

1 Introduction
Birds and mammals are two monophyletic groups belonging to tetrapod vertebrates. Although these two groups share a major metabolic trait, namely homeothermic regulation, they are not so closely related. Indeed, birds are issued from diapsid1 amniots and are a branch of theropod archosaurs (and then as such the non-extinct part of dinosaurs), whereas mammals are issued from synapsid amniots (Fig. 1). Among living species, the closest relatives of birds are crocodilians, and the sister-group of mammals is the aggregate of reptiles and birds [1].
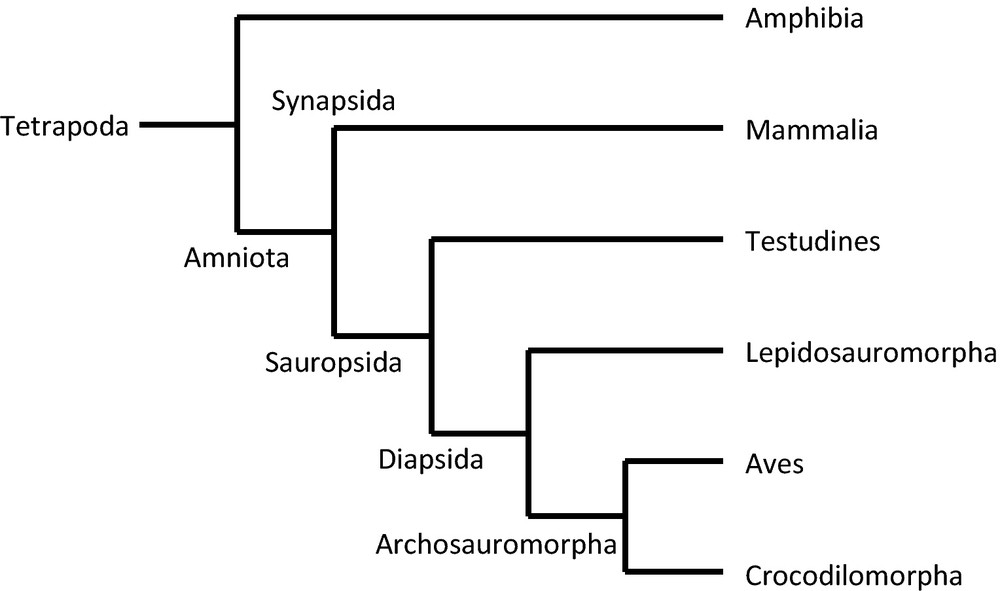
Phylogenetic tree of tetrapodes.
While domestic mammals are distributed in several orders of the mammalian phylogenetic tree (Artiodactyla, Lagomorpha, Carnivora, Perrissodactyla), almost all domestic birds (except pigeons, belonging to Columbiformes) belong to the Galloanserae basal phylogenetic taxon [2], grouping Galliformes (chicken, quail, turkey, guineafowl) and Anseriformes (duck, goose). Recently, many other avian species have been domesticated as pets or ornamental birds, with a focus on the characterization of color variants.
The history of domestic species can be decomposed in three distinct periods of time. The first and the longest one is the evolutionary phase, shared with other species before speciation, and along different routes after speciation. The most recent common ancestor of mammals and birds lived 300 million years ago. The most recent common ancestor between chicken and quail lived 40 million years ago. The second phase starts with the domestication of the species, several thousand years ago, and leading to diversification of domestic breeds. The last and recent phase of intensive selection for production traits applied to a subset of these breeds is shorter, with only few decades. Thus, on an evolutionary scale, domestication and, even more, strong selection for increased production levels represent a very short period of time, which is examined in more details in this review.
