functional competitive advantages. Finally, the modern genomes harbor in their actual chromosomal architecture traces of their evolutionary history notably concerning their specific pattern of ancestral chromosome fusions. A better understanding of the evolutionary processes (duplications and fusions) of plant chromosomes may allow developing in the next future appropriate tools aiming at accelerating and improving the evolution of modern species.
5 Comparative genomic-based tools
Evolutionary comparative genomics and derived paleogenomics studies provide a novel insight into the extent of gene content conservation between cereal genomes that can be used to: (i) define efficient strategies for genetic studies and gene isolation through the design of conserved orthologous marker sets [14]; and (ii) improve the accuracy of gene annotation through the alignment of conserved orthologous genes [4]. This is particularly useful for genomes for which physical map and whole genome sequence are not available yet, such as for the Triticeae.
To support these two applications, we developed an online user-friendly interface, called ‘Plant-Synteny’, to access comparative genomic and paleogenomic data (http://www.clermont.inra.fr/umr1095/PlantSynteny; [1]; Fig. 2A). The website based on ‘Narcisse’ browser (http://narcisse.toulouse.inra.fr/),provides access to the raw data (gene name, sequence, position, and alignment criteria) obtained from the synteny and duplication analyses as well as provides information about the non-redundant ancestral grass gene set that can be used as a platform for the development of COS (Conserved Orthologous Set) markers to support cross genome map-based cloning strategies (see [15] and [16] for synteny-based improvement of grain dietary fiber content and nitrogen use efficiency, respectively in bread wheat). This information can greatly increase the success rate of COS marker design because the selection of markers (genes) is not based on only one genome and applied to another with the risk that the locus of interest may have been subject to lineage-specific rearrangements not shared with the target species. It is shown that ‘Plant-Synteny’ simplify and accelerate the identification of candidate genes using the paleogenomic data allowing efficient translation of structural and functional information from models across grasses, i.e. translational genomics approach [14–16].
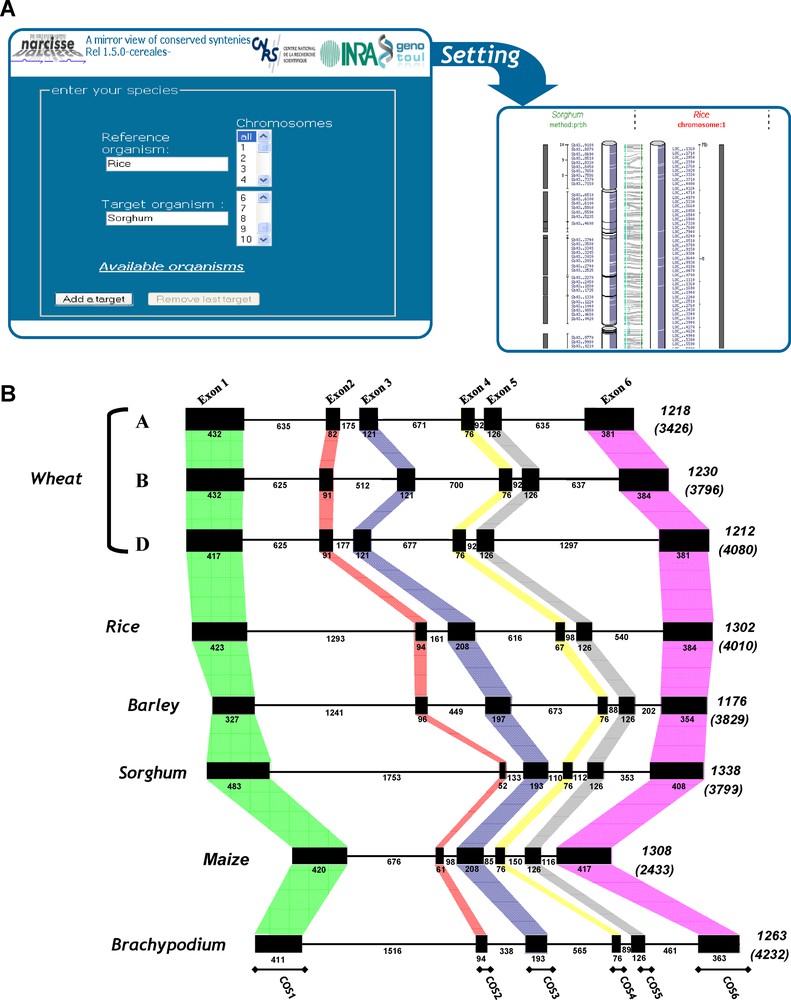
Synteny-based tools (updated from [4]).