2 Domestication and the Neolithic revolution
2.1 Wild Gallus species
Fossil remains of Gallus dated from the Pleistocene were found in the United Kingdom and named Gallus europaeus. Other remains found in Greece were attributed to Gallus aesculapi. These findings were interpreted as an indication of the existence of a western refuge subpopulation of Gallus that may have gone to extinction later on [3]. An Indian refuge subpopulation of junglefowls has been considered to be the major origin of modern Gallus but the contribution of a possible third refuge subpopulation in Asia was not excluded. Consequently, the wild ancestors for modern chickens have all originated from India and South-East Asia. Four living wild species of Gallus are known, namely Gallus gallus, Gallus sonneratii, Gallus varius and Gallus lafayetii that differ by their morphology and their geographical distribution in Asia. G. sonneratii has a grey plumage and is found in the southwest of the Indian continent. G. varius is found only on the island of Java, and is characterized by several morphological peculiarities including a single three-colored wattle (red, yellow, blue), the lack of indentations of the comb, two additional feathers on the tail and a greenish plumage color. It is the most distant from domestic chickens from the viewpoint of morphology. G. lafayetii is found only on the island of Sri Lanka and exhibits an orange-brown color of the breast with a purple spot on the top of the neck and a yellow spot on the comb. G. gallus is the closest to domestic chickens by its morphology and gives fertile offspring after crossing with domestic chickens, whereas crossing between domestic chickens and any of the three other wild species yields very poor hatchability and chick survival. Wild-type plumage color is very similar to that of some ancient breeds (Gauloise dorée, for instance) but wild G. gallus always exhibits blue shanks. This species lives in forests and covers a wide geographic area. It was tentatively subdivided into five subspecies based upon morphological differences and geographical distribution [4], G. gallus gallus (from South Vietnam, Cambodia, Thailand and Laos, with white earlobes), G. gallus spadiceus (from Myanmar, Thailand, Malaysia and China (Yunnan province), with red earlobes), G. gallus murghi (from North-east of India, with white earlobes), G. gallus bankiva (from Java and Sumatra, with red earlobes), and G. gallus jabouillei (from South China and North Vietnam, with red earlobes). The wild ancestor of the domestic chicken was considered for a long time to be the G. gallus species (i.e. the red junglefowl) alone, on the basis of morphology but also on the basis of protein polymorphism [5].
2.2 Domestication centres and migration of domestic chickens
Archeological findings have identified at least two main regions for chicken domestication. Early findings indicated that people from the Indus Valley (Harappan culture) were keeping domestic chickens about 2500 years BC [6]. Seals depicting fighting cocks and clay figurines of chickens were found in the Mohenjo-Daro city. Chicken bones were also found, exhibiting a larger size than those of the wild junglefowl. Leisure and game were probably the first motivation for chicken domestication, which must have taken place in agricultural regions where feed could be made available for chickens without competing with humans.
Much more ancient chicken remains were reported [7] in 16 Neolithic sites in Northern China (∼ < 6000 BC) and in 13 sites in Europe and Western Asia (yielding older bones than those of the Mohenjo-Daro city). Bone length was also larger than that of wild junglefowl, but smaller than that of domestic breeds. This study proposed that these bones originated from domestic chickens, since wild junglefowl was never reported to have inhabited North of China.
According to archeological findings and literature from the early 20th century, chickens reached Europe along two main trading routes: a northern route through China and Russia and a southern route through Persia and Greece [8]. An alternative scenario reports that the two routes started from Iran, one via the Mediterranean Sea, and the other via the Black Sea. The Mediterranean type of chicken is considered to be the most ancestral type of domestic chickens in Europe. Records from Greek life and myths support the fact that chickens reached Greece by 700 BC. At that time, some morphological variants can already be identified, such as the Rose Comb phenotype (R mutation) and the Muffs and Beard phenotype (MB mutation). Poultry keeping was well developed under the Romans, who adopted chickens as a food source but still used them for leisure, religion and divination.
Introduction of chickens in Africa is poorly documented. Excavation of chicken remains dated to about 500–800 AD was reported in Mali [9] but chickens may have been present in Africa well before [10]. A linguistic approach used to trace the reference to chickens in West Africa identified three major roots, suggesting that three introductions may have taken place, two across Central Africa from the east and one across the Sahara from the north [9]. In 1635 AD, the finding of chickens with black meat (typical of fibromelanosis, FM mutation) in Mozambique suggested direct introductions from India.
More recent archeological studies focused on Oceania and South America. Chicken bones have been recovered in several archeological sites in the Reef and Santa Cruz Islands in Near Oceania, one of which could be dated to 1400–900 BC [11]. The origin of Oceanic chickens is southeast Asia, but the distribution of chicken remains does not make possible to trace the transport from their mainland origin across archipelagos into Near Oceania. Regarding South America, a debate is still going on as to the presence of chickens in the pre-Columbian period, which would have come from Polynesia. This hypothesis has been supported by some studies [12] but questioned by others [13]. Direct dating of two chicken bones found in the El-Arenal site of Chile clearly showed their pre-Columbian origin. Furthermore, the possible dispersal mechanisms of chickens from Polynesia to the Americas were studied by computer simulations of seafaring, which showed that the probability of vessels reaching South America could reach 40% in the favorable season [14] whereas return voyages from Chile were not possible most times of the year. Thus, introduction of domestic chickens in South America is likely to have occurred from Polynesia. This can be related to the fact that the blue-egg shell mutation, O, is typical of the Araucana breed from South America and is also found in Chinese breeds but not in European breeds.
2.3 Molecular studies
2.3.1 Mitochondrial DNA
Most molecular studies of chicken domestication have used the polymorphism of mitochondrial DNA (mtDNA). Initial studies concluded to a monophyletic origin of the domestic chicken [15,16]. An extensive survey conducted on 834 mtDNA sequences from domestic chickens and 66 mtDNA sequences from four red junglefowl subspecies (mainly G. g. spadiceus and G. g. gallus and also G. g. jabouillei and G. g. bankiva) identified a total of 169 different haplotypes clustered in nine highly divergent clades, named A to I, seven of them including both wild and domestic individuals [17]. Clade E was the most ubiquitous, dominating in Europe, Middle East and India. A regional distribution was observed for the other clades. Here, A and B clades were mainly distributed in South China and Japan; C clade was mainly found in chickens from Japan and Southeast China; while F and G clades were only found in the northern part of Southeast China (Yunnan province). Clade D could be associated to the distribution of gamebirds, used for cockfighting. Clade D was the most frequent in red junglefowls. Clade H was found only in red junglefowls and clade I was mainly present in Vietnam. No breed-specific matrilineal clade was observed. The distribution patterns and expansion signatures supported the theory of multiple origins of domestication in South and Southeast Asia [17]. Additional data confirmed that domestication had occurred independently in different locations of Asia including India and established G. g. murghi as the most important ancestor of Indian domestic chickens [18]. Both studies [18,19] supported the obsoleteness of the subspecies status given to red junglefowls, except for G. g. bankiva that could be considered as a separate species. A later study with additional sampling concluded that clade D was also the second most frequent clade in India, after clade E and confirmed the geographic pattern of haplogroup distribution in Asia [19]. More recently, archeogenomic studies conducted in Oceania and South America [11–13] revealed also the ubiquitous presence of the E haplotype, which suggests that worldwide diffusion of domestic chickens took place from India.
Furthermore, interspecies hybridizations between the red junglefowl and G. sonneratii on one hand, and between G. sonneratii and G. lafayetii on the other hand, were suggested [20] by the analysis of the whole sequence of mtDNA and five autosomal loci (intron 9 of ornithine transcarbamylase, OTC, and four Chicken-Repeat-1 elements, CR1). Indeed, two grey junglefowls clustered in a clade with the red junglefowl and domestic chickens according to mtDNA. Another grey junglefowl clustered also with red junglefowl and domestic chickens according to OTC, but this pattern was not observed with CR1 elements. It should be noted that gene flow might still take place between wild junglefowls and village chickens in some regions [21]. Molecular markers and sequence analysis can be used to assess the genuine origin of junglefowl samples.