(A) Plant Synteny web site output layers – The layer at the left displays the entry page in which genomes (Triticeae, rice, maize, sorghum, Brachypodium) and specific chromosomes can be selected. The layer at the right side illustrates the detail information that can be obtained in selecting any orthologous relationship on the previous layer. (B) Comparative gene annotation. The SPA orthologous genes are illustrated with their exon (black boxes)-intron structures and sizes (numbers) for rice (LOC_Os07g08420), barley (X80068), maize (GRMZM2G015534_T03), Brachypodium (Bradi1g05480), sorghum (Sb02g004590) and the hexaploid bread wheat (SPA-A, -B, -D, respectively FM 242575, FM242576, FM242578) sequences. Total gene and coding sequence length (between brackets) are mentioned at the right end side of the genes.
Finally, the possibility of identifying genes and comparing their sequence within and between genomes provides strong support for genome annotation. Exon structure is conserved between cereals so that comparative genomics can help defining exon/intron boundaries. To this end, the SPA (for Storage Protein Activator) locus region, belonging to BZIP (Basic Leucine Zipper), located on chromosome group 1 in bread wheat, has been chosen as an example because of its importance in controlling seed storage protein accumulation and its sequence conservation in other cereals such as maize (Opaque 2), rice (RISBZ1-5) and barley (BLZ1-2) [17]. Fig. 2B illustrates the conservation of the SPA gene exon structure between rice, maize, barley, hexaploid bread wheat (three homoeologous copies) and Brachypodium. The SPA genes are composed of six exons and all of exon-intron junction sites obey the GT/AG rule of eukaryotic genes. The relative organization of the exons and introns is the same for the others SPA-like bZIP protein genes characterized to date in cereals, i.e. the number of exons and introns is conserved and individual introns occur at about the same sites for the maize, sorghum and barley SPA orthologs. Exon conservation allows the development of intron-spanning PCR-based primers located within conserved exons (see COS markers in Fig. 2B bottom). Our recent work allowed providing a large set of COS markers suitable for grass genome genotyping [14,15]. Such markers are highly transferable (as derived from a robust synteny relationship between cereals), highly polymorphic (as exploiting the largest source of polymorphism within introns, i.e. single nucleotide polymorphisms [SNP]) and co-dominant (as heterozygous haplotypes can be differentiated from homozygous ones) [14,15].
6 Perspectives
Whole genome sequencing projects in grasses including foxtail millet (www.jgi.doe.gov), banana (www.cns.fr), and the perspective of the barley and wheat genome sequences in the next decade (www.barleygenome.org, www.wheatgenome.org) will help to continue refining ancestral genome structure as well as the molecular mechanisms that have shaped the modern cereal species within 50–70 million years of speciation. Future insights into plant paleogenomics will also offer the opportunity to improve synteny-based tools such as COS markers and comparative gene annotation strategies.
Conflict of interest statement
The authors declare no conflict of interest regarding the current manuscript.
Acknowledgements
The work has been supported by grants from the Agence Nationale de la Recherche (Program ANRjc-PaleoCereal, ANR-09-JCJC-0058-01 and EXEGESE-BLE, ANR-05-BLANC-0258-01).