2.3.2 Whole genome approaches
The whole genome sequence of an inbred red junglefowl was published [22] and a new assembly has recently been made available [23] (http://www.ensembl.org/index.html; http://genome.ucsc.edu/). Knowing the genome sequence has triggered new and efficient approaches to explore the genome as an archive of chicken history and to search for footprints of domestication. Furthermore, the chicken genome exhibits a high rate of polymorphism, with about 1 single nucleotide polymorphism (SNP) every 200 bp (base pair) as determined by resequencing of the genome of three different breeds, which provides powerful tools for association studies [24].
2.3.2.1 Study of a simple phenotype: the yellow skin mutation
A recent study of the genetic determinism of the yellow skin phenotype yielded unexpected new insight on chicken domestication. Yellow skin is a phenotypic trait extensively used in many commercial lines, either layers or broilers, which is controlled by an autosomal recessive mutation. Mapping of this gene on a microchromosome (GGA24) led to the identification of a candidate gene BCDO2 coding an enzyme involved in the metabolism of carotenoids [25]. Further studies, aimed at identifying the identical-by-descent haplotype diagnostic of yellow skin breeds as compared to white skin breeds, revealed a surprisingly high degree of sequence divergence of 0.8% between yellow skin chickens and the reference sequence of G. gallus across the region of 24 kb including the BCDO2 gene. Resequencing this region in G. sonneratii showed that the yellow skin haplotype originated in fact from this species and not from G. gallus. Yellow skin breeds clustered with G. sonneratii in a phylogenetic analysis based upon the BCDO2 sequence, whereas white skin breeds clustered with G. gallus (Fig. 2). The only explanation for these results is that the yellow skin mutation in domestic chickens originates from G. sonneratii instead of G. gallus [25], which demonstrates a hybrid origin of the domestic chicken. Two introgression events probably took place as the White Leghorn breed and the Chinese Shek-ki breed are found on different branches of the phylogenetic tree (Fig. 2).
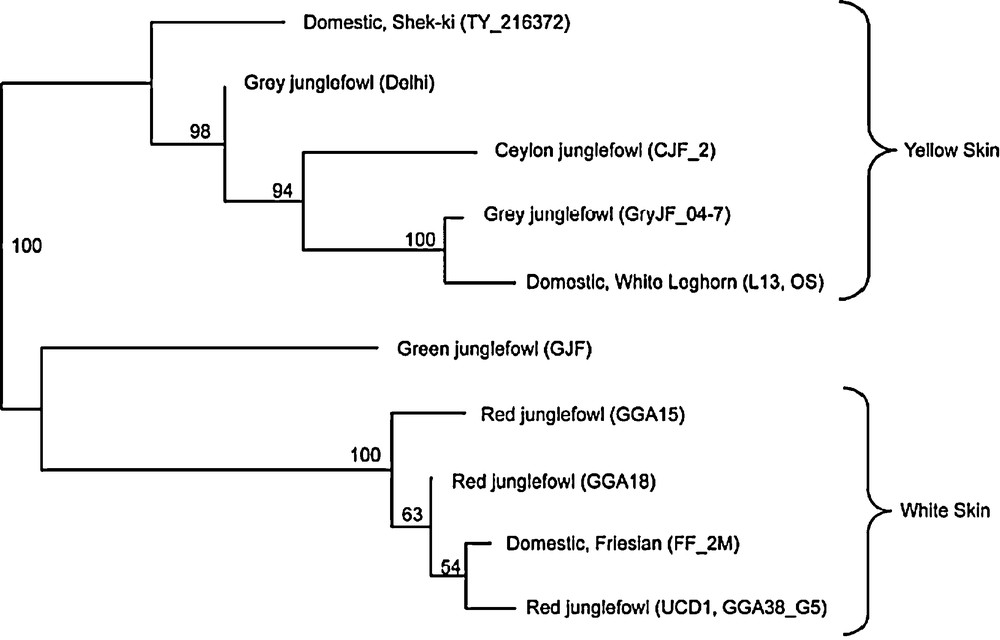
Phylogenetic tree of domestic chicken breeds and Gallus species established on the haplotype sequence of BCDO2 gene [25].
The possibility that other genomic regions have been inherited from G. sonneratii is still an open question, which could be answered by whole genome comparisons between wild species and domestic chickens.
2.3.2.2 Genome comparisons between wild ancestor and domestic chickens
The development of genetic maps in livestock species has made possible to identify genomic regions controlling the genetic variability of complex traits used in current breeding programs. The joint analysis of marker genotypes and performance data produces a list of quantitative traits loci (QTL), i.e. genomic regions associated with a significant share of the genetic variability of a trait. This may be done either within breed or in crossbred designs between breeds.
Through linkage analysis with microsatellite markers, a crossbred design was set up between the red junglefowl and a white-egg layer to identify QTL regions explaining important phenotypic differences between the wild ancestor and the domestic chicken selected for egg production [26]. This study identified some chromosomal regions susceptible to have played a major role in the switch from a wild junglefowl to a domestic chicken. One particular region on chromosome 1 was found to have a major effect on growth as well as pleiotropic effects on feed consumption, egg production and behavior. Yet, the resolutive power of such studies is limited by the size of the experimental design and the density in markers across the whole genome. Therefore, important work is still needed to identify the gene(s) likely to be responsible for the QTL effect.
Whole genome resequencing offers a very powerful approach that was recently applied in chickens by comparing the G. gallus wild population to a pool of commercial lines selected either for growth or egg production [27]. The rationale of the study was to compare the allelic frequency of polymorphic sites at the nucleotide level between chicken populations of different selection history, either layers or broilers. The analysis could be done along the whole genome with sliding windows of 40 kb (kilobase). The comparison conducted between all red junglefowls and all domestic chickens may be used to detect signatures of domestication in the genome, i.e. positions in the genome where allelic frequency has dramatically changed between wild fowls and domestic chickens. This approach was validated with the BCDO2 locus, which exhibited a very significant difference in allelic frequencies between G. gallus (white skin) and domestic (yellow skin) chickens. Three other chromosomal regions exhibited even stronger levels of differentiation between wild and domestic groups. Thus two were found in non-coding regions, either on chromosome 1 or on chromosome 2, and the third one matched to the gene coding the thyroid stimulating hormone receptor (TSHR) on chromosome 5 in which a non-synonymous nucleotide change in a strongly conserved region differs between a wild and a domestic allele. The functional consequences of this mutation are still poorly understood. It could affect the regulation of the thyreotroph axis in birds and may be related to reproduction traits or seasonality. Interestingly, a QTL regarding broodiness trait has been mapped in the very same region of chromosome 5 in a cross between the Chinese Silky breed and a commercial layer line [28]. The domestication signature identified on chromosome 1 did not match with the position of the major growth QTL previously detected in the cross between a red junglefowl and a white-egg layer and was considered to be a putative domestication QTL [26]. It is noted that these approaches are quite different since QTL detection is only relevant for the measured traits in a specific design, whereas the whole genome approach is based on the cumulated effects of different selection histories between populations. The challenge is now to understand the mechanisms underlying the 21 domestication signatures [27], which will require a good quality of gene annotation in the chicken.
3 Diversification of chicken breeds after domestication
3.1 Typology of domestic populations according to selection history
Genome diversity of current domestic chickens is a result of the founder effects at the time of domestication, the long-term domestication process, subsequent breed differentiation and recent strong selection for production (since 1950). The cumulated effect of domestication and subsequent selection by man has yielded an impressive phenotypic diversification of the chicken, both at the level of morphology and physiology. Four types of domestic populations may be considered depending on their selection history, namely traditional populations, standardized breeds, selected lines (either experimental or commercial) and experimental inbred lines.
Traditional populations represent the first step following domestication, based upon preferential breeding of birds exhibiting specific morphological features regarding comb size, plumage color or plumage distribution. Such variants generally have a negative selective value in the wild, but can be maintained in a controlled environment, where birds are more protected from predators. Such morphological variants can be found in ancient artifact showing chickens. In addition, cultural values could be attributed to typical features of the bird, which may explain the diversity of morphology observed within domestic chickens. As a consequence, populations of domestic chickens differ from the wild junglefowl by a large phenotypic variability due to the accumulation of mutations. Important color variability is generally observed in village chickens of Africa and Asia, as described for instance in Benin [29]. These populations still exhibit a high heterozygosity level for microsatellite markers, in the range of 0.55 to 0.7 [30], similar to values found for the wild G. gallus (Fig. 3).
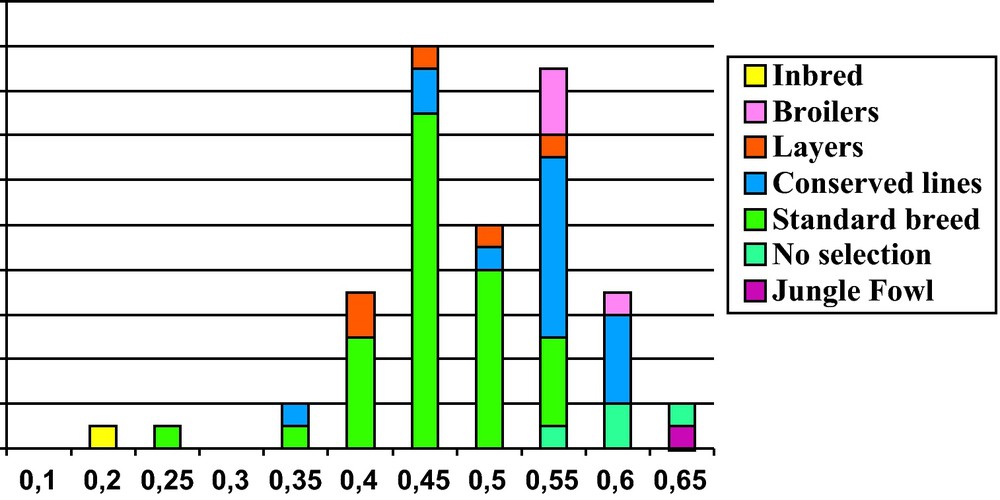
Distribution of heterozygosity values for 29 microsatellite markers in 65 populations with different management histories (colors) in chickens (data extracted from [30]).
Subsets of morphological variants were characteristic of the foundation of specific breeds, which led to the definition of a breed standard, based on an accurate phenotypic description. For instance, French breeds of the Burgundy region were characterized in the Middle Age by the autosomal barring phenotype consisting in a black stripe on a silver or gold feather color, due to the combination of two mutations, PG for the pattern gene and DB for the dark brown gene. Breeds of the Normandy region were characterized in the 16th century by variation in feather distribution with a crest on the head (CR mutation), the presence of muffs and bear (MB mutation) and with a typical comb shape, that is, the duplex comb. The buttercup mutation, which gives a crown-shape to the comb, is only found in three breeds which history can be traced back to the royal family who ruled Sicily as well as Normandy in the Middle Age. Game birds used for cockfighting exhibited a high frequency of the pea-comb mutation, P, which markedly decreases comb and wattle size. Such breeds fixed for a few phenotypic traits are still kept by fancy breeders in many countries nowadays. They represent a rich resource for genomic studies aimed at mapping and identifying causal mutations for their phenotypic features. They may also have been selected for performance, with low-selection intensity, most of them are dual-purpose breeds, showing moderately higher growth rate and egg production as compared to the wild junglefowl. Depending on the occurrence of bottlenecks in their history, these breeds may exhibit more or less variability at the genome level, as assessed with microsatellite markers. Heterozygosity for microsatellite markers could vary from 0.28 to 0.62 among European standard breeds [30].
The development of the quantitative genetics theory in the 20th century was applied with a high efficiency to develop experimental lines and commercial lines. Two categories of commercial lines have been selected, the broiler lines for meat production and the layer lines for egg production. These lines were derived from standard breeds. Experimental lines are generally used as model genotypes for research. An industrial broiler chicken reaches the weight of 2 kg in 35 days, a commercial layer reaches hardly 2 kg at the adulthood and lays 300 eggs per annum, whereas the red junglefowl does not reach the weight of 2 kg and does not lay more than 50 eggs per year. Industrial lines are kept in a highly controlled environment. Intensive selection started approximately 60 years ago (1 generation/year). One can thus speak of a rapid adaptation of the chicken to an environment more and more modified by man. This phenomenon results now in a greater erosion of genetic diversity in layers than in broilers. Assessment of genetic diversity of these lines with microsatellite markers [21,30] showed lowest values for white-egg layers (average heterozygosity He of 0.27–0.40), moderately low values for brown-egg layers (He from 0.41 to 0.50) and moderate to high values for broilers (He from 0.45 to 0.60). High density SNP genotyping indicates that 50% of ancestral genetic diversity is lost in commercial lines [31]. Currently, selection objectives are being revisited to answer market demands in terms of welfare and disease resistance, in addition to the usual breeding goals for yield and feed efficiency.
A global analysis of the genetic diversity of 65 chicken populations, differing by their selection histories and current management systems, identified six main clusters that corresponded to geographical origins and selection histories [32]. Multi-locus clustering was implemented with the STRUCTURE software [33] and applied to the genotyping data from 29 microsatellite markers. Geographical origins were either Asian or European and selection histories distinguished layers, either brown-egg or white-egg, and broilers. The genetic variation between populations was found to account for about 34% of the total genetic variation, 11% of the variation being between clusters and 23% being between populations within clusters. More recently, this approach was extended to a set of 2789 individuals from 85 populations, including African populations [34]. The highest probability was obtained for a clustering in three groups corresponding to an Asian cluster (As), a European cluster (Eu) including also the commercial white-egg layers, and an African/South European cluster (AfSe) including also the commercial broilers and brown-egg layers. Unbiased heterozygosity was highest in the As cluster (0.57), moderate in the AfSe cluster (0.54) and lowest in the Eu cluster (0.41). The Eu cluster also exhibited the largest deviation from Hardy-Weinberg equilibrium and the highest differentiation between populations within a cluster. The large number of fancy breeds kept in Europe, which exhibit important founder effects, can explain this result. A set of six breeds could not be clustered, whatever the number of groups tested (until 15), which may deserve further characterization with a higher marker density.
Such an overview could still be completed with South-American populations to be used for conservation issues. It makes possible to identify population subsets contributing the most to the global diversity of domestic chickens. It can also provide sampling rules for further molecular studies with high-density SNP chips.
3.2 Molecular characterization of morphological mutations
Domestic animals exhibit a wide range of coat or plumage color variation, whereas homogenous pigmentation is generally the case in wild species. Knowing the chicken genome has boosted the molecular identification of plumage color mutations, as well as other morphological mutations that have been selected along the domestication process. Since breeds separated for centuries may share a given mutation, genotyping a candidate region in subsets of breeds, carrying or not the mutation, makes the Identical-By-Descent mapping approach very efficient to narrow the mapping interval. Causal mutations have now been identified for about ten morphological mutations in chickens. A range of molecular defects has been observed documenting that nucleotide substitutions or short deletions in the coding sequence may cause a loss of function, intronic mutations may cause splicing defects, and indel polymorphisms or copy number variation may alter the regulation of gene expression. A few emblematic examples will be discussed here, illustrating homology between species for color genes.
The MC1R gene codes the melanocortin receptor 1 and controls the balance between eumelanin (black) and phaeomelanin (red) pigments over the body. Several nucleotide substitutions identified in the chicken MC1R gene are associated with different phenotypes, from the dominant extended black to the recessive yellow, with several intermediates [35–37]. A correlation between MC1R polymorphisms and coat color variation has also been observed in domestic mammals [38]. Interestingly, the study of MC1R polymorphism across a range of species of wild birds showed a correlation between the rate of amino acid change at this locus and the degree of sexual dichromatism, which has led to the conclusion that this gene is under sexual selection in birds [39]. Curiously, the same nucleotide substitution yielding to a constitutively active receptor has been associated to black color in mice and chickens [35], but also in quails [40], in the bananaquit [41], the lesser snow goose [42], and in swans [43]. It seems that MC1R has very few, if any, pleiotropic effects on functions other than pigmentation, which contributes to its high degree of variability.
The MLPH gene codes a structural protein of the melanosome. The same nucleotide substitution in exon 1 has been found in chickens carrying the recessive ‘lavender dilution’ mutation [44] and in the Griscelli III syndrome in humans. This site is also involved in the deletion causing the leaden diluted phenotype in mice. Mutations of this gene have also been associated with a recessive diluted phenotype in cats, dogs and mink. This example underlines the high degree of homology of color genes between mammals and birds and contributes to identify precise positions in the coding region with a high functional importance. Domestication has apparently favored diluted phenotypes for aesthetic and cultural purposes.
More complex mechanisms may also be encountered in color mutations such as the intronic retroviral insertion in the TYR gene causing recessive white plumage in chickens [45]. Most of the literature on the TYR gene deals with albinism and mutations in the coding sequence. In the chicken, the insertion of a full retrovirus of 7.4 kb in intron 4 modifies the gene expression pattern in a tissue-specific manner: a truncated mRNA is found in the skin, whereas a normal mRNA is found in the eye, thus explaining the normal pigmentation of the eye with a totally white plumage. This mutation has been favored in commercial lines of broilers, because the lack of pigmented feathers contributes to a better appearance of the carcass.
Other morphological mutations selected along domestication involve featherless appendages such as the comb. The Pea-comb dominant mutation is severely reducing comb size, which appears to be a selective advantage for cockfighting as well as for adaptation to cold climate. This mutation has been favored in a rather large number of chicken breeds. It was mapped to a small interval on chromosome 1 containing only one gene, SOX5, which plays a role in chondrocyte development and the production of extracellular matrix. Fine mapping of the mutation led to the identification of a copy number variation of an incomplete LINE element in a conserved non-coding sequence [46]. As a consequence, the gene is abnormally expressed in the cellular layers underneath the comb region at days 7 and 8 of embryo development and disturbs comb morphogenesis. This is a rare example showing the functional importance of conserved non-coding regions and the first occurrence of a viable mutation involving the SOX5 gene.
3.3 Selection footprints following domestication
The same approach used to detect domestication footprints in the genome was also quite successful to identify the candidate gene underlying a QTL responsible for the difference in body weight between two lines divergently selected on growth, which had been mapped on chicken chromosome 13 [47]. Whole genome resequencing of these chicken lines has revealed a deletion removing almost all the SH3RF2 gene associated with the allele responsible of increased growth [27]. The physiological consequence of this deletion is still unknown. Yet, as SH3RF2 is normally expressed in brain and muscle, it probably plays a role in the limitation of muscle development. This result underlines the power of the whole genome approach to provide a more accurate identification of genome regions involved in selection response compared to QTL detection experiments.
4 Conclusion
The genome is an archive of population history, and the development of genomics and high throughput sequencing is now providing efficient tools to improve our understanding of chicken domestication. These tools are all the more useful that large scale sampling of chicken populations is available across continents, and across time periods with ancient DNA studies. The fact that ancestor species are still available for sampling in Asia is also a very useful contribution to the study of chicken domestication. The identification of causal mutations for major differences in color or morphology between domestic chickens and wild Gallus is progressing rapidly and provides interesting case studies of the genetic determinism of complex phenotypes. Molecular mechanisms explaining major shifts in chicken domestication and selection are starting to be understood but very few results have been obtained yet regarding traits such as behavior, growth or reproduction, which have been modified by domestication and subsequent selection. It is hoped that combining resequencing with careful phenotyping of current populations and functional analysis of candidate genes will allow major progress in that direction.
Conflict of interest statement
The authors declared no conflict of interest.
1 Diapsid and synapsid refer to cranial anatomical differences.